
Dexmedetomidine Reduces Presynaptic γ-Aminobutyric Acid Release and Prolongs Postsynaptic Responses in Layer 5 Pyramidal Neurons in the Primary Somatosensory Cortex of Mice


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
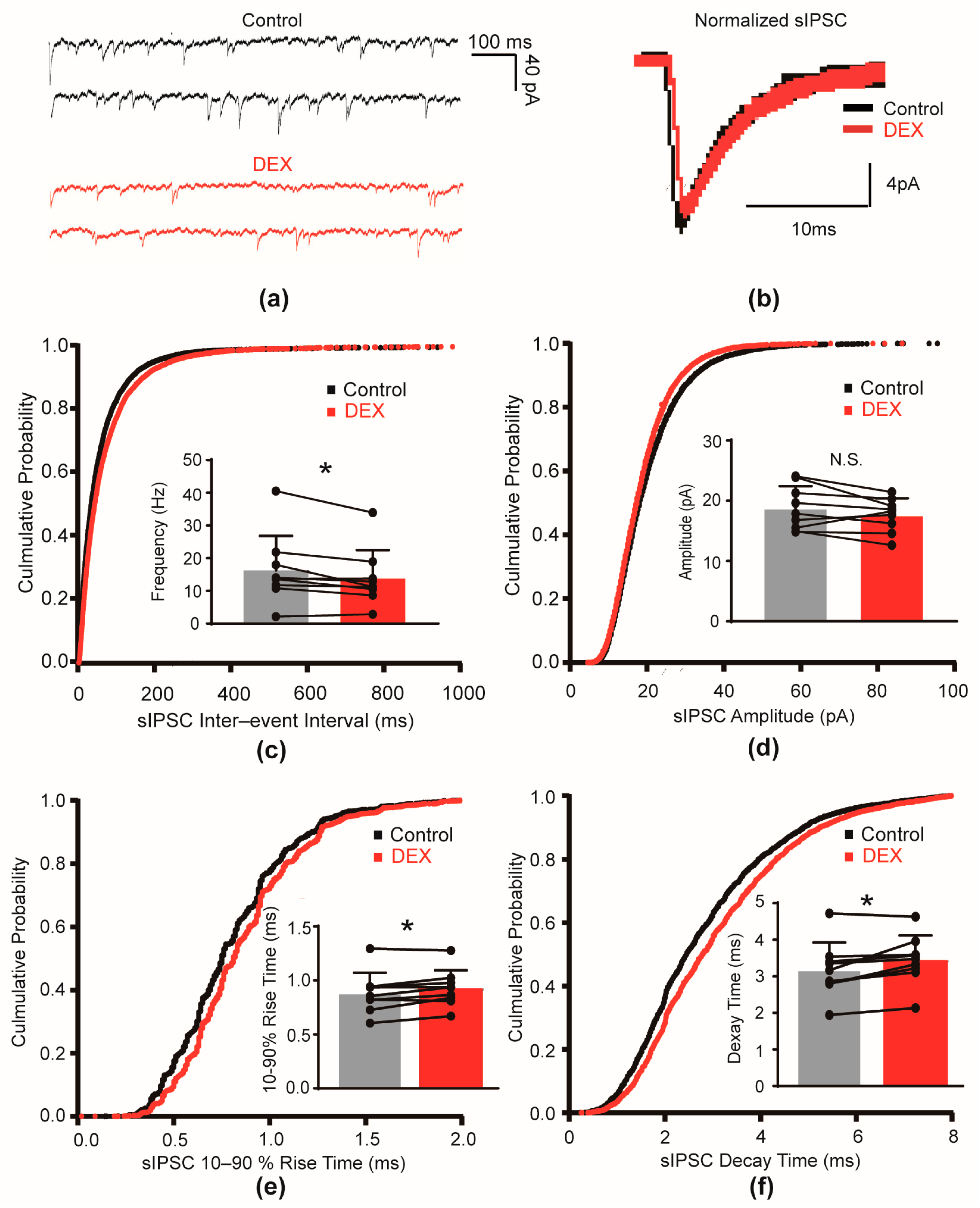
2.1. DEX Decreased the Frequency and Slowed the Kinetics of sIPSCs
2.2. DEX Prolonged the Decay Time of mIPSCs
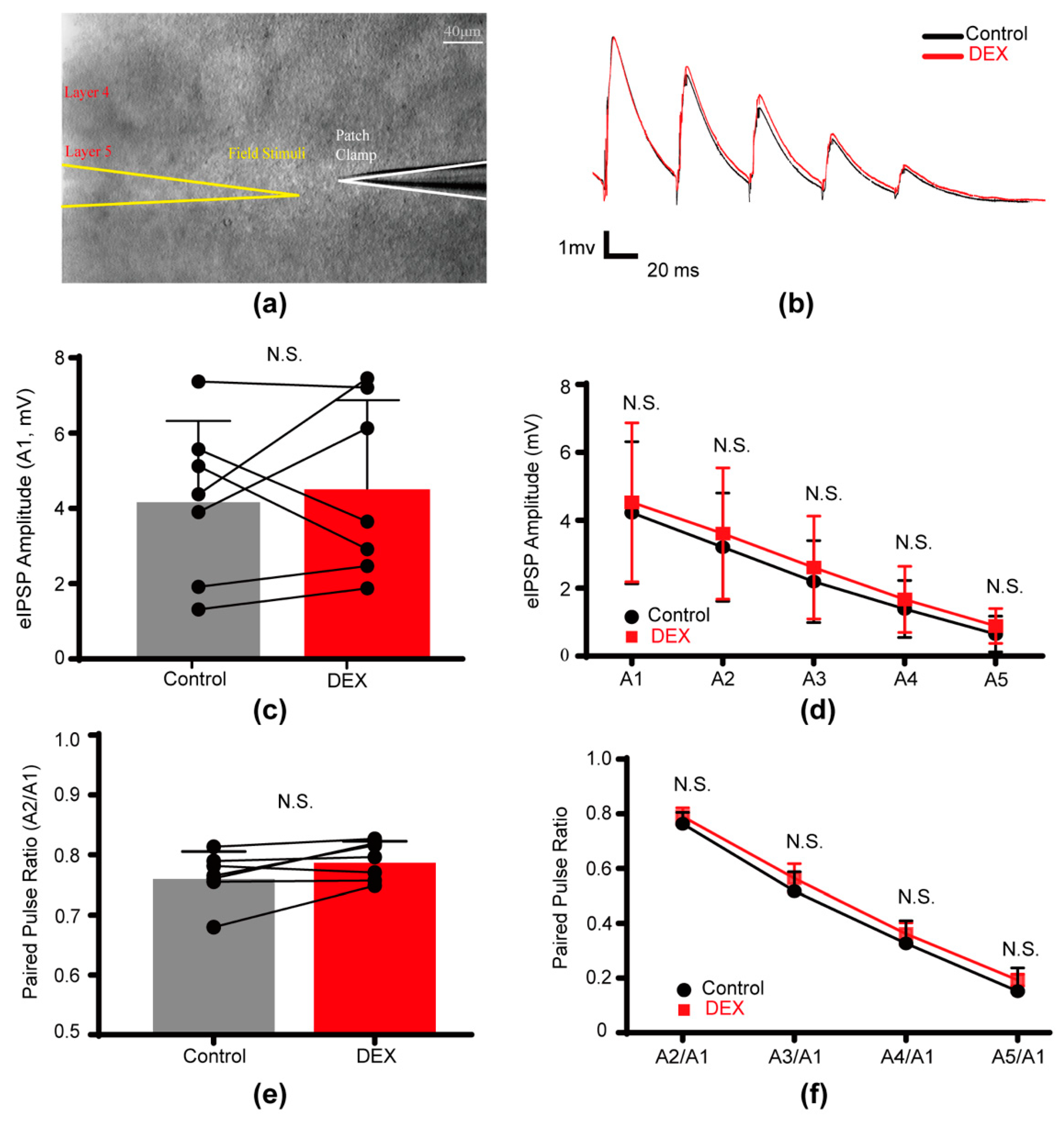
2.3. DEX Had No Effect on Evoked Inhibitory Postsynaptic Potentials
3. Discussion
4. Materials and Methods
4.1. Slice Preparation
4.2. Electrophysiological Recordings
4.3. Immunohistochemistry
4.4. Data Analysis
4.5. Statistical Analysis
4.6. Drugs and Chemicals
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DEX | Dexmedetomidine |
| α2-AR | α2-adrenoceptor |
| GABA | γ-aminobutyric acid |
| GABAA receptor | GABAAR |
| S1 | Primary somatosensory cortex |
| sIPSC | Spontaneous inhibitory postsynaptic currents |
| mIPSC | Miniature inhibitory postsynaptic current |
| eIPSP | Evoked inhibitory postsynaptic potential |
| PPR | Paired-pulse ratio |
References
- Liu, X.; Li, Y.; Kang, L.; Wang, Q. Recent Advances in the Clinical Value and Potential of Dexmedetomidine. J. Inflamm. Res. 2021, 14, 7507–7527. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Franks, N.P.; Wisden, W. Sleep and Sedative States Induced by Targeting the Histamine and Noradrenergic Systems. Front. Neural Circuits 2018, 12, 4. [Google Scholar] [CrossRef]
- Zhang, Z.; Ferretti, V.; Güntan, İ.; Moro, A.; Steinberg, E.A.; Ye, Z.; Zecharia, A.Y.; Yu, X.; Vyssotski, A.L.; Brickley, S.G.; et al. Neuronal ensembles sufficient for recovery sleep and the sedative actions of α2 adrenergic agonists. Nat. Neurosci. 2015, 18, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Miracca, G.; Yu, X.; Harding, E.C.; Miao, A.; Yustos, R.; Vyssotski, A.L.; Franks, N.P.; Wisden, W. Galanin Neurons Unite Sleep Homeostasis and α2-Adrenergic Sedation. Curr. Biol. 2019, 29, 3315–3322.e3. [Google Scholar] [CrossRef] [PubMed]
- Qiu, G.; Wu, Y.; Yang, Z.; Li, L.; Zhu, X.; Wang, Y.; Sun, W.; Dong, H.; Li, Y.; Hu, J. Dexmedetomidine Activation of Dopamine Neurons in the Ventral Tegmental Area Attenuates the Depth of Sedation in Mice. Anesthesiology 2020, 133, 377–392. [Google Scholar] [CrossRef]
- Wang, K.; Wang, Y.; Zhang, T.; Chang, B.; Fu, D.; Chen, X. The Role of Intravenous Anesthetics for Neuro: Protection or Toxicity? Neurosci. Bull. 2024, 41, 107–130. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.S.; Kaneshwaran, K.; Lei, G.; Mostafa, F.; Wang, J.; Lecker, I.; Avramescu, S.; Xie, Y.F.; Chan, N.K.; Fernandez-Escobar, A.; et al. Dexmedetomidine Prevents Excessive γ-Aminobutyric Acid Type A Receptor Function After Anesthesia. Anesthesiology 2018, 129, 477–489. [Google Scholar] [CrossRef]
- Modolo, J.; Hassan, M.; Wendling, F.; Benquet, P. Decoding the circuitry of consciousness: From local microcircuits to brain-scale networks. Netw. Neurosci. 2020, 4, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.; Suk, K.; Lee, M.G.; Jang, I.S. α(2A) adrenoceptor-mediated presynaptic inhibition of GABAergic transmission in rat tuberomammillary nucleus neurons. J. Neurochem. 2013, 125, 832–842. [Google Scholar] [CrossRef]
- Sharp, D.B.; Wang, X.; Mendelowitz, D. Dexmedetomidine decreases inhibitory but not excitatory neurotransmission to cardiac vagal neurons in the nucleus ambiguus. Brain Res. 2014, 1574, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, D.M.; Kavalali, E.T. Differential regulation of spontaneous and evoked neurotransmitter release at central synapses. Curr. Opin. Neurobiol. 2011, 21, 275–282. [Google Scholar] [CrossRef]
- Roth, F.C.; Hu, H. An axon-specific expression of HCN channels catalyzes fast action potential signaling in GABAergic interneurons. Nat. Commun. 2020, 11, 2248. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Kobayashi, M. Opposite Roles in Short-Term Plasticity for N-Type and P/Q-Type Voltage-Dependent Calcium Channels in GABAergic Neuronal Connections in the Rat Cerebral Cortex. J. Neurosci. 2018, 38, 9814–9828. [Google Scholar] [CrossRef]
- Chiu, K.M.; Lin, T.Y.; Lu, C.W.; Wang, S.J. Inhibitory effect of glutamate release from rat cerebrocortical nerve terminals by α2 adrenoceptor agonist dexmedetomidine. Eur. J. Pharmacol. 2011, 670, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Salgado, H.; Garcia-Oscos, F.; Martinolich, L.; Hall, S.; Restom, R.; Tseng, K.Y.; Atzori, M. Pre- and postsynaptic effects of norepinephrine on γ-aminobutyric acid-mediated synaptic transmission in layer 2/3 of the rat auditory cortex. Synapse 2012, 66, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Salgado, H.; Treviño, M.; Atzori, M. Layer- and area-specific actions of norepinephrine on cortical synaptic transmission. Brain Res. 2016, 1641 Pt B, 163–176. [Google Scholar] [CrossRef]
- Roychowdhury, S.; Zwierzchowski, A.N.; Garcia-Oscos, F.; Olguin, R.C.; Delgado, R.S.; Atzori, M. Layer- and area-specificity of the adrenergic modulation of synaptic transmission in the rat neocortex. Neurochem. Res. 2014, 39, 2377–2384. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, R.; Lee, S.; Rudy, B. GABAergic Interneurons in the Neocortex: From Cellular Properties to Circuits. Neuron 2016, 91, 260–292. [Google Scholar] [CrossRef] [PubMed]
- Feldmeyer, D.; Qi, G.; Emmenegger, V.; Staiger, J.F. Inhibitory interneurons and their circuit motifs in the many layers of the barrel cortex. Neuroscience 2018, 368, 132–151. [Google Scholar] [CrossRef]
- Kim, D.; Roh, H.; Lee, H.M.; Kim, S.J.; Im, M. Localization of hyperpolarization-activated cyclic nucleotide-gated channels in the vertebrate retinas across species and their physiological roles. Front. Neuroanat. 2024, 18, 1385932. [Google Scholar]
- Farrant, M.; Nusser, Z. Variations on an inhibitory theme: Phasic and tonic activation of GABA(A) receptors. Nat. Rev. Neurosci. 2005, 6, 215–229. [Google Scholar] [CrossRef]
- Hinkle, D.J.; Macdonald, R.L. Beta subunit phosphorylation selectively increases fast desensitization and prolongs deactivation of alpha1beta1gamma2L and alpha1beta3gamma2L GABA(A) receptor currents. J. Neurosci. 2003, 23, 11698–11710. [Google Scholar] [CrossRef]
- Houston, C.M.; Bright, D.P.; Sivilotti, L.G.; Beato, M.; Smart, T.G. Intracellular chloride ions regulate the time course of GABA-mediated inhibitory synaptic transmission. J. Neurosci. 2009, 29, 10416–10423. [Google Scholar] [CrossRef] [PubMed]
- Eyre, M.D.; Renzi, M.; Farrant, M.; Nusser, Z. Setting the time course of inhibitory synaptic currents by mixing multiple GABA(A) receptor α subunit isoforms. J. Neurosci. 2012, 32, 5853–5867. [Google Scholar] [CrossRef]
- Luscher, B.; Fuchs, T.; Kilpatrick, C.L. GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 2011, 70, 385–409. [Google Scholar] [CrossRef]
- Liu, L.; Luo, Z.; Mai, Y.; Lu, Y.; Sun, Z.; Chen, J.; Zeng, T.; Chen, L.; Liu, Z.; Yang, H.; et al. Dexmedetomidine relieves inflammatory pain by enhancing GABAergic synaptic activity in pyramidal neurons of the anterior cingulate cortex. Neuropharmacology 2023, 240, 109710. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.J.; Amato, A.; Connolly, C.N.; Benke, D.; Moss, S.J.; Smart, T.G. Adjacent phosphorylation sites on GABAA receptor beta subunits determine regulation by cAMP-dependent protein kinase. Nat. Neurosci. 1998, 1, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Morrow, D.H.; Nathanson, A.J.; Henley, J.M.; Wilkinson, K.A.; Moss, S.J. Phosphorylation on Ser-359 of the α2 subunit in GABA type A receptors down-regulates their density at inhibitory synapses. J. Biol. Chem. 2020, 295, 12330–12342. [Google Scholar] [CrossRef]
- Kelz, M.B.; Mashour, G.A. The Biology of General Anesthesia from Paramecium to Primate. Curr. Biol. 2019, 29, R1199–R1210. [Google Scholar] [CrossRef] [PubMed]
- Fu, B.; Wang, Y.; Yang, H.; Yu, T. Effects of Etomidate on GABAergic and Glutamatergic Transmission in Rat Thalamocortical Slices. Neurochem. Res. 2016, 41, 3181–3191. [Google Scholar] [CrossRef]
- Koyanagi, Y.; Oi, Y.; Yamamoto, K.; Koshikawa, N.; Kobayashi, M. Fast-spiking cell to pyramidal cell connections are the most sensitive to propofol-induced facilitation of GABAergic currents in rat insular cortex. Anesthesiology 2014, 121, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, J.S.; Scanziani, M. How inhibition shapes cortical activity. Neuron 2011, 72, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Bharioke, A.; Munz, M.; Brignall, A.; Kosche, G.; Eizinger, M.F.; Ledergerber, N.; Hillier, D.; Gross-Scherf, B.; Conzelmann, K.K.; Macé, E.; et al. General anesthesia globally synchronizes activity selectively in layer 5 cortical pyramidal neurons. Neuron 2022, 110, 2024–2040.e10. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Tellez, N.; Iqbal, F.; Pehar, M.; Casas-Ortiz, A.; Rice, T.; Syed, N.I. Dexmedetomidine does not compromise neuronal viability, synaptic connectivity, learning and memory in a rodent model. Sci. Rep. 2021, 11, 16153. [Google Scholar] [CrossRef]
- Guo, F.; Ding, Y.; Yu, X.; Cai, X. Effect of dexmedetomidine, midazolam, and propofol on lipopolysaccharide-stimulated dendritic cells. Exp. Ther. Med. 2018, 15, 5487–5494. [Google Scholar] [CrossRef] [PubMed]
- Llorca, A.; Deogracias, R. Origin, Development, and Synaptogenesis of Cortical Interneurons. Front. Neurosci. 2022, 16, 929469. [Google Scholar] [CrossRef]
- Fan, S.; Cheng, X.; Zhang, P.; Wang, Y.; Wang, L.; Cheng, J. The α2 Adrenoceptor Agonist and Sedative/Anaesthetic Dexmedetomidine Excites Diverse Neuronal Types in the Ventrolateral Preoptic Area of Male Mice. ASN Neuro 2023, 15, 17590914231191016. [Google Scholar] [CrossRef]
- Funai, Y.; Pickering, A.E.; Uta, D.; Nishikawa, K.; Mori, T.; Asada, A.; Imoto, K.; Furue, H. Systemic dexmedetomidine augments inhibitory synaptic transmission in the superficial dorsal horn through activation of descending noradrenergic control: An in vivo patch-clamp analysis of analgesic mechanisms. PAIN 2014, 155, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Agmon, A.; Connors, B.W. Thalamocortical responses of mouse somatosensory (barrel) cortexin vitro. Neuroscience 1991, 41, 365–379. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, B.; Tang, J.; Huang, Y. Dexmedetomidine Reduces Presynaptic γ-Aminobutyric Acid Release and Prolongs Postsynaptic Responses in Layer 5 Pyramidal Neurons in the Primary Somatosensory Cortex of Mice. Int. J. Mol. Sci. 2025, 26, 1931. https://doi.org/10.3390/ijms26051931
Tang B, Tang J, Huang Y. Dexmedetomidine Reduces Presynaptic γ-Aminobutyric Acid Release and Prolongs Postsynaptic Responses in Layer 5 Pyramidal Neurons in the Primary Somatosensory Cortex of Mice. International Journal of Molecular Sciences. 2025; 26(5):1931. https://doi.org/10.3390/ijms26051931
Chicago/Turabian StyleTang, Bo, Jiali Tang, and Yuguang Huang. 2025. "Dexmedetomidine Reduces Presynaptic γ-Aminobutyric Acid Release and Prolongs Postsynaptic Responses in Layer 5 Pyramidal Neurons in the Primary Somatosensory Cortex of Mice" International Journal of Molecular Sciences 26, no. 5: 1931. https://doi.org/10.3390/ijms26051931
APA StyleTang, B., Tang, J., & Huang, Y. (2025). Dexmedetomidine Reduces Presynaptic γ-Aminobutyric Acid Release and Prolongs Postsynaptic Responses in Layer 5 Pyramidal Neurons in the Primary Somatosensory Cortex of Mice. International Journal of Molecular Sciences, 26(5), 1931. https://doi.org/10.3390/ijms26051931