
Increased Kindlin-2 via SMURF1 Inhibition Attenuates Endothelial Permeability and Acute Lung Injury


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
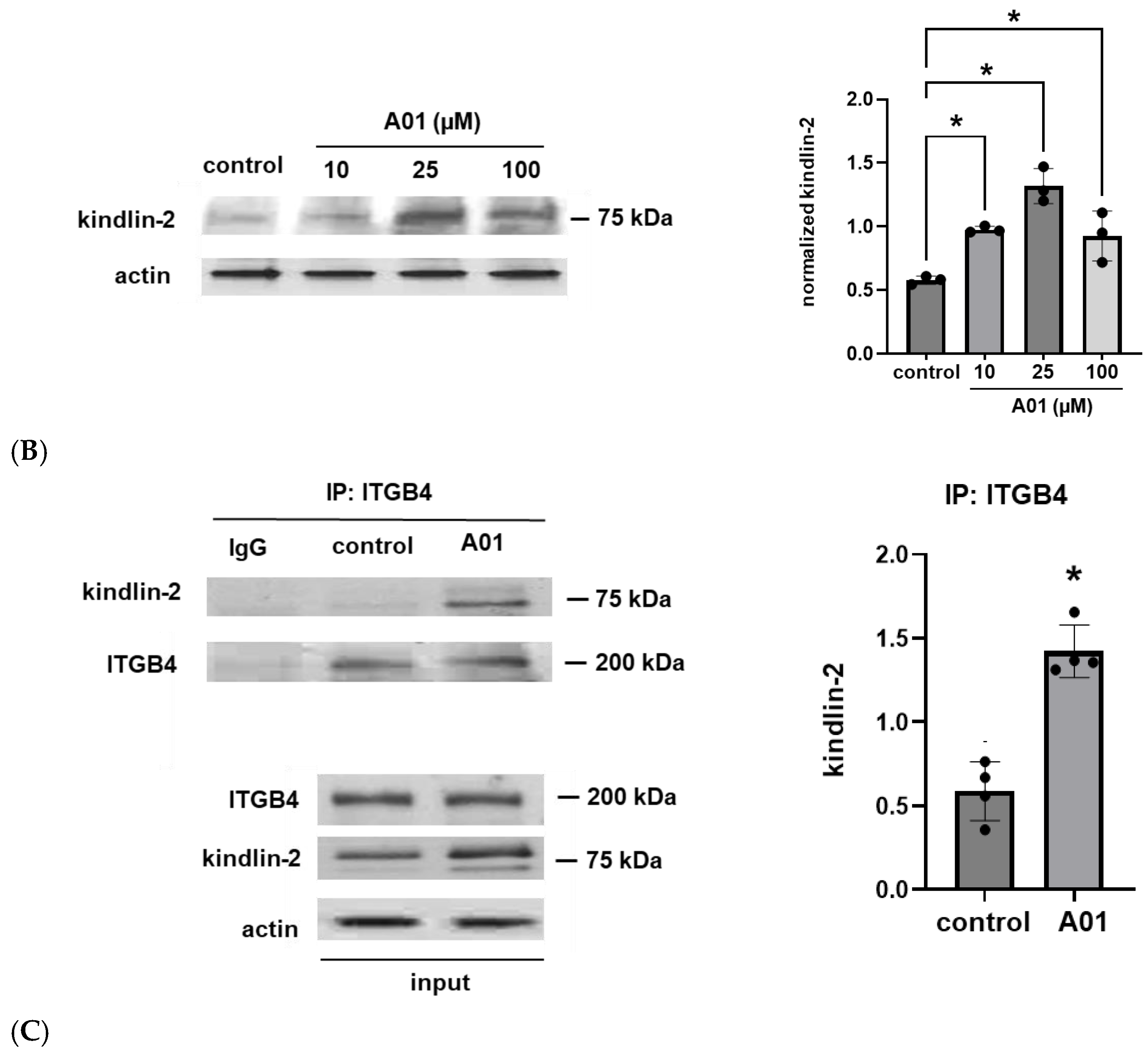
2.1. Association of Kindlin-2 and ITGB4 in Human Lung Endothelial Cells (EC) and Effects of SMURF1 Inhibition on Kindlin-2 Expression
2.2. The Effects of Kindlin-2 Depletion on Human Lung EC Barrier Function
2.3. The Effects of Kindlin-2 Augmentation via SMURF1 Inhibition on Human Lung EC Barrier Function
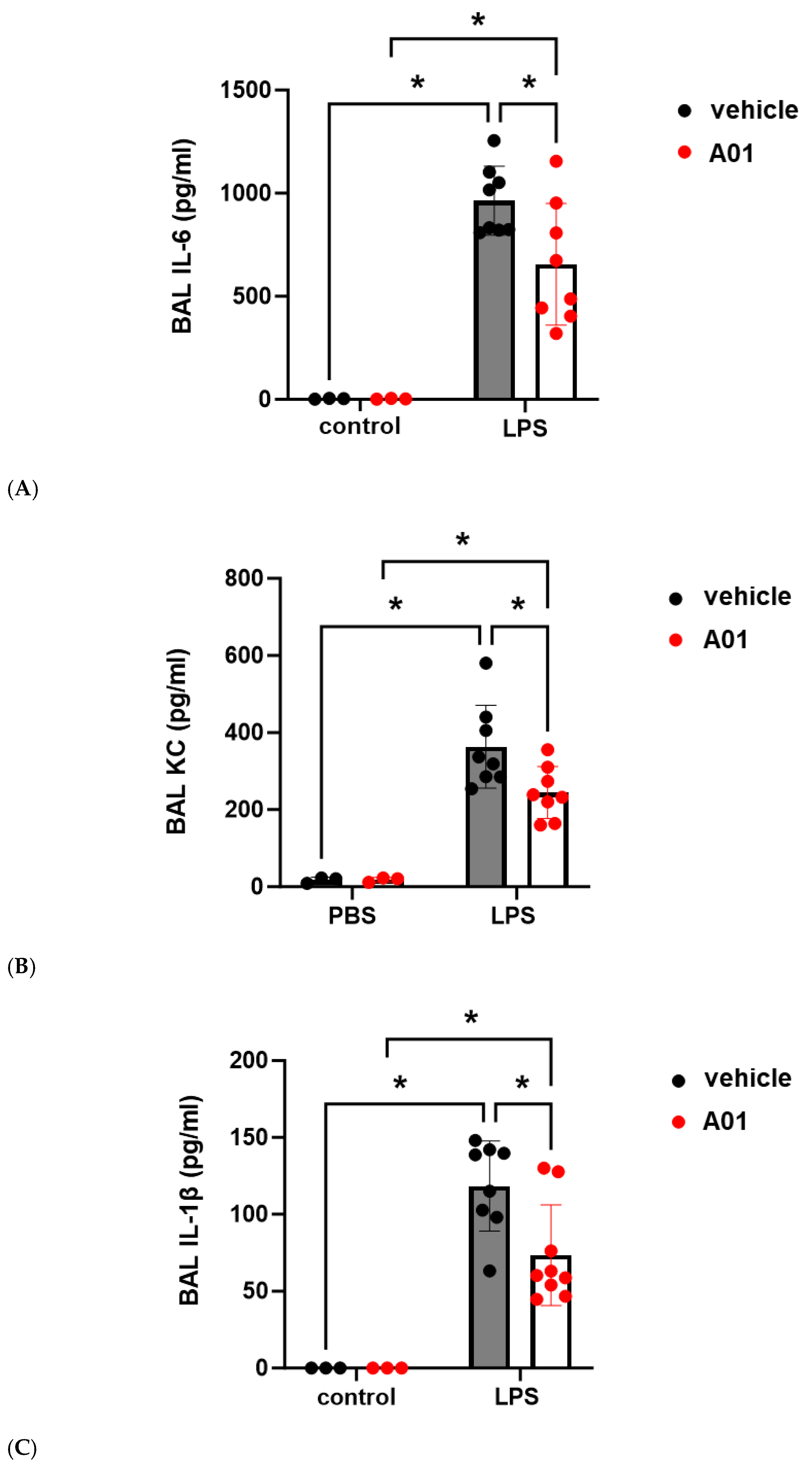
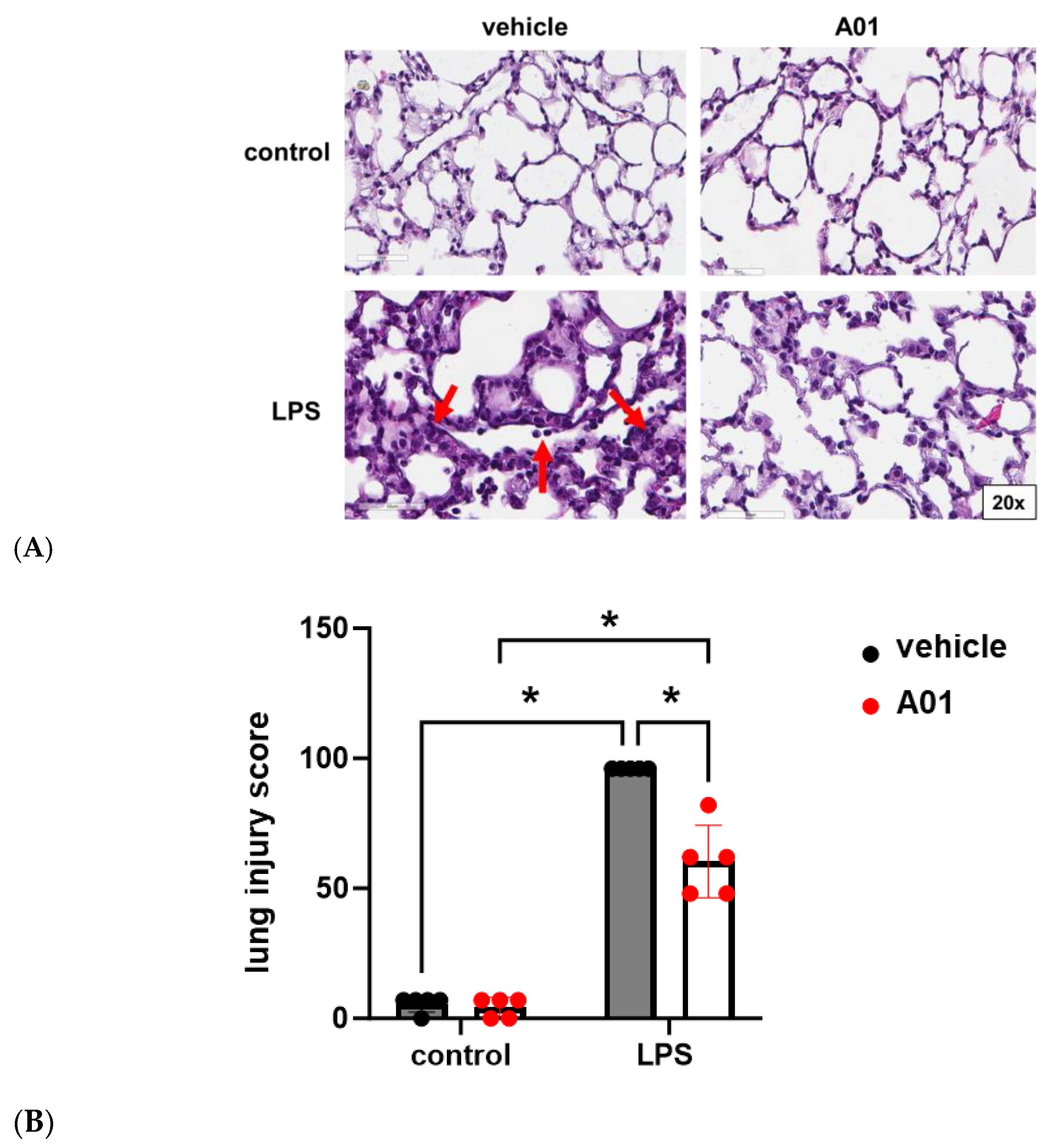
2.4. SMURF1 Inhibition via A01 in Murine ALI
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Endothelial Cell Culture
4.3. Endothelial Cell (EC) siRNA Transfection
4.4. Immunoblotting and Immunoprecipitation
4.5. Transendothelial Electrical Resistance (TER) Measurement
4.6. Transwell Permeability Assay
4.7. Murine Acute Lung Injury (ALI) Model
4.8. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALI | acute lung injury |
| BAL | bronchoalveolar lavage |
| EC | endothelial cells |
| FAK | focal adhesion kinase |
| FAK | focal adhesion kinase |
| ITGB1 | integrin β1 |
| ITGB3 | integrin β3 |
| ITGB4 | integrin β4 |
| LPS | lipopolysaccharide |
| siRNA | silencing RNA |
| SMURF1 | smad ubiquitination regulatory factor-1 |
| TER | transendothelial electrical resistance |
References
- Chen, W.; Sammani, S.; Mitra, S.; Ma, S.F.; Garcia, J.G.; Jacobson, J.R. Critical role for integrin-beta4 in the attenuation of murine acute lung injury by simvastatin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L279–L285. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Garcia, J.G.; Jacobson, J.R. Integrin beta4 attenuates SHP-2 and MAPK signaling and reduces human lung endothelial inflammatory responses. J. Cell. Biochem. 2010, 110, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Epshtein, Y.; Ni, X.; Dull, R.O.; Cress, A.E.; Garcia, J.G.; Jacobson, J.R. Role of Integrin beta4 in Lung Endothelial Cell Inflammatory Responses to Mechanical Stress. Sci. Rep. 2015, 5, 16529. [Google Scholar]
- Hogervorst, F.; Kuikman, I.; von dem Borne, A.E.; Sonnenberg, A. Cloning and sequence analysis of beta-4 cDNA: An integrin subunit that contains a unique 118 kd cytoplasmic domain. EMBO J. 1990, 9, 765–770. [Google Scholar] [CrossRef]
- Niessen, C.M.; Hulsman, E.H.; Oomen, L.C.; Kuikman, I.; Sonnenberg, A. A minimal region on the integrin beta4 subunit that is critical to its localization in hemidesmosomes regulates the distribution of HD1/plectin in COS-7 cells. J. Cell Sci. 1997, 110 Pt 15, 1705–1716. [Google Scholar] [CrossRef]
- Tai, Y.L.; Lai, I.R.; Peng, Y.J.; Ding, S.T.; Shen, T.L. Activation of focal adhesion kinase through an interaction with beta4 integrin contributes to tumorigenicity of colon cancer. FEBS Lett. 2016, 590, 1826–1837. [Google Scholar] [CrossRef]
- Theodosiou, M.; Widmaier, M.; Bottcher, R.T.; Rognoni, E.; Veelders, M.; Bharadwaj, M.; Lambacher, A.; Austen, K.; Muller, D.J.; Zent, R.; et al. Kindlin-2 cooperates with talin to activate integrins and induces cell spreading by directly binding paxillin. Elife 2016, 5, e10130. [Google Scholar] [CrossRef]
- Zhu, L.; Plow, E.F.; Qin, J. Initiation of focal adhesion assembly by talin and kindlin: A dynamic view. Protein Sci. 2021, 30, 531–542. [Google Scholar] [CrossRef]
- Harburger, D.S.; Bouaouina, M.; Calderwood, D.A. Kindlin-1 and -2 directly bind the C-terminal region of beta integrin cytoplasmic tails and exert integrin-specific activation effects. J. Biol. Chem. 2009, 284, 11485–11497. [Google Scholar] [CrossRef]
- Rognoni, E.; Ruppert, R.; Fassler, R. The kindlin family: Functions, signaling properties and implications for human disease. J. Cell Sci. 2016, 129, 17–27. [Google Scholar] [CrossRef]
- Bottcher, R.T.; Veelders, M.; Rombaut, P.; Faix, J.; Theodosiou, M.; Stradal, T.E.; Rottner, K.; Zent, R.; Herzog, F.; Fassler, R. Kindlin-2 recruits paxillin and Arp2/3 to promote membrane protrusions during initial cell spreading. J. Cell Biol. 2017, 216, 3785–3798. [Google Scholar] [CrossRef] [PubMed]
- Pluskota, E.; Bledzka, K.M.; Bialkowska, K.; Szpak, D.; Soloviev, D.A.; Jones, S.V.; Verbovetskiy, D.; Plow, E.F. Kindlin-2 interacts with endothelial adherens junctions to support vascular barrier integrity. J. Physiol. 2017, 595, 6443–6462. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Wang, X.; Zhan, J.; Chen, Y.; Fang, W.; Zhang, L.; Zhang, H. Smurf1 inhibits integrin activation by controlling Kindlin-2 ubiquitination and degradation. J. Cell Biol. 2017, 216, 1455–1471. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, C.; Zhang, X.; Xing, G.; Lu, K.; Gu, Y.; He, F.; Zhang, L. Selective small molecule compounds increase BMP-2 responsiveness by inhibiting Smurf1-mediated Smad1/5 degradation. Sci. Rep. 2014, 4, 4965. [Google Scholar] [CrossRef]
- Dong, Y.; Ma, G.; Hou, X.; Han, Y.; Ding, Z.; Tang, W.; Chen, L.; Chen, Y.; Zhou, B.; Rao, F.; et al. Kindlin-2 controls angiogenesis through modulating Notch1 signaling. Cell Mol. Life Sci. 2023, 80, 223. [Google Scholar] [CrossRef]
- Pluskota, E.; Dowling, J.J.; Gordon, N.; Golden, J.A.; Szpak, D.; West, X.Z.; Nestor, C.; Ma, Y.Q.; Bialkowska, K.; Byzova, T.; et al. The integrin coactivator kindlin-2 plays a critical role in angiogenesis in mice and zebrafish. Blood 2011, 117, 4978–4987. [Google Scholar] [CrossRef]
- Sidibe, A.; Mykuliak, V.V.; Zhang, P.; Hytonen, V.P.; Wu, J.; Wehrle-Haller, B. Acetyl-NPKY of integrin-beta1 binds KINDLIN2 to control endothelial cell proliferation and junctional integrity. iScience 2024, 27, 110129. [Google Scholar] [CrossRef]
- Ma, Y.Q.; Qin, J.; Wu, C.; Plow, E.F. Kindlin-2 (Mig-2): A co-activator of beta3 integrins. J. Cell Biol. 2008, 181, 439–446. [Google Scholar] [CrossRef]
- van der Bijl, I.; Nawaz, K.; Kazlauskaite, U.; van Stalborch, A.M.; Tol, S.; Jimenez Orgaz, A.; van den Bout, I.; Reinhard, N.R.; Sonnenberg, A.; Margadant, C. Reciprocal integrin/integrin antagonism through kindlin-2 and Rho GTPases regulates cell cohesion and collective migration. Matrix Biol. 2020, 93, 60–78. [Google Scholar] [CrossRef]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef]
- Ephstein, Y.; Singleton, P.A.; Chen, W.; Wang, L.; Salgia, R.; Kanteti, P.; Dudek, S.M.; Garcia, J.G.; Jacobson, J.R. Critical role of S1PR1 and integrin beta4 in HGF/c-Met-mediated increases in vascular integrity. J. Biol. Chem. 2013, 288, 2191–2200. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Ying, S.X.; Zhang, G.M.; Li, C.; Cheng, S.Y.; Deng, C.X.; Zhang, Y.E. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 2005, 121, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Mathew, R.; Huang, J.; Farahani, R.; Peng, H.; Olson, S.C.; Etlinger, J.D. Smurf1 ubiquitin ligase causes downregulation of BMP receptors and is induced in monocrotaline and hypoxia models of pulmonary arterial hypertension. Exp. Biol. Med. 2010, 235, 805–813. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F. Therapeutic Potential of Smurf-1 Inhibitors for the Treatment of Pulmonary Arterial Hypertension (PAH). ACS Med. Chem. Lett. 2016, 7, 878–880. [Google Scholar] [CrossRef]
- Li, D.; Wei, T.T.; Cai, J.; Xie, T.H.; Yao, Y.; Zhu, L. Smurf1: A possible therapeutic target in dry age-related macular degeneration. Exp. Eye Res. 2023, 233, 109549. [Google Scholar] [CrossRef]
- Chong, P.A.; Lin, H.; Wrana, J.L.; Forman-Kay, J.D. Coupling of tandem Smad ubiquitination regulatory factor (Smurf) WW domains modulates target specificity. Proc. Natl. Acad. Sci. USA 2010, 107, 18404–18409. [Google Scholar] [CrossRef]
- Hakanpaa, L.; Kiss, E.A.; Jacquemet, G.; Miinalainen, I.; Lerche, M.; Guzman, C.; Mervaala, E.; Eklund, L.; Ivaska, J.; Saharinen, P. Targeting beta1-integrin inhibits vascular leakage in endotoxemia. Proc. Natl. Acad. Sci. USA 2018, 115, E6467. [Google Scholar] [CrossRef]
- Liu, C.; Xu, J.; Fan, J.; Liu, C.; Xie, W.; Kong, H. DPP-4 exacerbates LPS-induced endothelial cells inflammation via integrin-alpha5beta1/FAK/AKT signaling. Exp. Cell Res. 2024, 435, 113909. [Google Scholar] [CrossRef]
- Tong, Y.; Bao, C.; Xu, Y.Q.; Tao, L.; Zhou, Y.; Zhuang, L.; Meng, Y.; Zhang, H.; Xue, J.; Wang, W.; et al. The beta3/5 Integrin-MMP9 Axis Regulates Pulmonary Inflammatory Response and Endothelial Leakage in Acute Lung Injury. J. Inflamm. Res. 2021, 14, 5079–5094. [Google Scholar] [CrossRef]
- Wan, H.; Xie, T.; Xu, Q.; Hu, X.; Xing, S.; Yang, H.; Gao, Y.; He, Z. Thy-1 depletion and integrin beta3 upregulation-mediated PI3K-Akt-mTOR pathway activation inhibits lung fibroblast autophagy in lipopolysaccharide-induced pulmonary fibrosis. Lab. Invest. 2019, 99, 1636–1649. [Google Scholar] [CrossRef]
- Tang, W.; Ding, Z.; Gao, H.; Yan, Q.; Liu, J.; Han, Y.; Hou, X.; Liu, Z.; Chen, L.; Yang, D.; et al. Targeting Kindlin-2 in adipocytes increases bone mass through inhibiting FAS/PPARgamma/FABP4 signaling in mice. Acta Pharm. Sin. B 2023, 13, 4535–4552. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Yan, Q.; Wang, D.; Lai, Y.; Zhou, B.; Zhang, Q.; Jin, W.; Lin, S.; Lei, Y.; Ma, L.; et al. Focal adhesion protein Kindlin-2 regulates bone homeostasis in mice. Bone Res. 2020, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.T.; Xiong, D.Y.; Xiao, J.N.; Deng, L.; Liu, W.; Tang, S.Y. Kindlin-2 protects pancreatic beta cells through inhibiting NLRP3 inflammasome activation in diabetic mice. Biochem. Biophys. Res. Commun. 2022, 614, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Kansakar, U.; Markovic, V.; Sossey-Alaoui, K. Role of Kindlin-2 in cancer progression and metastasis. Ann. Transl. Med. 2020, 8, 901. [Google Scholar] [CrossRef]
- Garcia, J.G.; Liu, F.; Verin, A.D.; Birukova, A.; Dechert, M.A.; Gerthoffer, W.T.; Bamberg, J.R.; English, D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J. Clin. Invest. 2001, 108, 689–701. [Google Scholar] [CrossRef]
- Chen, W.; Sharma, R.; Rizzo, A.N.; Siegler, J.H.; Garcia, J.G.; Jacobson, J.R. Role of claudin-5 in the attenuation of murine acute lung injury by simvastatin. Am. J. Respir. Cell Mol. Biol. 2014, 50, 328–336. [Google Scholar] [CrossRef]
- Meyer, N.J.; Huang, Y.; Singleton, P.A.; Sammani, S.; Moitra, J.; Evenoski, C.L.; Husain, A.N.; Mitra, S.; Moreno-Vinasco, L.; Jacobson, J.R.; et al. GADD45a is a novel candidate gene in inflammatory lung injury via influences on Akt signaling. FASEB J. 2009, 23, 1325–1337. [Google Scholar] [CrossRef]
- Matute-Bello, G.; Downey, G.; Moore, B.B.; Groshong, S.D.; Matthay, M.A.; Slutsky, A.S.; Kuebler, W.M. An official American Thoracic Society workshop report: Features and measurements of experimental acute lung injury in animals. Am. J. Respir. Cell Mol. Biol. 2011, 44, 725–738. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.; Epshtein, Y.; Vagts, C.; Cress, A.E.; Jacobson, J.R. Increased Kindlin-2 via SMURF1 Inhibition Attenuates Endothelial Permeability and Acute Lung Injury. Int. J. Mol. Sci. 2025, 26, 1880. https://doi.org/10.3390/ijms26051880
Chen W, Epshtein Y, Vagts C, Cress AE, Jacobson JR. Increased Kindlin-2 via SMURF1 Inhibition Attenuates Endothelial Permeability and Acute Lung Injury. International Journal of Molecular Sciences. 2025; 26(5):1880. https://doi.org/10.3390/ijms26051880
Chicago/Turabian StyleChen, Weiguo, Yulia Epshtein, Christen Vagts, Anne E. Cress, and Jeffrey R. Jacobson. 2025. "Increased Kindlin-2 via SMURF1 Inhibition Attenuates Endothelial Permeability and Acute Lung Injury" International Journal of Molecular Sciences 26, no. 5: 1880. https://doi.org/10.3390/ijms26051880
APA StyleChen, W., Epshtein, Y., Vagts, C., Cress, A. E., & Jacobson, J. R. (2025). Increased Kindlin-2 via SMURF1 Inhibition Attenuates Endothelial Permeability and Acute Lung Injury. International Journal of Molecular Sciences, 26(5), 1880. https://doi.org/10.3390/ijms26051880