
A Closer Look into Autoimmune Liver Diseases

 , , , and
, , , and {kind=link}
Abstract
1. Introduction
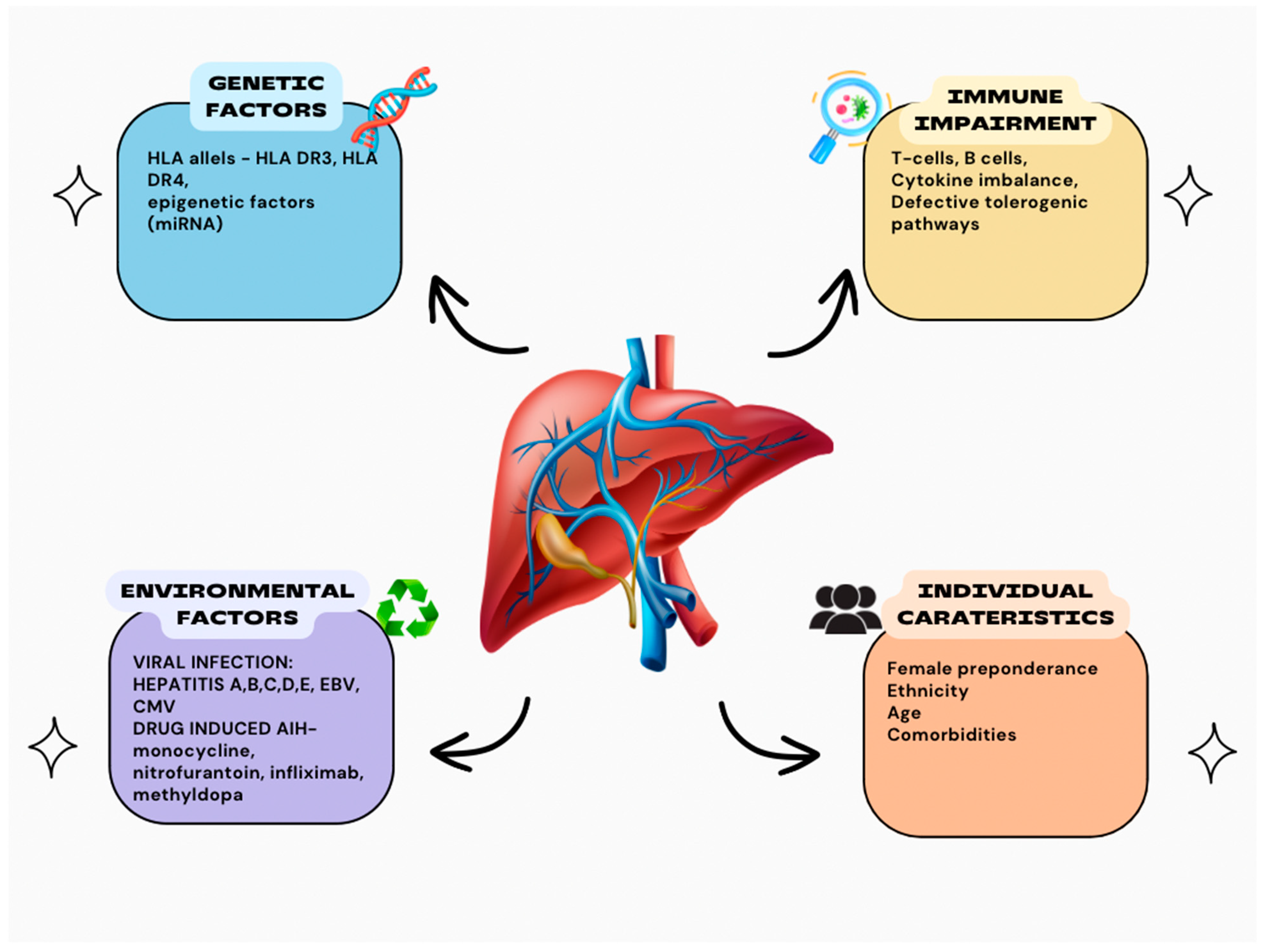
2. Autoimmune Hepatitis
3. Genetics
4. Immune Dysregulation
5. Environmental Factors
6. Therapy for Autoimmune Hepatitis
7. Primary Biliary Cholangitis
8. Genetics and Epigenetics
9. Immunology
10. Treatment of PBC
11. Primary Sclerosing Cholangitis
12. Pathogenesis of PSC
13. Genetic Factors
14. PSC-IBD Association
15. A Closer Look into Existing Medical Treatments and Future Treatment Modalities
16. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Heo, N.-Y.; Kim, H. Epidemiology and updated management for autoimmune liver disease. Clin. Mol. Hepatol. 2023, 29, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Janik, M.K.; Wunsch, E.; Milkiewicz, P. Variants of autoimmune liver diseases: How to diagnose and treat them? Pol. Achives Intern. Med. 2023, 133, 16408. [Google Scholar] [CrossRef] [PubMed]
- Ricciuto, A.; Kamath, B.M.; Hirschfield, G.M.; Trivedi, P.J. Primary sclerosing cholangitis and overlap features of autoimmune hepatitis: A coming of age or an age-ist problem? J. Hepatol. 2023, 79, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.L.; Danford, C.J.; Patwardhan, V.; Bonder, A. Treatment of Overlap Syndromes in Autoimmune Liver Disease: A Systematic Review and Meta-Analysis. J. Clin. Med. 2020, 9, 1449. [Google Scholar] [CrossRef]
- Jepsen, P.; Gronbaek, L.; Vilstrup, H. Worldwide incidence of autoimmune liver disease. Dig Dis. 2015, 33, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Zachou, K.; Arvaniti, P.; Lyberopoulou, A.; Dalekos, G.N. Impact of genetic and environmental factors on autoimmune hepatitis. J. Transl. Autoimmun. 2021, 4, 100125. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.X.; Huang, L.X.; Wang, X.X.; Wang, Z.L.; Li, X.H.; Feng, B. Exploring the Pathogenesis of Autoimmune Liver Diseases from the Heterogeneity of Target Cells. J. Clin. Transl. Hepatol. 2024, 12, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D. How genetic risk contributes to autoimmune liver disease. In Seminars in Immunopathology; Springer: Berlin/Heidelberg, Germany, 2022; Volume 44, pp. 397–410. [Google Scholar] [CrossRef]
- Gerussi, A.; Carbone, M.; Asselta, R.; Invernizzi, P. Genetics of Autoimmune Liver Diseases; Springer: Cham, Switzerland, 2020; pp. 69–85. [Google Scholar] [CrossRef]
- Invernizzi, F.; Cilla, M.; Trapani, S.; Guarino, M.; Cossiga, V.; Gambato, M.; Morelli, M.C.; Morisco, F.; Burra, P.; Floreani, A. Gender and Autoimmune Liver Diseases: Relevant Aspects in Clinical Practice. J. Pers. Med. 2022, 12, 925. [Google Scholar] [CrossRef]
- Richardson, N.; Wootton, G.E.; Bozward, A.G.; Oo, Y.H. Challenges and opportunities in achieving effective regulatory T cell therapy in autoimmune liver disease. Semin. Immunopathol. 2022, 44, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Horst, A.K.; Kumashie, K.G.; Neumann, K.; Diehl, L.; Tiegs, G. Antigen presentation, autoantibody production, and therapeutic targets in autoimmune liver disease. Cell. Mol. Immunol. 2021, 18, 92–111. [Google Scholar] [CrossRef] [PubMed]
- Czaja, A.J. Incorporating the Molecular Mimicry of Environmental Antigens into the Causality of Autoimmune Hepatitis. Dig. Dis. Sci. 2023, 68, 2824–2842. [Google Scholar] [CrossRef] [PubMed]
- Floreani, A.; Gabbia, D.; de Martin, S. Are Gender Differences Important for Autoimmune Liver Diseases? Life 2024, 14, 500. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, D.; Carbone, M. Prognostic models and autoimmune liver diseases. Best Pract. Res. Clin. Gastroenterol. 2023, 67, 101878. [Google Scholar] [CrossRef]
- Rabiee, A.; Silveira, M.G. Primary sclerosing cholangitis. Transl. Gastroenterol. Hepatol. 2021, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Ma, J.; Zhao, C.; Tai, W. Reasons why women are more likely to develop primary biliary cholangitis. Heliyon 2024, 10, e25634. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Neuberger, J.M. Autoimmune liver disease, autoimmunity and liver transplantation. J. Hepatol. 2014, 60, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Richardson, N.; Ng, S.T.H.; Wraith, D.C. Antigen-Specific Immunotherapy for Treatment of Autoimmune Liver Diseases. Front. Immunol. 2020, 11, 1586. [Google Scholar] [CrossRef] [PubMed]
- Bernal, R.B.; Medina-Morales, E.; Goyes, D.; Patwardhan, V.; Bonder, A. Management of Autoimmune Liver Diseases after Liver Transplantation. Transplantology 2021, 2, 162–182. [Google Scholar] [CrossRef]
- Harputluoglu, M.; Caliskan, A.R.; Akbulut, S. Autoimmune hepatitis and liver transplantation: Indications, and recurrent and de novo autoimmune hepatitis. World J. Transplant. 2022, 12, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Engel, B.; Taubert, R.; Jaeckel, E.; Manns, M.P. The future of autoimmune liver diseases—Understanding pathogenesis and improving morbidity and mortality. Liver Int. 2020, 40, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Sirbe, C.; Simu, G.; Szabo, I.; Grama, A.; Pop, T.L. Pathogenesis of Autoimmune Hepatitis-Cellular and Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 13578. [Google Scholar] [CrossRef] [PubMed]
- Vergani, D.; Mieli-Vergani, G. Autoimmune hepatitis: Diagnostic criteria and serological testing. Clin. Liver Dis. 2014, 3, 38–41. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Longhi, M.S.; Mieli-Vergani, G.; Vergani, D. Autoimmune hepatitis. Curr. Pediatr. Rev. 2014, 10, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Urzúa, Á.; Pizarro, C.; Gajardo, A.; Poniachik, R.; Pavez, C.; Cattaneo, M.; Brahm, J.; Carreño, L.; Poniachik, J. Autoimmune Hepatitis with Acute Presentation: Clinical, Biochemical, and Histological Features of 126 Patients. Can. J. Gastroenterol. Hepatol. 2022, 2022, 6470847. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Liu, Y.; Shang, Y.; Han, Z.; Han, Y. Clinical characteristics and treatment outcomes of acute severe autoimmune hepatitis. BMC Gastroenterol. 2021, 21, 93. [Google Scholar] [CrossRef] [PubMed]
- Vuerich, M.; Wang, N.; Kalbasi, A.; Graham, J.J.; Longhi, M.S. Dysfunctional Immune Regulation in Autoimmune Hepatitis: From Pathogenesis to Novel Therapies. Front. Immunol. 2021, 12, 746436. [Google Scholar] [CrossRef]
- de Boer, Y.S.; van Gerven, N.M.; Zwiers, A.; Verwer, B.J.; van Hoek, B.; van Erpecum, K.J.; Beuers, U.; van Buuren, H.R.; Drenth, J.P.; Ouden, J.W.D.; et al. Genome-Wide Association Study Identifies Variants Associated with Autoimmune Hepatitis Type 1. Gastroenterology 2014, 147, 443–452. [Google Scholar] [CrossRef]
- Strettell, M.D.; Donaldson, P.T.; Thomson, L.J.; Santrach, P.J.; Moore, S.B.; Czaja, A.J.; Williams, R. Allelic basis for HLA-encoded susceptibility to type 1 autoimmune hepatitis. Gastroenterology 1997, 112, 2028–2035. [Google Scholar] [CrossRef]
- Donaldson, P.T.; Doherty, D.G.; Hayllar, K.M.; McFarlane, I.G.; Johnson, P.J.; Williams, R. Susceptibility to autoimmune chronic active hepatitis: Human leukocyte antigens DR4 and A1-B8-DR3 are independent risk factors. Hepatology 1991, 13, 701–706. [Google Scholar] [CrossRef]
- Muratori, P.; Czaja, A.J.; Muratori, L.; Pappas, G.; Maccariello, S.; Cassani, F.; Granito, A.; Ferrari, R.; Mantovani, V.; Lenzi, M.; et al. Genetic distinctions between autoimmune hepatitis in Italy and North America. World J. Gastroenterol. 2005, 11, 1862–1866. [Google Scholar] [CrossRef]
- Umemura, T.; Katsuyama, Y.; Yoshizawa, K.; Kimura, T.; Joshita, S.; Komatsu, M.; Matsumoto, A.; Tanaka, E.; Ota, M. Human leukocyte antigen class II haplotypes affect clinical characteristics and progression of type 1 autoimmune hepatitis in Japan. PLoS ONE 2014, 9, e100565. [Google Scholar] [CrossRef] [PubMed]
- Cancado, E.L.R.; Goldbaum-Crescente, J.; Terrabuio, D.R.B. HLA-related genetic susceptibility in autoimmune hepatitis according to autoantibody profile. Front. Immunol. 2022, 13, 1032591. [Google Scholar] [CrossRef]
- Bala, S.; Marcos, M.; Kodys, K.; Csak, T.; Catalano, D.; Mandrekar, P.; Szabo, G. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor α (TNFα) production via increased mRNA half-life in alcoholic liver disease. J. Biol. Chem. 2011, 286, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Bala, S. MicroRNAs in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Muratori, L.; Longhi, M.S. The interplay between regulatory and effector T cells in autoimmune hepatitis: Implications for innovative treatment strategies. J. Autoimmun. 2013, 46, 74–80. [Google Scholar] [CrossRef]
- Sgamato, C.; Rocco, A.; Compare, D.; Minieri, S.; Marchitto, S.A.; Maurea, S.; Nardone, G. Autoimmune liver diseases and SARS-CoV-2. World J. Gastroenterol. 2023, 29, 1838–1851. [Google Scholar] [CrossRef]
- Marjot, T.; Buescher, G.; Sebode, M.; Barnes, E.; Barritt, A.S., 4th; Armstrong, M.J.; Baldelli, L.; Kennedy, J.; Mercer, C.; Ozga, A.K.; et al. SARS-CoV-2 infection in patients with autoimmune hepatitis. J. Hepatol. 2021, 74, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Schultheiß, C.; Steinmann, S.; Lohse, A.W.; Binder, M. B cells in autoimmune hepatitis: Bystanders or central players? Semin. Immunopathol. 2022, 44, 411–427. [Google Scholar] [CrossRef]
- Muratori, L.; Lohse, A.W.; Lenzi, M. Diagnosis and management of autoimmune hepatitis. BMJ 2023, 380, e070201. [Google Scholar] [CrossRef] [PubMed]
- Mack, C.L.; Adams, D.; Assis, D.N.; Kerkar, N.; Manns, M.P.; Mayo, M.J.; Vierling, J.M.; Alsawas, M.; Murad, M.H.; Czaja, A.J. Diagnosis and Management of Autoimmune Hepatitis in Adults and Children: 2019 Practice Guidance and Guidelines From the American Association for the Study of Liver Diseases. Hepatology 2020, 72, 671–722. [Google Scholar] [CrossRef] [PubMed]
- Sucher, E.; Sucher, R.; Gradistanac, T.; Brandacher, G.; Schneeberger, S.; Berg, T. Autoimmune Hepatitis-Immunologically Triggered Liver Pathogenesis-Diagnostic and Therapeutic Strategies. J. Immunol. Res. 2019, 2019, 9437043. [Google Scholar] [CrossRef] [PubMed]
- Baven-Pronk, M.A.; Hew, J.M., Jr.; Biewenga, M.; Tushuizen, M.E.; van den Berg, A.P.; Bouma, G.; Brouwer, J.T.; van Hoek, B. Calcineurin Inhibitors in the Treatment of Adult Autoimmune Hepatitis: A Systematic Review. J. Clin. Transl. Hepatol. 2022, 10, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.; Samant, H. Primary Biliary Cholangitis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. [Google Scholar] [PubMed]
- Trivella, J.; John, B.V.; Levy, C. Primary biliary cholangitis: Epidemiology, prognosis, and treatment. Hepatol. Commun. 2023, 7, e0179. [Google Scholar] [CrossRef]
- Lleo, A.; Maroni, L.; Glaser, S.; Alpini, G.; Marzioni, M. Role of cholangiocytes in primary biliary cirrhosis. Semin. Liver Dis. 2014, 34, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Selmi, C.; Mayo, M.J.; Bach, N.; Ishibashi, H.; Invernizzi, P.; Gish, R.G.; Gordon, S.C.; Wright, H.I.; Zweiban, B.; Podda, M.; et al. Primary biliary cirrhosis in monozygotic and dizygotic twins: Genetics, epigenetics, and environment. Gastroenterology 2004, 127, 485–492. [Google Scholar] [CrossRef]
- Umemura, T.; Ota, M. Genetic factors affect the etiology, clinical characteristics and outcome of autoimmune hepatitis. Clin. J. Gastroenterol. 2015, 8, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zheng, H.; Tian, Q.B.; Rui, M.N.; Liu, D.W. HLA-DR polymorphism and primary biliary cirrhosis: Evidence from a meta-analysis. Arch. Med. Res. 2014, 45, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Alric, L.; Fort, M.; Izopet, J.; Vinel, J.P.; Charlet, J.P.; Selves, J.; Puel, J.; Pascal, J.P.; Duffaut, M.; Abbal, M. Genes of the major histocompatibility complex class II influence the outcome of hepatitis C virus infection. Gastroenterology 1997, 113, 1675–1681. [Google Scholar] [CrossRef] [PubMed]
- Bontkes, H.J.; de Gruijl, T.D.; Walboomers, J.M.; Schiller, J.T.; Dillner, J.; Helmerhorst, T.J.; Verheijen, R.H.; Scheper, R.J.; Meijer, C.J. Immune responses against human papillomavirus (HPV) type 16 virus-like particles in a cohort study of women with cervical intraepithelial neoplasia. II. Systemic but not local IgA responses correlate with clearance of HPV-16. J. Gen. Virol. 1999, 80 Pt 2, 409–417. [Google Scholar] [CrossRef]
- Thursz, M.R.; Kwiatkowski, D.; Allsopp, C.E.; Greenwood, B.M.; Thomas, H.C.; Hill, A.V. Association between an MHC class II allele and clearance of hepatitis B virus in the Gambia. N. Engl. J. Med. 1995, 332, 1065–1069. [Google Scholar] [CrossRef] [PubMed]
- Joshita, S.; Umemura, T.; Tanaka, E.; Ota, M. Genetics and epigenetics in the pathogenesis of primary biliary cholangitis. Clin. J. Gastroenterol. 2018, 11, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Gerussi, A.; Paraboschi, E.M.; Cappadona, C.; Caime, C.; Binatti, E.; Cristoferi, L.; Asselta, R.; Invernizzi, P. The Role of Epigenetics in Primary Biliary Cholangitis. Int. J. Mol. Sci. 2022, 23, 4873. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Liu, X.; Xu, C.; Lu, Y.; Xie, G.; Lu, Y.; Gu, X.; Walker, E.J.; Jing, K.; Juran, B.D.; et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N. Engl. J. Med. 2009, 360, 2544–2555, Erratum in N. Engl. J. Med. 2009, 360, 2797–2798. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pulickal, A.S.; Hambleton, S.; Callaghan, M.J.; Moore, C.E.; Goulding, J.; Goodsall, A.; Baretto, R.; Lammas, D.A.; Anderson, S.T.; Levin, M.; et al. Biliary cirrhosis in a child with inherited interleukin-12 deficiency. J. Trop. Pediatr. 2008, 54, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Qiao, L.; Wang, B. Primary biliary cirrhosis is a generalized autoimmune epithelitis. Int. J. Mol. Sci. 2015, 16, 6432–6446. [Google Scholar] [CrossRef] [PubMed]
- Gulamhusein, A.F.; Hirschfield, G.M. Pathophysiology of primary biliary cholangitis. Best Pract. Res. Clin. Gastroenterol. 2018, 34, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Rigopoulou, E.I.; Bogdanos, D.P. Role of autoantibodies in the clinical management of primary biliary cholangitis. World J. Gastroenterol. 2023, 29, 1795–1810. [Google Scholar] [CrossRef] [PubMed]
- Lleo, A.; Selmi, C.; Invernizzi, P.; Podda, M.; Coppel, R.L.; Mackay, I.R.; Gores, G.J.; Ansari, A.A.; van de Water, J.; Gershwin, M.E. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology 2009, 49, 871–879. [Google Scholar] [CrossRef]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2019, 69, 394–419. [Google Scholar] [CrossRef] [PubMed]
- Saare, M.; Hämarik, U.; Venta, R.; Panarina, M.; Zucchelli, C.; Pihlap, M.; Remm, A.; Kisand, K.; Toots, U.; Möll, K.; et al. SP140L, an Evolutionarily Recent Member of the SP100 Family, Is an Autoantigen in Primary Biliary Cirrhosis. J. Immunol. Res. 2015, 2015, 526518. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.D.; Yao, T.T.; Wang, Y.; Wang, G.Q. Treatment of primary biliary cholangitis with ursodeoxycholic acid, prednisolone and immunosuppressants in patients not responding to ursodeoxycholic acid alone and the prognostic indicators. Clin. Res. Hepatol. Gastroenterol. 2020, 44, 874–884. [Google Scholar] [CrossRef]
- Granito, A.; Yang, W.H.; Muratori, L.; Lim, M.J.; Nakajima, A.; Ferri, S.; Pappas, G.; Quarneti, C.; Bianchi, F.B.; Bloch, D.B.; et al. PML nuclear body component Sp140 is a novel autoantigen in primary biliary cirrhosis. Am. J. Gastroenterol. 2010, 105, 125–131. [Google Scholar] [CrossRef]
- Nakamura, M.; Kondo, H.; Mori, T.; Komori, A.; Matsuyama, M.; Ito, M.; Takii, Y.; Koyabu, M.; Yokoyama, T.; Migita, K.; et al. Anti-gp210 and anti-centromere antibodies are different risk factors for the progression of primary biliary cirrhosis. Hepatology 2007, 45, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Melero, S.; Spirlì, C.; Zsembery, A.; Medina, J.F.; Joplin, R.E.; Duner, E.; Zuin, M.; Neuberger, J.M.; Prieto, J.; Strazzabosco, M. Defective regulation of cholangiocyte Cl-/HCO3(-) and Na+/H+ exchanger activities in primary biliary cirrhosis. Hepatology 2002, 35, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, S.; Wenniger, L.M.d.B.; Paulusma, C.C.; van Vliet, S.J.; Jefferson, D.M.; Elferink, R.P.O.; Beuers, U. A biliary HCO3-umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology 2012, 55, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; He, X.; Rojas, M.; Leung, P.S.C.; Gao, L. Mechanism-based target therapy in primary biliary cholangitis: Opportunities before liver cirrhosis? Front. Immunol. 2023, 14, 1184252. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Choi, J.; Chen, Y.; Invernizzi, P.; Yang, G.; Zhang, W.; Shao, T.; Jordan, F.; Nemeria, N.S.; Coppel, R.L.; et al. Coli and the etiology of human PBC: Antimitochondrial antibodies and spreading determinants. Hepatology 2021, 75, 266–279. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, M.; Moritoki, Y.; Lian, Z.-X.; Zhang, W.; Yoshida, K.; Wakabayashi, K.; Yang, G.-X.; Nakatani, T.; Vierling, J.; Lindor, K.; et al. Biochemical and immunologic effects of rituximab in patients with primary biliary cirrhosis and an incomplete response to ursodeoxycholic acid. Hepatology 2011, 55, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shao, T.; Leung, P.S.; Tsuneyama, K.; Heuer, L.; Young, H.A.; Ridgway, W.M.; Gershwin, M.E. Dual b-cell targeting therapy ameliorates autoimmune cholangitis. J. Autoimmun. 2022, 132, 102897. [Google Scholar] [CrossRef]
- Aron, J.H.; Bowlus, C.L. The immunobiology of primary sclerosing cholangitis. Semin. Immunopathol. 2009, 31, 383–397. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef]
- Abbas, N.; Quraishi, M.N.; Trivedi, P. Emerging drugs for the treatment of primary sclerosing cholangitis. Curr. Opin. Pharmacol. 2022, 62, 23–35. [Google Scholar] [CrossRef]
- O’Mahony, C.A.; Vierling, J.M. Etiopathogenesis of primary sclerosing cholangitis. Semin. Liver Dis. 2006, 26, 3–21. [Google Scholar] [CrossRef] [PubMed]
- van Munster, K.N.; Bergquist, A.; Ponsioen, C.Y. Inflammatory bowel disease and primary sclerosing cholangitis: One disease or two? J. Hepatol. 2024, 80, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Dyson, J.K.; Beuers, U.; Jones, D.E.J.; Lohse, A.W.; Hudson, M. Primary sclerosing cholangitis. Lancet 2018, 391, 2547–2559. [Google Scholar] [CrossRef]
- Sjöblom, N.; Boyd, S.; Kautiainen, H.; Arola, J.; Färkkilä, M. Novel histological scoring for predicting disease outcome in primary sclerosing cholangitis. Histopathology 2022, 81, 192–204. [Google Scholar] [CrossRef]
- Reich, M.; Spomer, L.; Klindt, C.; Fuchs, K.; Stindt, J.; Deutschmann, K.; Höhne, J.; Liaskou, E.; Hov, J.R.; Karlsen, T.H.; et al. Downregulation of TGR5 (GPBAR1) in biliary epithelial cells contributes to the pathogenesis of sclerosing cholangitis. J. Hepatol. 2021, 75, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; Ali, A.H.; Lindor, K.D. Primary Sclerosing Cholangitis, Part 1: Epidemiology, Etiopathogenesis, Clinical Features, and Treatment. Gastroenterol. Hepatol. 2018, 14, 293–304. [Google Scholar] [PubMed] [PubMed Central]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygård, S.; Vesterhus, M.; Høivik, M.L.; Trøseid, M.; Marschall, H.-U.; Schrumpf, E.; Moum, B.; et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef]
- Little, R.; Wine, E.; Kamath, B.M.; Griffiths, A.M.; Ricciuto, A. Gut microbiome in primary sclerosing cholangitis: A review. World J. Gastroenterol. 2020, 26, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Karlsen, T.H. Genetics of primary sclerosing cholangitis and pathophysiological implications. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Hurley, E.H.; Park, Y.; Ko, S. Primary sclerosing cholangitis (PSC) and inflammatory bowel disease (IBD): A condition exemplifying the crosstalk of the gut-liver axis. Exp. Mol. Med. 2023, 55, 1380–1387. [Google Scholar] [CrossRef]
- Goode, E.C.; Rushbrook, S.M. A review of the medical treatment of primary sclerosing cholangitis in the 21st century. Ther. Adv. Chronic Dis. 2016, 7, 68–85. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Talwalkar, J.A.; Lindor, K.D. Role of the microbiota and antibiotics in primary sclerosing cholangitis. BioMed Res. Int. 2013, 2013, 389537. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Filipovic, B.; Marjanovic-Haljilji, M.; Blagojevic, D.; Dragovic, M.; Krsmanovic, E.; Matovic, A.; Panic, N.; Kiurski, S.; Zagorac, Z.; Milanovic, M.; et al. A Closer Look into Autoimmune Liver Diseases. Int. J. Mol. Sci. 2025, 26, 1863. https://doi.org/10.3390/ijms26051863
Filipovic B, Marjanovic-Haljilji M, Blagojevic D, Dragovic M, Krsmanovic E, Matovic A, Panic N, Kiurski S, Zagorac Z, Milanovic M, et al. A Closer Look into Autoimmune Liver Diseases. International Journal of Molecular Sciences. 2025; 26(5):1863. https://doi.org/10.3390/ijms26051863
Chicago/Turabian StyleFilipovic, Branka, Marija Marjanovic-Haljilji, Dragana Blagojevic, Milica Dragovic, Emilija Krsmanovic, Ana Matovic, Natasa Panic, Stanimir Kiurski, Zagor Zagorac, Miljan Milanovic, and et al. 2025. "A Closer Look into Autoimmune Liver Diseases" International Journal of Molecular Sciences 26, no. 5: 1863. https://doi.org/10.3390/ijms26051863
APA StyleFilipovic, B., Marjanovic-Haljilji, M., Blagojevic, D., Dragovic, M., Krsmanovic, E., Matovic, A., Panic, N., Kiurski, S., Zagorac, Z., Milanovic, M., Markovic, O., Djokovic, A., Glisic, T., Dragasevic, S., & Popovic, D. (2025). A Closer Look into Autoimmune Liver Diseases. International Journal of Molecular Sciences, 26(5), 1863. https://doi.org/10.3390/ijms26051863