
The Novel H10N3 Avian Influenza Virus Triggers Lethal Cytokine Storm by Activating Multiple Forms of Programmed Cell Death in Mammalian Lungs

 ,
,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
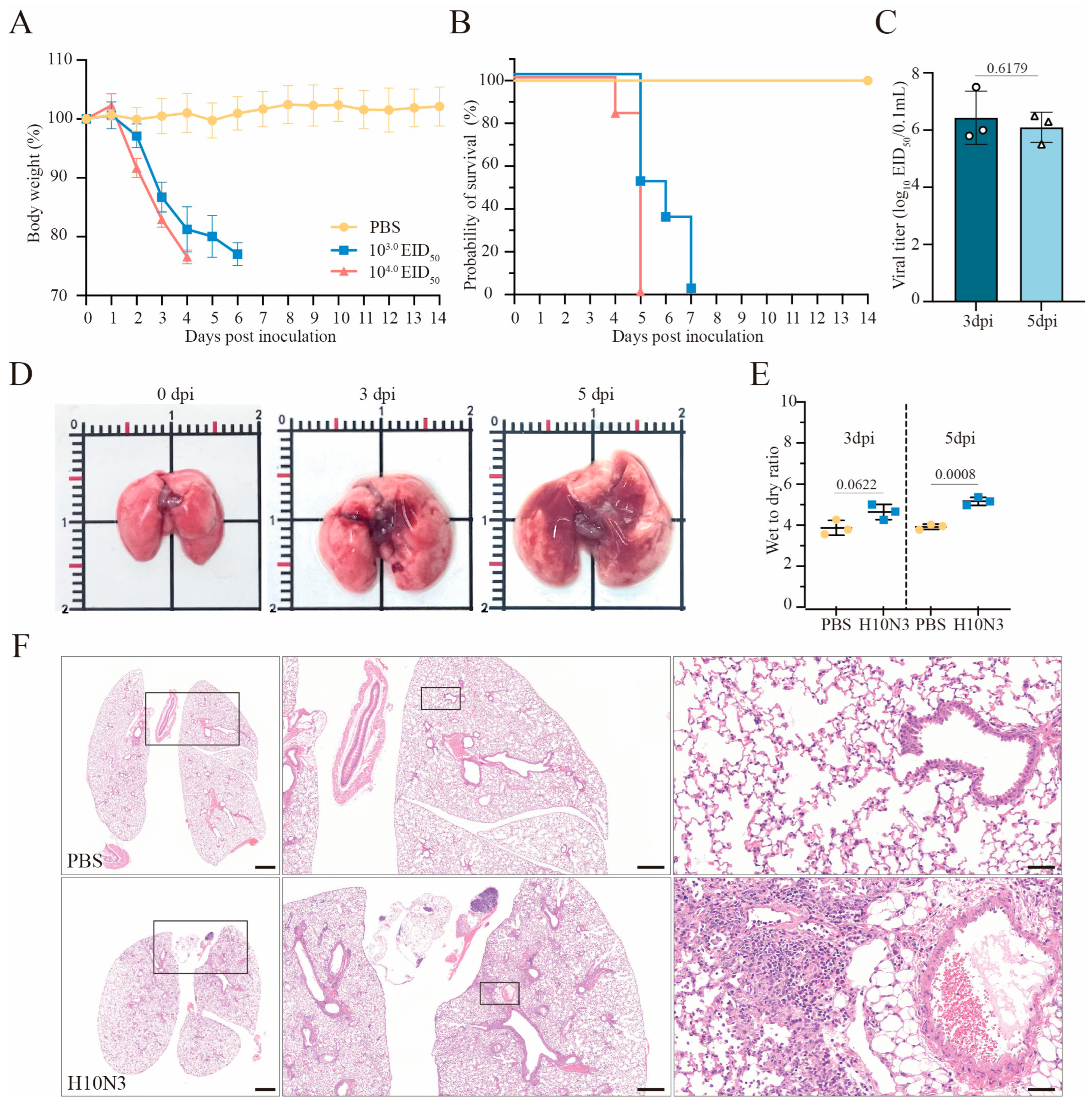
2.1. H10N3 Infection Causes Mouse Mortality and Severe Pulmonary Damage
2.2. H10N3 Infection Induces Significant Cytokine Storm
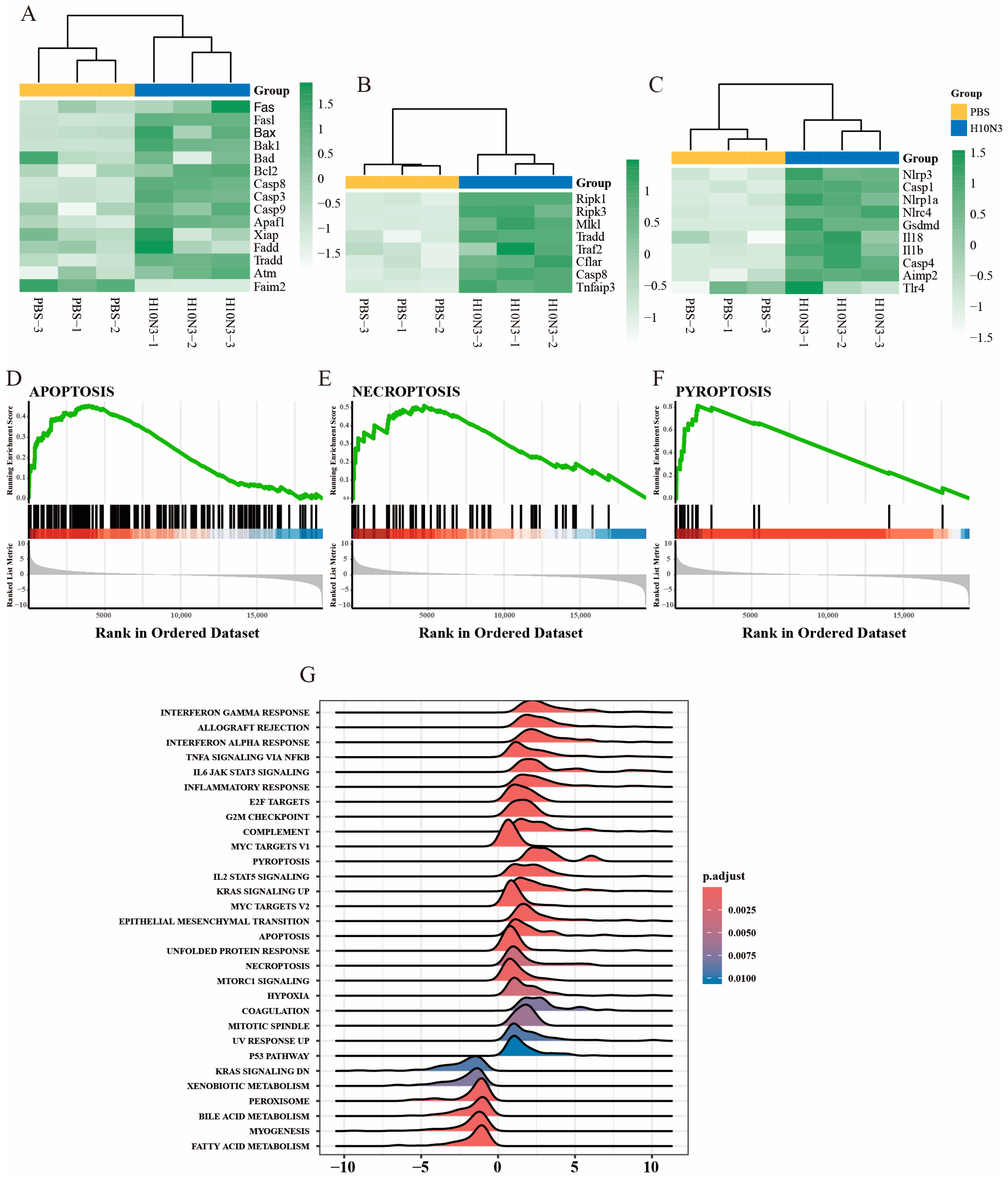
2.3. H10N3 Infection Activates Multiple Forms of Cell Death
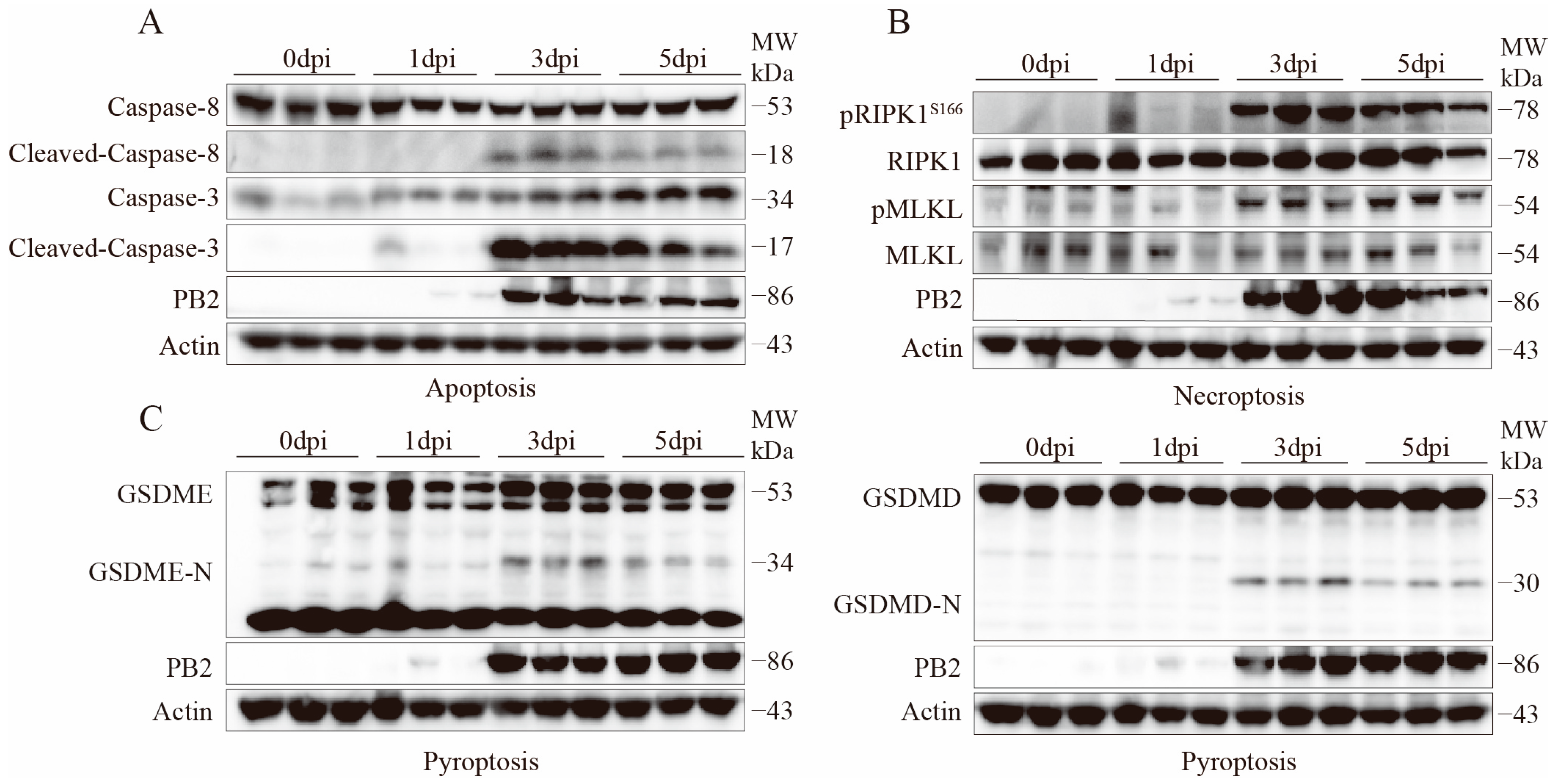
2.4. Activation Validation of Cell Death Marker Proteins
3. Discussion
4. Materials and Methods
4.1. Virus
4.2. Mouse Challenge Experiments
4.3. RNA Detection, Library Construction, and Sequencing
4.4. Gene Set Enrichment Analysis (GSEA)
4.5. Assessment of Inflammatory Mediators in Bronchoalveolar Lavage Fluid
4.6. Antibodies
4.7. Western Blot
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meng, F.; Chen, Y.; Song, Z.; Zhong, Q.; Zhang, Y.; Qiao, C.; Yan, C.; Kong, H.; Liu, L.; Li, C.; et al. Continued evolution of the Eurasian avian-like H1N1 swine influenza viruses in China. Sci. China Life Sci. 2023, 66, 269–282. [Google Scholar] [CrossRef]
- Cui, P.; Shi, J.; Yan, C.; Wang, C.; Zhang, Y.; Zhang, Y.; Xing, X.; Chen, Y.; Zhang, J.; Liu, L.; et al. Analysis of avian influenza A (H3N8) viruses in poultry and their zoonotic potential, China, September 2021 to May 2022. Eurosurveillance 2023, 28, 2200871. [Google Scholar] [CrossRef] [PubMed]
- Xing, X.; Shi, J.; Cui, P.; Yan, C.; Zhang, Y.; Zhang, Y.; Wang, C.; Chen, Y.; Zeng, X.; Tian, G.; et al. Evolution and biological characterization of H5N1 influenza viruses bearing the clade 2.3.2.1 hemagglutinin gene. Emerg. Microbes Infect. 2024, 13, 2284294. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shi, J.; Deng, G.; Guo, J.; Zeng, X.-Y.; He, X.; Kong, H.; Gu, C.; Li, X.; Liu, J.; et al. H7N9 influenza viruses are transmissible in ferrets by respiratory droplet. Science 2013, 341, 410–414. [Google Scholar] [CrossRef]
- Liu, K.; Qi, X.; Bao, C.; Wang, X.; Liu, X. Novel H10N3 avian influenza viruses: A potential threat to public health. Lancet Microbe 2024, 5, e417. [Google Scholar] [CrossRef] [PubMed]
- Eisfeld, A.J.; Biswas, A.; Guan, L.; Gu, C.; Maemura, T.; Trifkovic, S.; Wang, T.; Babujee, L.; Dahn, R.; Halfmann, P.J.; et al. Pathogenicity and transmissibility of bovine H5N1 influenza virus. Nature 2024, 633, 426–432. [Google Scholar] [CrossRef]
- Gu, C.; Maemura, T.; Guan, L.; Eisfeld, A.J.; Biswas, A.; Kiso, M.; Uraki, R.; Ito, M.; Trifkovic, S.; Wang, T.; et al. A human isolate of bovine H5N1 is transmissible and lethal in animal models. Nature 2024, 636, 711–718. [Google Scholar] [CrossRef]
- Lin, T.-H.; Zhu, X.; Wang, S.; Zhang, D.; McBride, R.; Yu, W.; Babarinde, S.; Paulson, J.C.; Wilson, I.A. A single mutation in bovine influenza H5N1 hemagglutinin switches specificity to human receptors. Science 2024, 386, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Moncla, L.; Dudas, G.; VanInsberghe, D.; Sukhova, K.; Lloyd-Smith, J.O.; Worobey, M.; Lowen, A.C.; Nelson, M.I. The global H5N1 influenza panzootic in mammals. Nature 2025, 637, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Yang, J.; Wang, L.; Ran, L.; Gao, G.F. Ecology and evolution of avian influenza viruses. Curr. Biol. 2024, 34, R716–R721. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Sun, H.; Gao, F.; Luo, K.; Huang, Z.; Tong, Q.; Song, H.; Han, Q.; Liu, J.; Lan, Y.; et al. Human infection of avian influenza A H3N8 virus and the viral origins: A descriptive study. Lancet Microbe 2022, 3, e824–e834. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.F. From “A”IV to “Z”IKV: Attacks from Emerging and Re-emerging Pathogens. Cell 2018, 172, 1157–1159. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.J.; Wu, Y.; Bi, Y.; Shi, W.; Wang, D.; Shi, Y.; Gao, G.F. Emerging HxNy Influenza A Viruses. Cold Spring Harb Perspect Med. 2022, 12, a038406. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.C.; Weng, S.S.; Xue, F.; Wu, X.; Xu, T.M.; Zhang, W.H. First human infection by a novel avian influenza A(H7N4) virus. J. Infect. 2018, 77, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Qiu, H.; Hao, S.; Zhu, F.; Huang, Y.; Xu, K.; Yu, H.; Wang, D.; Zhou, L.; Dai, Q.; et al. Human Infection with an Avian-Origin Influenza A (H10N3) Virus. N. Engl. J. Med. 2022, 386, 1087–1088. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, Z.; Wang, M.; Pan, X.; Jiang, X. Second Identified Human Infection With the Avian Influenza Virus H10N3: A Case Report. Ann. Intern. Med. 2023, 176, 429–431. [Google Scholar] [CrossRef]
- WHO. Human Infection with Avian Influenza a Viruses 2024. Available online: https://www.who.int (accessed on 31 December 2024).
- Gao, R.; Zheng, H.; Liu, K.; Ji, Z.; Cai, M.; Gu, M.; Hu, J.; Liu, X.; Hu, S.; Wang, X.; et al. Genesis, evolution and host species distribution of influenza A (H10N3) virus in China. J. Infect. 2021, 83, 607–635. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Ding, P.; Pei, Y.; Gao, R.; Han, W.; Zheng, H.; Ji, Z.; Cai, M.; Gu, J.; Li, X.; et al. Emergence of a novel reassortant avian influenza virus (H10N3) in Eastern China with high pathogenicity and respiratory droplet transmissibility to mammals. Sci. China Life Sci. 2022, 65, 1024–1035. [Google Scholar] [CrossRef]
- D’Elia, R.V.; Harrison, K.; Oyston, P.C.; Lukaszewski, R.A.; Clark, G.C. Targeting the “cytokine storm” for therapeutic benefit. Clin. Vaccine Immunol. 2013, 20, 319–327. [Google Scholar] [CrossRef]
- Guo, X.J.; Thomas, P.G. New fronts emerge in the influenza cytokine storm. Semin. Immunopathol. 2017, 39, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhou, Y.H.; Yang, Z.Q. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell. Mol. Immunol. 2016, 13, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R. The role of cytokine responses during influenza virus pathogenesis and potential therapeutic options. Curr. Top. Microbiol. Immunol. 2015, 386, 3–22. [Google Scholar] [PubMed]
- Karki, R.; Kanneganti, T.D. The ’cytokine storm’: Molecular mechanisms and therapeutic prospects. Trends Immunol. 2021, 42, 681–705. [Google Scholar] [CrossRef]
- Zhi, Y.; Wei, J.; Liu, Z.; Zhang, Q.; Zhang, T.; Hu, G. Inhibitory effects of Belamcanda extract on inflammatory response and antiviral mechanism in H9N2 Avian influenza virus: Insights from in vitro and in vivo studies. Poult. Sci. 2024, 103, 103885. [Google Scholar] [CrossRef] [PubMed]
- El-Hack, M.E.A.; El-Saadony, M.T.; Alqhtani, A.H.; Swelum, A.A.; Salem, H.M.; Elbestawy, A.R.; Noreldin, A.E.; Babalghith, A.O.; Khafaga, A.F.; Hassan, M.I.; et al. The relationship among avian influenza, gut microbiota and chicken immunity: An updated overview. Poult. Sci. 2022, 101, 102021. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, X.; Wang, X. The immune system of chicken and its response to H9N2 avian influenza virus. Vet. Q. 2023, 43, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Subbarao Malireddi, R.K.; et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168.e17. [Google Scholar] [CrossRef] [PubMed]
- Bedient, L.; Pokharel, S.M.; Chiok, K.R.; Mohanty, I.; Beach, S.S.; Miura, T.A.; Bose, S. Lytic Cell Death Mechanisms in Human Respiratory Syncytial Virus-Infected Macrophages: Roles of Pyroptosis and Necroptosis. Viruses 2020, 12, 932. [Google Scholar] [CrossRef] [PubMed]
- Liang, K.; Barnett, K.C.; Hsu, M.; Chou, W.C.; Bais, S.S.; Riebe, K.; Xie, Y.; Nguyen, T.T.; Oguin, T.H.; Vannella, K.M.; et al. Initiator cell death event induced by SARS-CoV-2 in the human airway epithelium. Sci. Immunol. 2024, 9, eadn0178. [Google Scholar] [CrossRef] [PubMed]
- Orzalli, M.H.; Kagan, J.C. Apoptosis and Necroptosis as Host Defense Strategies to Prevent Viral Infection. Trends Cell Biol. 2017, 27, 800–809. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Frank, D.; Vince, J.E. Pyroptosis versus necroptosis: Similarities, differences, and crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef]
- Ye, K.; Chen, Z.; Xu, Y. The double-edged functions of necroptosis. Cell Death Dis. 2023, 14, 163. [Google Scholar] [CrossRef]
- Malik, G.; Zhou, Y. Innate Immune Sensing of Influenza A Virus. Viruses. 2020, 12, 755. [Google Scholar] [CrossRef] [PubMed]
- Eng, V.V.; Wemyss, M.A.; Pearson, J.S. The diverse roles of RIP kinases in host-pathogen interactions. Semin. Cell Dev. Biol. 2021, 109, 125–143. [Google Scholar] [CrossRef] [PubMed]
- Belkhelfa, M.; Rafa, H.; Medjeber, O.; Arroul-Lammali, A.; Behairi, N.; Abada-Bendib, M.; Makrelouf, M.; Belarbi, S.; Masmoudi, A.N.; Tazir, M.; et al. IFN-γ and TNF-α are involved during Alzheimer disease progression and correlate with nitric oxide production: A study in Algerian patients. J. Interferon. Cytokine Res. 2014, 34, 839–847. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.D.; Chau, T.N.B.; Hoang, D.M.; Chau, N.V.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.A.; Szretter, K.J.; Katz, J.M.; Mizgerd, J.P.; Tumpey, T.M. Mice lacking both TNF and IL-1 receptors exhibit reduced lung inflammation and delay in onset of death following infection with a highly virulent H5N1 virus. J. Infect. Dis. 2010, 202, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Kam, K.-Q.; Yung, C.F.; Cui, L.; Lin, R.T.P.; Mak, T.M.; Maiwald, M.; Li, J.; Chong, C.Y.; Nadua, K.; Tan, N.W.H.; et al. A Well Infant With Coronavirus Disease 2019 With High Viral Load. Clin. Infect. Dis. 2020, 71, 847–849. [Google Scholar] [CrossRef] [PubMed]
- Peiris, J.; Chu, C.; Cheng, V.; Chan, K.; Hung, I.; Poon, L.; Law, K.; Tang, B.; Hon, T.; Chan, C.; et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: A prospective study. Lancet 2003, 361, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Boyd, D.F.; Nikhar, S.; Zhang, T.; Siokas, I.; Van de Velde, L.-A.; Gaevert, J.; Meliopoulos, V.; Thapa, B.; Rodriguez, D.A.; et al. Necroptosis blockade prevents lung injury in severe influenza. Nature 2024, 628, 835–843. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, W.; Yi, W.; Yang, L.; Bi, X.; Lin, Y.; Deng, W.; Dong, J.; Li, M.; Xie, Y. Pathway of Cell Death and Its Role in Virus Infection. Viral Immunol. 2022, 35, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, Q.; Tang, Y.D.; Zhai, J.; Hu, W.; Zheng, C. When ferroptosis meets pathogenic infections. Trends Microbiol. 2023, 31, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Wang, X.; Hao, X.; Gao, R.; Lu, X.; Yang, W.; Chen, Y.; Hu, J.; Gu, M.; Liu, X.; et al. The Novel H10N3 Avian Influenza Virus Triggers Lethal Cytokine Storm by Activating Multiple Forms of Programmed Cell Death in Mammalian Lungs. Int. J. Mol. Sci. 2025, 26, 1977. https://doi.org/10.3390/ijms26051977
Wang X, Wang X, Hao X, Gao R, Lu X, Yang W, Chen Y, Hu J, Gu M, Liu X, et al. The Novel H10N3 Avian Influenza Virus Triggers Lethal Cytokine Storm by Activating Multiple Forms of Programmed Cell Death in Mammalian Lungs. International Journal of Molecular Sciences. 2025; 26(5):1977. https://doi.org/10.3390/ijms26051977
Chicago/Turabian StyleWang, Xin, Xiyue Wang, Xiaojuan Hao, Ruyi Gao, Xiaolong Lu, Wenhao Yang, Yu Chen, Jiao Hu, Min Gu, Xiaowen Liu, and et al. 2025. "The Novel H10N3 Avian Influenza Virus Triggers Lethal Cytokine Storm by Activating Multiple Forms of Programmed Cell Death in Mammalian Lungs" International Journal of Molecular Sciences 26, no. 5: 1977. https://doi.org/10.3390/ijms26051977
APA StyleWang, X., Wang, X., Hao, X., Gao, R., Lu, X., Yang, W., Chen, Y., Hu, J., Gu, M., Liu, X., Hu, S., Liu, K., Wang, X., & Liu, X. (2025). The Novel H10N3 Avian Influenza Virus Triggers Lethal Cytokine Storm by Activating Multiple Forms of Programmed Cell Death in Mammalian Lungs. International Journal of Molecular Sciences, 26(5), 1977. https://doi.org/10.3390/ijms26051977