
Development and Characterization of Three Novel FGFR Inhibitor Resistant Cervical Cancer Cell Lines to Help Drive Cervical Cancer Research

 ,
,  ,
,  and
and
Abstract

1. Introduction
2. Results
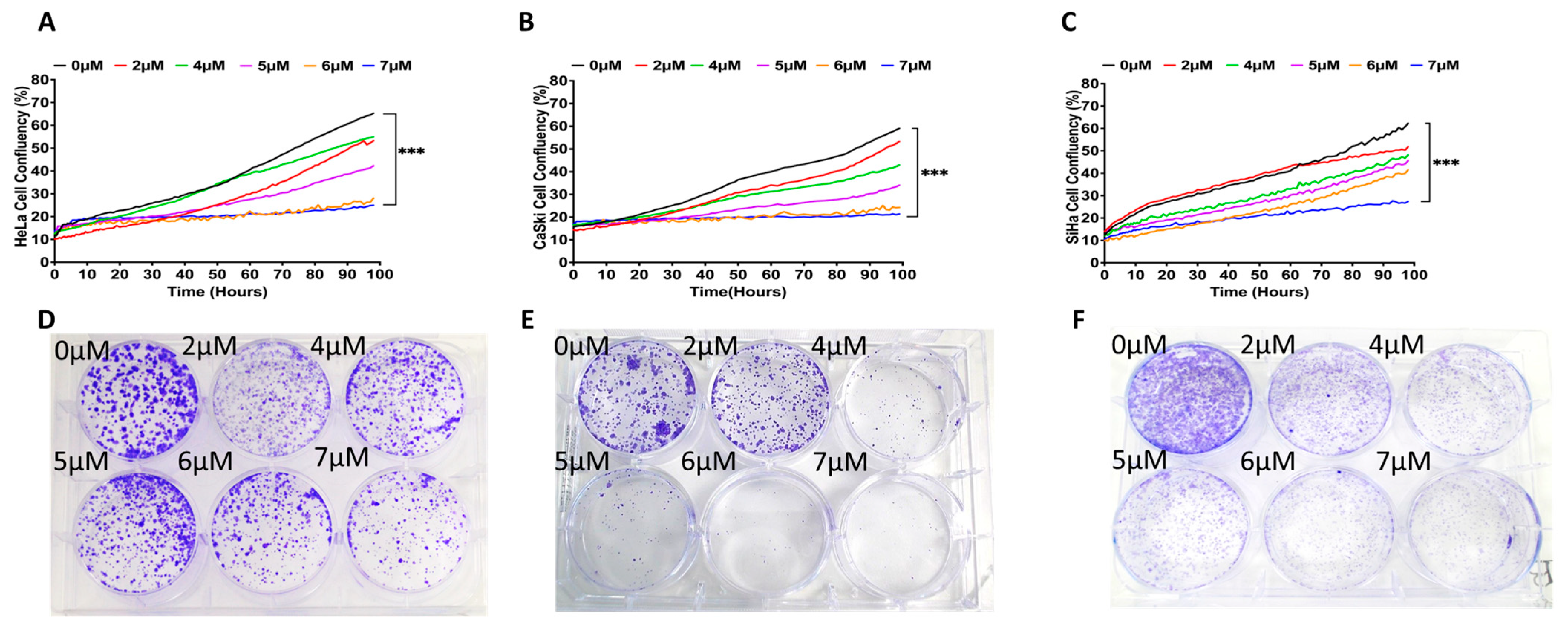
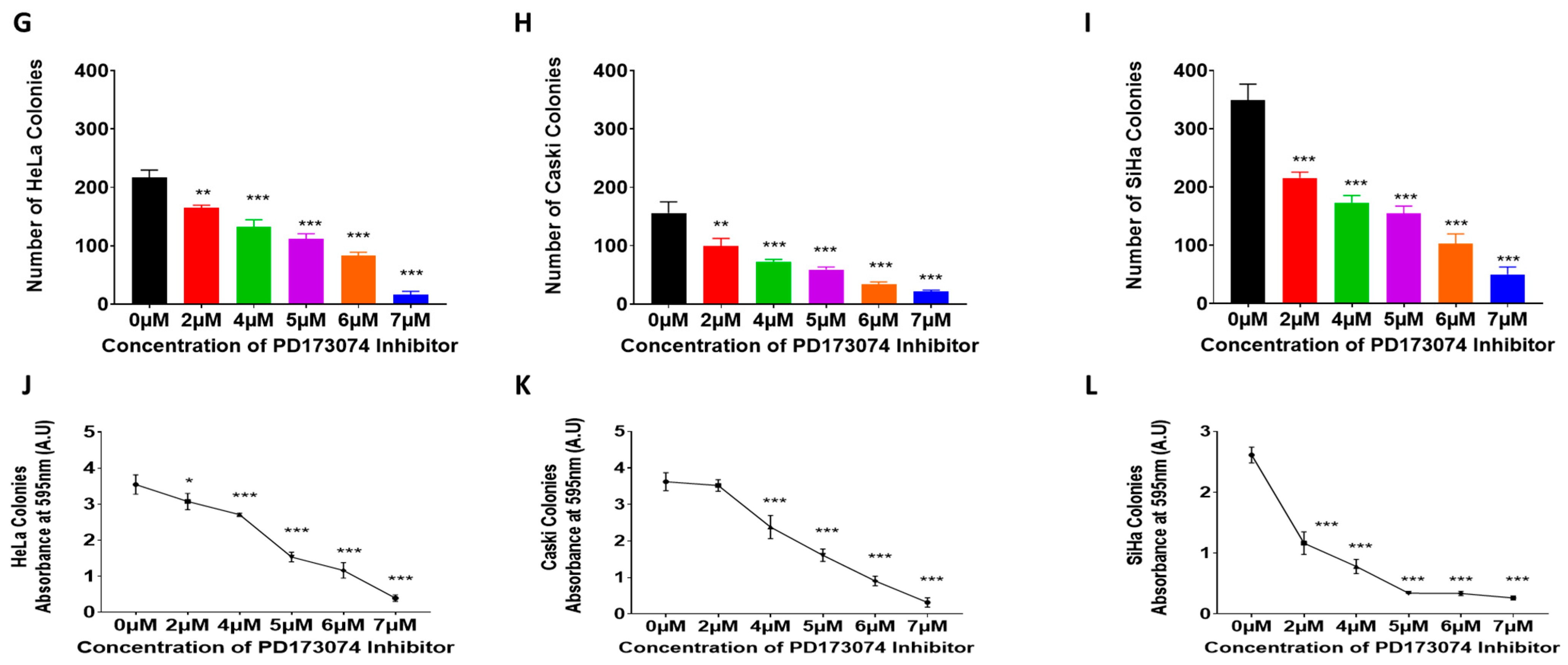
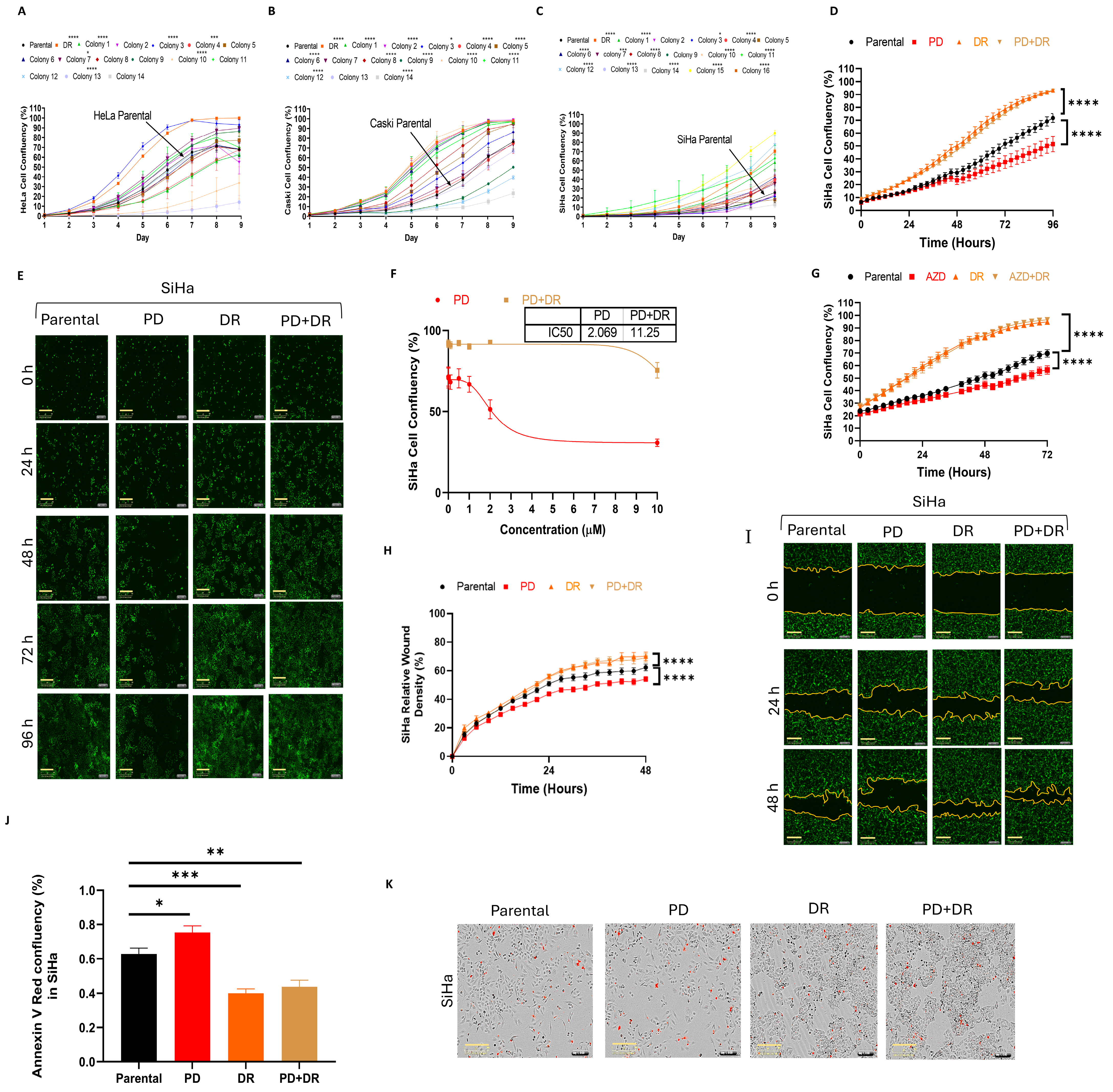
2.1. Establishing PD173074-Resistant Cervical Cancer Cell Lines
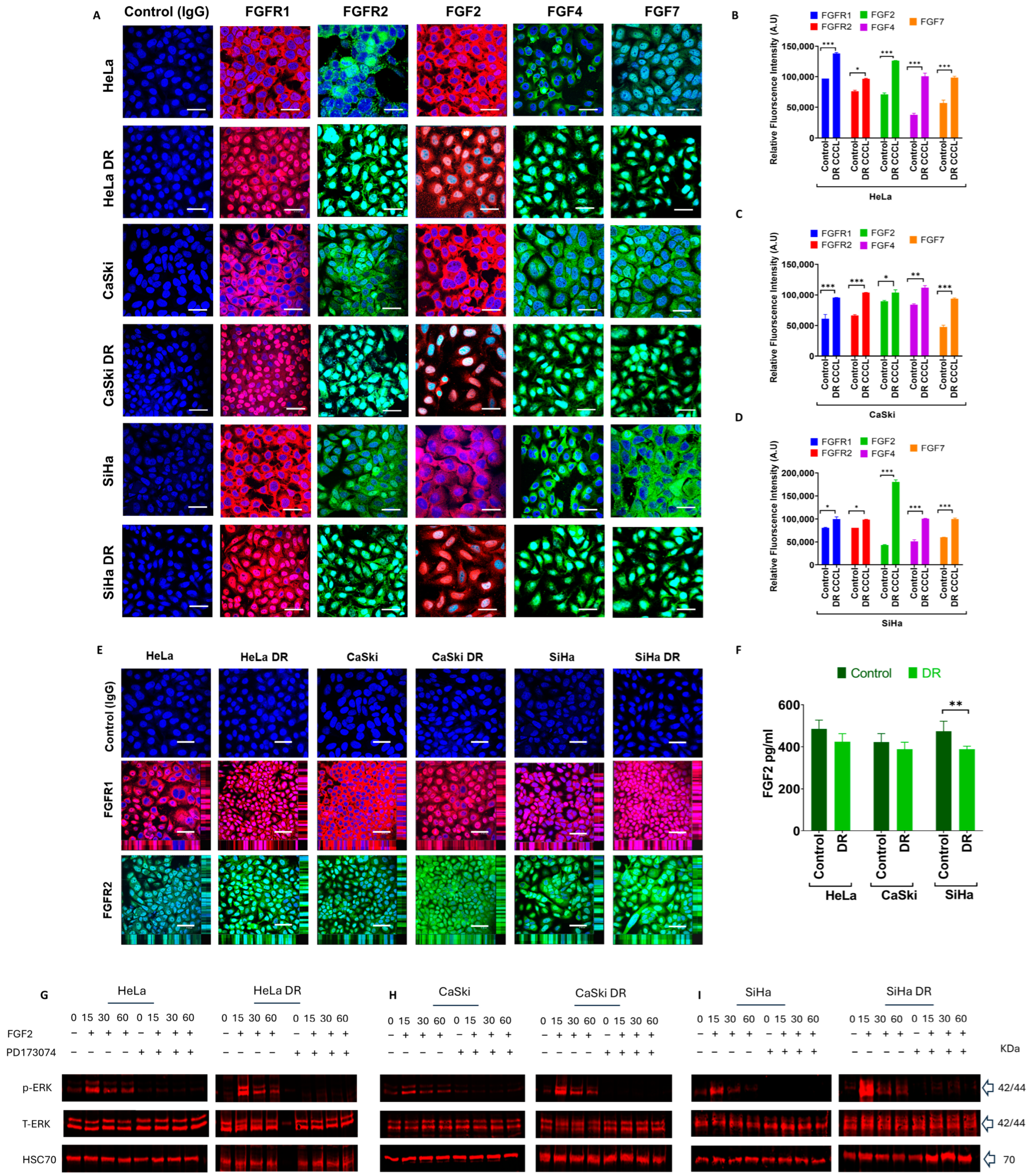
2.2. Profiling FGF(R) Protein Expression, Secretion, and Activation in PD173074-Resistant Cell Lines
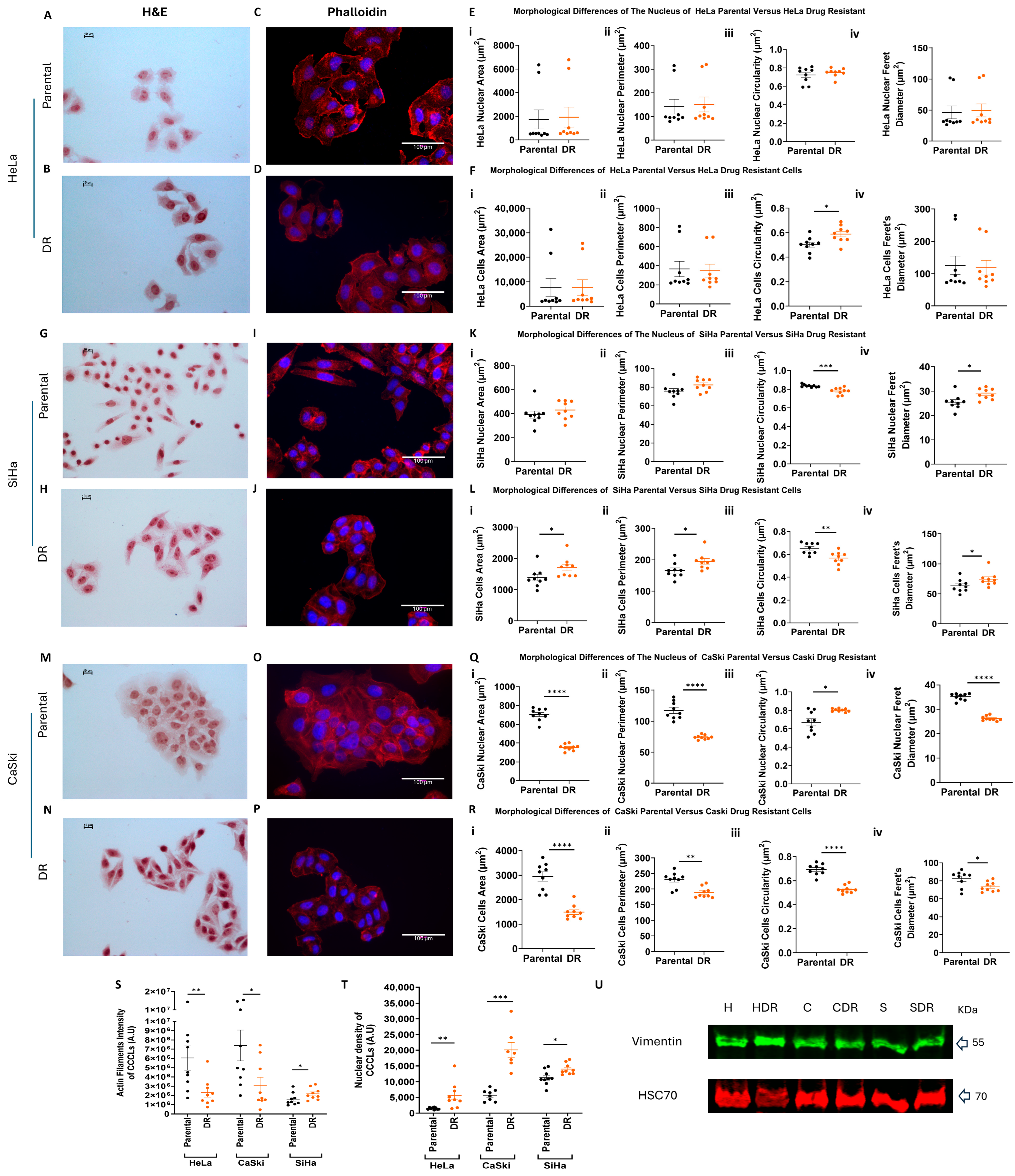
2.3. Morphological and Functional Characteristics of PD173074-Resistant Cervical Cancer Cell Lines
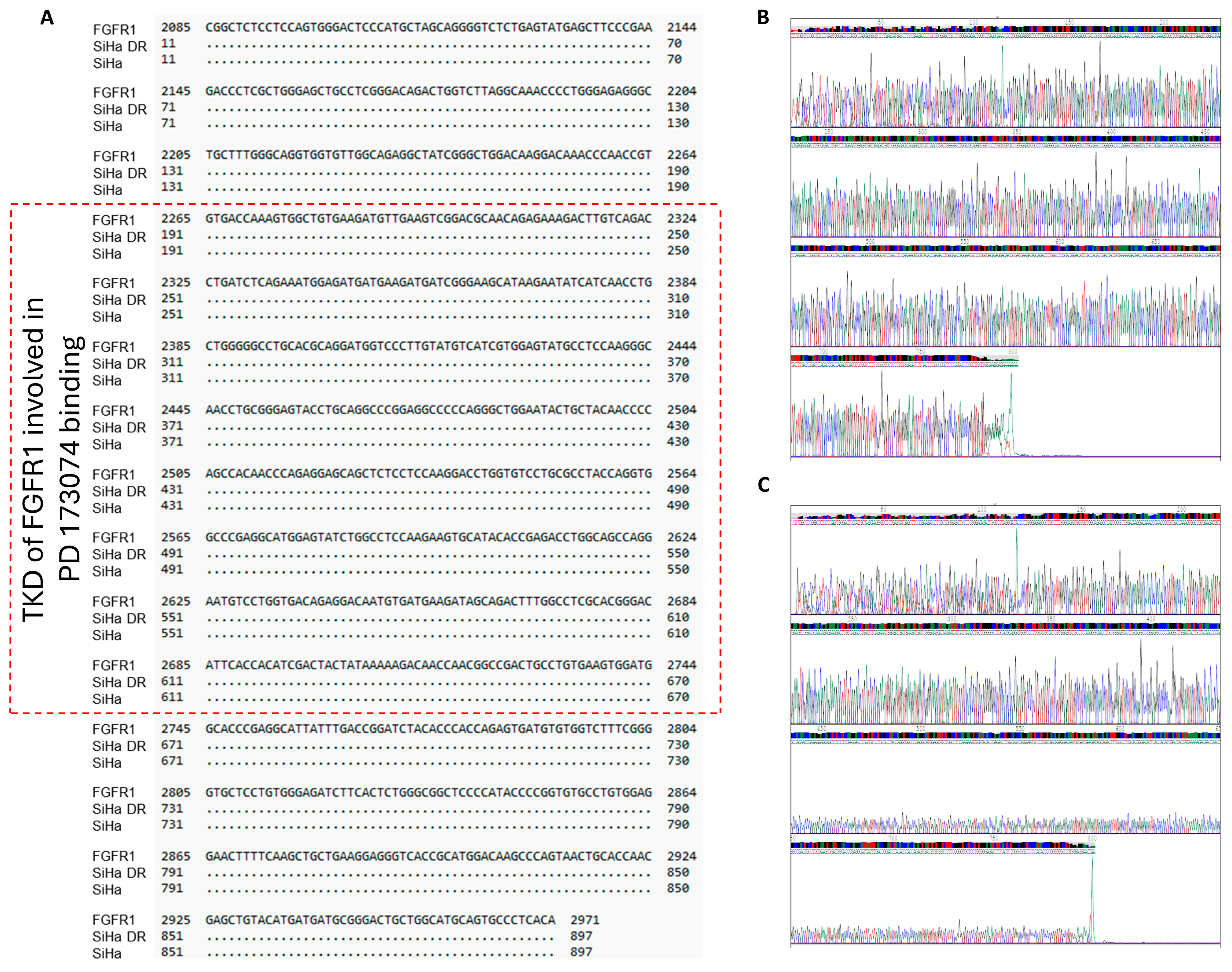
2.4. No Mutations Exist in the FGFR1 TK Domain of the Three PD173074-Resistant Cervical Cancer Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Establishment of PD173074-Resistant Cell Lines
4.3. PD173074-Resistant Colony Formation
4.4. Functional Studies
4.4.1. Cell Proliferation
4.4.2. Lateral Migration
4.4.3. Apoptosis
4.5. Western Blot Analysis
4.6. Immunocytochemistry and H&E Staining
4.7. ELISA
4.8. Polymerase Chain Reaction (PCR) and Sanger Sequencing
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Ala | Alanine |
| Asp | Aspartic acid |
| ATP | Adenosine triphosphate |
| AZD | AZD4547 |
| Bcl-2 | B-cell lymphoma 2 |
| Bax | Bcl-2-associated protein x |
| BSA | Bovine serum albumin |
| CCCLs | Cervical cancer cell lines |
| HCCCLs | Human cervical cancer cell lines |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DR | Drug resistant |
| ECM | Extracellular matrix |
| EGFR | Epidermal growth factor receptor |
| ERK | Extracellular signal-regulated kinases |
| EMT | Epithelial-mesenchymal transition |
| ER positive | Oestrogen receptor positive |
| FBS | Fetal bovine serum |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| HPV | Human papillomavirus |
| IgG | immunoglobulin G |
| Ile | Isoleucine |
| ICC | Immunocytochemistry |
| Glu | Glutamic acid |
| KIT | Stem cell factor receptor |
| Lys | Lysine |
| MAPK | Mitogen-activated protein kinase |
| MET | Mesenchymal Epithelial Transition |
| Met | Methionine |
| TK | Tyrosine kinase |
| TKD | Tyrosine kinase domain |
| TKIs | Tyrosine kinase inhibitors |
| TME | Tumour microenvironment |
| PBS | Phosphate buffered saline |
| PBST | Phosphate buffered saline tween 20 |
| PD | PD173074 |
| PDGFR | Platelet-derived growth factor receptor |
| P-ERK | Phospho Extracellular signal-regulated kinase |
| PI3K/AKT | Phosphoinositol-3-kinase/protein kinase B |
| Phe | Phenylalanine |
| RET | Rearranged during transfection |
| RTKs | Receptors tyrosine kinase |
| VAL | Valine |
| VEGFR | Vascular endothelial growth factor receptor |
References
- World Health Organization. Cervical Cancer. 2020. Available online: https://www.who.int/health-topics/cervical-cancer#tab=tab_1 (accessed on 1 November 2024).
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Pruski, D.; Millert-Kalinska, S.; Klemenska, P.; Jach, R.; Przybylski, M. Clinical use of the Onclarity test with extended HPV genotyping and phenotyping in patients with suspected squamous intraepithelial lesions. Ginekol. Pol. 2024, 95, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Wang, L.; Zhang, C.; Hong, Z.; Han, Z. Cervical cancer heterogeneity: A constant battle against viruses and drugs. Biomark. Res. 2022, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- George, I.A.; Chauhan, R.; Dhawale, R.; Iyer, R.; Limaye, S.; Sankaranarayanan, R.; Venkataramanan, R.; Kumar, P. Insights into therapy resistance in cervical cancer. Adv. Cancer Biol.-Metastasis 2022, 6, 100074. [Google Scholar] [CrossRef]
- Bhattacharjee, R.; Dey, T.; Kumar, L.; Kar, S.; Sarkar, R.; Ghorai, M.; Malik, S.; Jha, N.K.; Vellingiri, B.; Kesari, K.K.; et al. Cellular landscaping of cisplatin resistance in cervical cancer. Biomed. Pharmacother. 2022, 153, 113345. [Google Scholar] [CrossRef]
- Masadah, R.; Rauf, S.; Pratama, M.Y.; Tiribelli, C.; Pascut, D. The Role of microRNAs in the Cisplatin- and Radio-Resistance of Cervical Cancer. Cancers 2021, 13, 1168. [Google Scholar] [CrossRef]
- Tan, X.; Wang, X.; Liao, X.; Wang, X.; Jiang, Z.; Liang, W.; Cao, C.; Gong, D.; Hu, Z.; Tian, X. Downregulation of VPS13C promotes cisplatin resistance in cervical cancer by upregulating GSTP1. iScience 2023, 26, 107315. [Google Scholar] [CrossRef]
- Bou Antoun, N.; Chioni, A.M. Dysregulated Signalling Pathways Driving Anticancer Drug Resistance. Int. J. Mol. Sci. 2023, 24, 12222. [Google Scholar] [CrossRef]
- Kaemmerer, E.; Loessner, D.; Avery, V.M. Addressing the tumour microenvironment in early drug discovery: A strategy to overcome drug resistance and identify novel targets for cancer therapy. Drug Discov. Today 2021, 26, 663–676. [Google Scholar] [CrossRef]
- Roy, S.; Kumaravel, S.; Sharma, A.; Duran, C.L.; Bayless, K.J.; Chakraborty, S. Hypoxic tumor microenvironment: Implications for cancer therapy. Exp. Biol. Med. 2020, 245, 1073–1086. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, T.; Su, W.; Dou, Z.; Zhao, D.; Jin, X.; Lei, H.; Wang, J.; Xie, X.; Cheng, B.; et al. Mutant p53 in cancer: From molecular mechanism to therapeutic modulation. Cell Death Dis. 2022, 13, 974. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, W.; Li, Y.; Fu, Y.; Xu, C.; Yu, M. Polyphyllin I Sensitizes Cisplatin-Resistant Human Cervical Cancer Cells to Cisplatin Treatment. Nutr. Cancer 2024, 76, 656–665. [Google Scholar] [CrossRef]
- Kuno, I.; Takayanagi, D.; Asami, Y.; Murakami, N.; Matsuda, M.; Shimada, Y.; Hirose, S.; Kato, M.K.; Komatsu, M.; Hamamoto, R.; et al. TP53 mutants and non-HPV16/18 genotypes are poor prognostic factors for concurrent chemoradiotherapy in locally advanced cervical cancer. Sci. Rep. 2021, 11, 19261. [Google Scholar] [CrossRef]
- Heerboth, S.; Lapinska, K.; Snyder, N.; Leary, M.; Rollinson, S.; Sarkar, S. Use of epigenetic drugs in disease: An overview. Genet. Epigenet. 2014, 6, 9–19. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Wang, N.; Ma, T.; Yu, B. Targeting epigenetic regulators to overcome drug resistance in cancers. Signal Transduct. Target. Ther. 2023, 8, 69. [Google Scholar] [CrossRef]
- Wajapeyee, N.; Gupta, R. Epigenetic Alterations and Mechanisms That Drive Resistance to Targeted Cancer Therapies. Cancer Res. 2021, 81, 5589–5595. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Liang, X.J. Overcoming drug efflux-based multidrug resistance in cancer with nanotechnology. Chin. J. Cancer 2012, 31, 100–109. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Pastan, I.H. The Role of Multidrug Resistance Efflux Pumps in Cancer: Revisiting a JNCI Publication Exploring Expression of the MDR1 (P-glycoprotein) Gene. J. Natl. Cancer Inst. 2015, 107, djv222. [Google Scholar] [CrossRef]
- Li, L.Y.; Guan, Y.D.; Chen, X.S.; Yang, J.M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2020, 11, 629266. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed]
- Boshuizen, J.; Peeper, D.S. Rational Cancer Treatment Combinations: An Urgent Clinical Need. Mol. Cell 2020, 78, 1002–1018. [Google Scholar] [CrossRef] [PubMed]
- Tomuleasa, C.; Tigu, A.B.; Munteanu, R.; Moldovan, C.S.; Kegyes, D.; Onaciu, A.; Gulei, D.; Ghiaur, G.; Einsele, H.; Croce, C.M. Therapeutic advances of targeting receptor tyrosine kinases in cancer. Signal Transduct. Target. Ther. 2024, 9, 201. [Google Scholar] [CrossRef] [PubMed]
- Sudhesh Dev, S.; Zainal Abidin, S.A.; Farghadani, R.; Othman, I.; Naidu, R. Receptor Tyrosine Kinases and Their Signaling Pathways as Therapeutic Targets of Curcumin in Cancer. Front. Pharmacol. 2021, 12, 772510. [Google Scholar] [CrossRef]
- Ebrahimi, N.; Fardi, E.; Ghaderi, H.; Palizdar, S.; Khorram, R.; Vafadar, R.; Ghanaatian, M.; Rezaei-Tazangi, F.; Baziyar, P.; Ahmadi, A.; et al. Receptor tyrosine kinase inhibitors in cancer. Cell. Mol. Life Sci. 2023, 80, 104. [Google Scholar] [CrossRef]
- Ou, X.; Gao, G.; Habaz, I.A.; Wang, Y. Mechanisms of resistance to tyrosine kinase inhibitor-targeted therapy and overcoming strategies. MedComm 2024, 5, e694. [Google Scholar] [CrossRef]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181. [Google Scholar] [CrossRef]
- Laestander, C.; Engstrom, W. Role of fibroblast growth factors in elicitation of cell responses. Cell Prolif. 2014, 47, 3–11. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Marie, P.J. FGF signaling pathways in endochondral and intramembranous bone development and human genetic disease. Genes Dev. 2002, 16, 1446–1465. [Google Scholar] [CrossRef]
- Chioni, A.M.; Grose, R.P. Biological Significance and Targeting of the FGFR Axis in Cancer. Cancers 2021, 13, 5681. [Google Scholar] [CrossRef]
- Kommalapati, A.; Tella, S.H.; Borad, M.; Javle, M.; Mahipal, A. FGFR Inhibitors in Oncology: Insight on the Management of Toxicities in Clinical Practice. Cancers 2021, 13, 2968. [Google Scholar] [CrossRef] [PubMed]
- Patel, T.H.; Marcus, L.; Horiba, M.N.; Donoghue, M.; Chatterjee, S.; Mishra-Kalyani, P.S.; Schuck, R.N.; Li, Y.; Zhang, X.; Fourie Zirkelbach, J.; et al. FDA Approval Summary: Pemigatinib for Previously Treated, Unresectable Locally Advanced or Metastatic Cholangiocarcinoma with FGFR2 Fusion or Other Rearrangement. Clin. Cancer Res. 2023, 29, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, A.; Vogel, A. Futibatinib: Second EMA approval for FGFR inhibitor in cholangiocarcinoma. ESMO Open 2023, 8, 102049. [Google Scholar] [CrossRef]
- Mahmood, H.A.; Tomas Bort, E.; Walker, A.J.; Grose, R.P.; Chioni, A.M. FGF signalling facilitates cervical cancer progression. FEBS J. 2022, 289, 3440–3456. [Google Scholar] [CrossRef]
- Terai, H.; Soejima, K.; Yasuda, H.; Nakayama, S.; Hamamoto, J.; Arai, D.; Ishioka, K.; Ohgino, K.; Ikemura, S.; Sato, T.; et al. Activation of the FGF2-FGFR1 autocrine pathway: A novel mechanism of acquired resistance to gefitinib in NSCLC. Mol. Cancer Res. 2013, 11, 759–767. [Google Scholar] [CrossRef]
- Coleman, S.J.; Chioni, A.M.; Ghallab, M.; Anderson, R.K.; Lemoine, N.R.; Kocher, H.M.; Grose, R.P. Nuclear translocation of FGFR1 and FGF2 in pancreatic stellate cells facilitates pancreatic cancer cell invasion. EMBO Mol. Med. 2014, 6, 467–481. [Google Scholar] [CrossRef]
- Chioni, A.M.; Grose, R. FGFR1 cleavage and nuclear translocation regulates breast cancer cell behavior. J. Cell Biol. 2012, 197, 801–817. [Google Scholar] [CrossRef]
- Suh, J.; Kim, D.H.; Kim, S.J.; Cho, N.C.; Lee, Y.H.; Jang, J.H.; Surh, Y.J. Nuclear Localization of Fibroblast Growth Factor Receptor 1 in Breast Cancer Cells Interacting with Cancer Associated Fibroblasts. J. Cancer Prev. 2022, 27, 68–76. [Google Scholar] [CrossRef]
- Servetto, A.; Kollipara, R.; Formisano, L.; Lin, C.C.; Lee, K.M.; Sudhan, D.R.; Gonzalez-Ericsson, P.I.; Chatterjee, S.; Guerrero-Zotano, A.; Mendiratta, S.; et al. Nuclear FGFR1 Regulates Gene Transcription and Promotes Antiestrogen Resistance in ER(+) Breast Cancer. Clin. Cancer Res. 2021, 27, 4379–4396. [Google Scholar] [CrossRef]
- Iams, W.T.; Lovly, C.M. Molecular Pathways: Clinical Applications and Future Direction of Insulin-like Growth Factor-1 Receptor Pathway Blockade. Clin. Cancer Res. 2015, 21, 4270–4277. [Google Scholar] [CrossRef] [PubMed]
- Malchers, F.; Ercanoglu, M.; Schutte, D.; Castiglione, R.; Tischler, V.; Michels, S.; Dahmen, I.; Bragelmann, J.; Menon, R.; Heuckmann, J.M.; et al. Mechanisms of Primary Drug Resistance in FGFR1-Amplified Lung Cancer. Clin. Cancer Res. 2017, 23, 5527–5536. [Google Scholar] [CrossRef] [PubMed]
- Yue, S.; Li, Y.; Chen, X.; Wang, J.; Li, M.; Chen, Y.; Wu, D. FGFR-TKI resistance in cancer: Current status and perspectives. J. Hematol. Oncol. 2021, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.T.; Kohsaka, S.; Ikegami, M.; Ikeuchi, H.; Ueno, T.; Li, K.; Beyett, T.S.; Koyama, T.; Shimizu, T.; Yamamoto, N.; et al. Comprehensive functional evaluation of variants of fibroblast growth factor receptor genes in cancer. NPJ Precis. Oncol. 2021, 5, 66. [Google Scholar] [CrossRef]
- Chell, V.; Balmanno, K.; Little, A.S.; Wilson, M.; Andrews, S.; Blockley, L.; Hampson, M.; Gavine, P.R.; Cook, S.J. Tumour cell responses to new fibroblast growth factor receptor tyrosine kinase inhibitors and identification of a gatekeeper mutation in FGFR3 as a mechanism of acquired resistance. Oncogene 2013, 32, 3059–3070. [Google Scholar] [CrossRef]
- Bunney, T.D.; Wan, S.; Thiyagarajan, N.; Sutto, L.; Williams, S.V.; Ashford, P.; Koss, H.; Knowles, M.A.; Gervasio, F.L.; Coveney, P.V.; et al. The Effect of Mutations on Drug Sensitivity and Kinase Activity of Fibroblast Growth Factor Receptors: A Combined Experimental and Theoretical Study. EBioMedicine 2015, 2, 194–204. [Google Scholar] [CrossRef]
- Szymczyk, J.; Sluzalska, K.D.; Materla, I.; Opalinski, L.; Otlewski, J.; Zakrzewska, M. FGF/FGFR-Dependent Molecular Mechanisms Underlying Anti-Cancer Drug Resistance. Cancers 2021, 13, 5796. [Google Scholar] [CrossRef]
- Mahapatra, S.; Jonniya, N.A.; Koirala, S.; Ursal, K.D.; Kar, P. The FGF/FGFR signalling mediated anti-cancer drug resistance and therapeutic intervention. J. Biomol. Struct. Dyn. 2023, 41, 13509–13533. [Google Scholar] [CrossRef]
- Cheng, W.; Wang, M.; Tian, X.; Zhang, X. An overview of the binding models of FGFR tyrosine kinases in complex with small molecule inhibitors. Eur. J. Med. Chem. 2017, 126, 476–490. [Google Scholar] [CrossRef]
- Rabal, O.; Schneider, G.; Borrell, J.I.; Teixido, J. Structure-based virtual screening of FGFR inhibitors: Cross-decoys and induced-fit effect. BioDrugs 2007, 21, 31–45. [Google Scholar] [CrossRef]
- Lu, X.; Smaill, J.B.; Patterson, A.V.; Ding, K. Discovery of Cysteine-targeting Covalent Protein Kinase Inhibitors. J. Med. Chem. 2022, 65, 58–83. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, M.; Froum, S.; Hamby, J.M.; Schroeder, M.C.; Panek, R.L.; Lu, G.H.; Eliseenkova, A.V.; Green, D.; Schlessinger, J.; Hubbard, S.R. Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO J. 1998, 17, 5896–5904. [Google Scholar] [CrossRef] [PubMed]
- Klein, T.; Vajpai, N.; Phillips, J.J.; Davies, G.; Holdgate, G.A.; Phillips, C.; Tucker, J.A.; Norman, R.A.; Scott, A.D.; Higazi, D.R.; et al. Structural and dynamic insights into the energetics of activation loop rearrangement in FGFR1 kinase. Nat. Commun. 2015, 6, 7877. [Google Scholar] [CrossRef] [PubMed]
- Goyal, L.; Saha, S.K.; Liu, L.Y.; Siravegna, G.; Leshchiner, I.; Ahronian, L.G.; Lennerz, J.K.; Vu, P.; Deshpande, V.; Kambadakone, A.; et al. Polyclonal Secondary FGFR2 Mutations Drive Acquired Resistance to FGFR Inhibition in Patients with FGFR2 Fusion-Positive Cholangiocarcinoma. Cancer Discov. 2017, 7, 252–263. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, C.; Lu, G.; Hu, Z.; Chen, Q.; Du, X. FGF/FGFR signaling pathway involved resistance in various cancer types. J. Cancer 2020, 11, 2000–2007. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef]
- van der Wekken, A.J.; Saber, A.; Hiltermann, T.J.; Kok, K.; van den Berg, A.; Groen, H.J. Resistance mechanisms after tyrosine kinase inhibitors afatinib and crizotinib in non-small cell lung cancer, a review of the literature. Crit. Rev. Oncol. Hematol. 2016, 100, 107–116. [Google Scholar] [CrossRef]
- Tang, C.; Schafranek, L.; Watkins, D.B.; Parker, W.T.; Moore, S.; Prime, J.A.; White, D.L.; Hughes, T.P. Tyrosine kinase inhibitor resistance in chronic myeloid leukemia cell lines: Investigating resistance pathways. Leuk. Lymphoma 2011, 52, 2139–2147. [Google Scholar] [CrossRef]
- Gandhi, J.; Zhang, J.; Xie, Y.; Soh, J.; Shigematsu, H.; Zhang, W.; Yamamoto, H.; Peyton, M.; Girard, L.; Lockwood, W.W.; et al. Alterations in genes of the EGFR signaling pathway and their relationship to EGFR tyrosine kinase inhibitor sensitivity in lung cancer cell lines. PLoS ONE 2009, 4, e4576. [Google Scholar] [CrossRef]
- Plunder, S.; Danesin, C.; Glise, B.; Ferreira, M.A.; Merino-Aceituno, S.; Theveneau, E. Modelling variability and heterogeneity of EMT scenarios highlights nuclear positioning and protrusions as main drivers of extrusion. Nat. Commun. 2024, 15, 7365. [Google Scholar] [CrossRef] [PubMed]
- Ogden, S.; Carys, K.; Ahmed, I.; Bruce, J.; Sharrocks, A.D. Regulatory chromatin rewiring promotes metabolic switching during adaptation to oncogenic receptor tyrosine kinase inhibition. Oncogene 2022, 41, 4808–4822. [Google Scholar] [CrossRef] [PubMed]
- Mishra, J.; Chakraborty, S.; Niharika; Roy, A.; Manna, S.; Baral, T.; Nandi, P.; Patra, S.K. Mechanotransduction and epigenetic modulations of chromatin: Role of mechanical signals in gene regulation. J. Cell. Biochem. 2024, 125, e30531. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Saha, P.; Samanta, A.; Bishayee, A. Emerging Concepts of Hybrid Epithelial-to-Mesenchymal Transition in Cancer Progression. Biomolecules 2020, 10, 1561. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.T.; Yang, M.H. Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms. Cells 2020, 9, 623. [Google Scholar] [CrossRef]
- Canciello, A.; Cervero-Varona, A.; Peserico, A.; Mauro, A.; Russo, V.; Morrione, A.; Giordano, A.; Barboni, B. “In medio stat virtus”: Insights into hybrid E/M phenotype attitudes. Front. Cell Dev. Biol. 2022, 10, 1038841. [Google Scholar] [CrossRef]
- Sample, R.A.; Nogueira, M.F.; Mitra, R.D.; Puram, S.V. Epigenetic regulation of hybrid epithelial-mesenchymal cell states in cancer. Oncogene 2023, 42, 2237–2248. [Google Scholar] [CrossRef]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2019, 194, 161–184. [Google Scholar] [CrossRef]
- Augimeri, G.; Gonzalez, M.E.; Paoli, A.; Eido, A.; Choi, Y.; Burman, B.; Djomehri, S.; Karthikeyan, S.K.; Varambally, S.; Buschhaus, J.M.; et al. A hybrid breast cancer/mesenchymal stem cell population enhances chemoresistance and metastasis. JCI Insight 2023, 8, 18. [Google Scholar] [CrossRef]
- Bontemps, I.; Lallemand, C.; Biard, D.; Dechamps, N.; Kortulewski, T.; Bourneuf, E.; Siberchicot, C.; Boussin, F.; Chevillard, S.; Campalans, A.; et al. Loss of CD24 promotes radiation- and chemo-resistance by inducing stemness properties associated with a hybrid E/M state in breast cancer cells. Oncol. Rep. 2023, 49, 4. [Google Scholar] [CrossRef]
- Bornes, L.; Belthier, G.; van Rheenen, J. Epithelial-to-Mesenchymal Transition in the Light of Plasticity and Hybrid E/M States. J. Clin. Med. 2021, 10, 2403. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S.; et al. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Jolly, M.K.; Balamurugan, K. Hypoxia, partial EMT and collective migration: Emerging culprits in metastasis. Transl. Oncol. 2020, 13, 100845. [Google Scholar] [CrossRef]
- Huang, Y.; Hong, W.; Wei, X. The molecular mechanisms and therapeutic strategies of EMT in tumor progression and metastasis. J. Hematol. Oncol. 2022, 15, 129. [Google Scholar] [CrossRef]
- Coban, B.; Bergonzini, C.; Zweemer, A.J.M.; Danen, E.H.J. Metastasis: Crosstalk between tissue mechanics and tumour cell plasticity. Br. J. Cancer 2021, 124, 49–57. [Google Scholar] [CrossRef]
- Leggett, S.E.; Hruska, A.M.; Guo, M.; Wong, I.Y. The epithelial-mesenchymal transition and the cytoskeleton in bioengineered systems. Cell Commun. Signal. 2021, 19, 32. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef]
- Datta, A.; Deng, S.; Gopal, V.; Yap, K.C.; Halim, C.E.; Lye, M.L.; Ong, M.S.; Tan, T.Z.; Sethi, G.; Hooi, S.C.; et al. Cytoskeletal Dynamics in Epithelial-Mesenchymal Transition: Insights into Therapeutic Targets for Cancer Metastasis. Cancers 2021, 13, 1882. [Google Scholar] [CrossRef]
- Williams, E.D.; Gao, D.; Redfern, A.; Thompson, E.W. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nat. Rev. Cancer 2019, 19, 716–732. [Google Scholar] [CrossRef]
- Santamaria, P.G.; Moreno-Bueno, G.; Cano, A. Contribution of Epithelial Plasticity to Therapy Resistance. J. Clin. Med. 2019, 8, 676. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Wang, P.; Toh, A.; Thompson, E.W. New Insights Into the Role of Phenotypic Plasticity and EMT in Driving Cancer Progression. Front. Mol. Biosci. 2020, 7, 71. [Google Scholar] [CrossRef] [PubMed]
- Bhat, G.R.; Sethi, I.; Sadida, H.Q.; Rah, B.; Mir, R.; Algehainy, N.; Albalawi, I.A.; Masoodi, T.; Subbaraj, G.K.; Jamal, F.; et al. Cancer cell plasticity: From cellular, molecular, and genetic mechanisms to tumor heterogeneity and drug resistance. Cancer Metastasis Rev. 2024, 43, 197–228. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Pierce, S.E.; Kroeger, C.; Stover, D.G.; Pattabiraman, D.R.; Thiru, P.; Liu Donaher, J.; Reinhardt, F.; Chaffer, C.L.; Keckesova, Z.; et al. Integrin-beta4 identifies cancer stem cell-enriched populations of partially mesenchymal carcinoma cells. Proc. Natl. Acad. Sci. USA 2017, 114, E2337–E2346. [Google Scholar] [CrossRef]
- Kyuno, D.; Takasawa, A.; Kikuchi, S.; Takemasa, I.; Osanai, M.; Kojima, T. Role of tight junctions in the epithelial-to-mesenchymal transition of cancer cells. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183503. [Google Scholar] [CrossRef]
- Gibson, S.V.; Madzharova, E.; Tan, A.C.; Allen, M.D.; Keller, U.A.D.; Louise Jones, J.; Carter, E.P.; Grose, R.P. ADAMTS3 restricts cancer invasion in models of early breast cancer progression through enhanced fibronectin degradation. Matrix Biol. 2023, 121, 74–89. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Template Strand Length | Start | Stop | Product Length |
|---|---|---|---|---|
| TKD FGFR Forward (set 1) TAGGCAAACCCCTGGGAGA | 19 | 9 | 27 | 802 |
| TKD FGFR Reverse (set 1) AAGGTGGGTCTCTGTGAGGG | 20 | 810 | 791 | |
| TKD FGFR Forward (set 2) TGCATCCATGAACTCTGGGG | 20 | 5 | 24 | 825 |
| TKD FGFR Reverse (set 2) AGTTCCTCCACAGGCACAC | 19 | 829 | 811 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bou Antoun, N.; Afshan Mahmood, H.-T.-N.; Walker, A.J.; Modjtahedi, H.; Grose, R.P.; Chioni, A.-M. Development and Characterization of Three Novel FGFR Inhibitor Resistant Cervical Cancer Cell Lines to Help Drive Cervical Cancer Research. Int. J. Mol. Sci. 2025, 26, 1799. https://doi.org/10.3390/ijms26051799
Bou Antoun N, Afshan Mahmood H-T-N, Walker AJ, Modjtahedi H, Grose RP, Chioni A-M. Development and Characterization of Three Novel FGFR Inhibitor Resistant Cervical Cancer Cell Lines to Help Drive Cervical Cancer Research. International Journal of Molecular Sciences. 2025; 26(5):1799. https://doi.org/10.3390/ijms26051799
Chicago/Turabian StyleBou Antoun, Nauf, Hiba-Tun-Noor Afshan Mahmood, Anthony J. Walker, Helmout Modjtahedi, Richard P. Grose, and Athina-Myrto Chioni. 2025. "Development and Characterization of Three Novel FGFR Inhibitor Resistant Cervical Cancer Cell Lines to Help Drive Cervical Cancer Research" International Journal of Molecular Sciences 26, no. 5: 1799. https://doi.org/10.3390/ijms26051799
APA StyleBou Antoun, N., Afshan Mahmood, H.-T.-N., Walker, A. J., Modjtahedi, H., Grose, R. P., & Chioni, A.-M. (2025). Development and Characterization of Three Novel FGFR Inhibitor Resistant Cervical Cancer Cell Lines to Help Drive Cervical Cancer Research. International Journal of Molecular Sciences, 26(5), 1799. https://doi.org/10.3390/ijms26051799