
In Vivo and In Vitro Models of Hepatic Fibrosis for Pharmacodynamic Evaluation and Pathology Exploration
Abstract
1. Introduction
2. In Vivo Hf Models
2.1. HF Animal Models Induced by Chemicals
2.1.1. HF Animal Model Induced by Ethanol
2.1.2. HF Animal Model Induced by Carbon Tetrachloride (CCl4)
2.1.3. HF Animal Model Induced by TAA
2.1.4. HF Animal Model Induced by DMN
2.2. Immunological HF Models
2.2.1. Virus-Induced HF Animal Models
2.2.2. ConA-Induced HF Animal Models
2.2.3. Schistosoma-Induced HF Animal Models
2.2.4. Porcine Serum-Induced HF Animal Models
2.3. Establishment of HF Animal Models by Surgical Operation
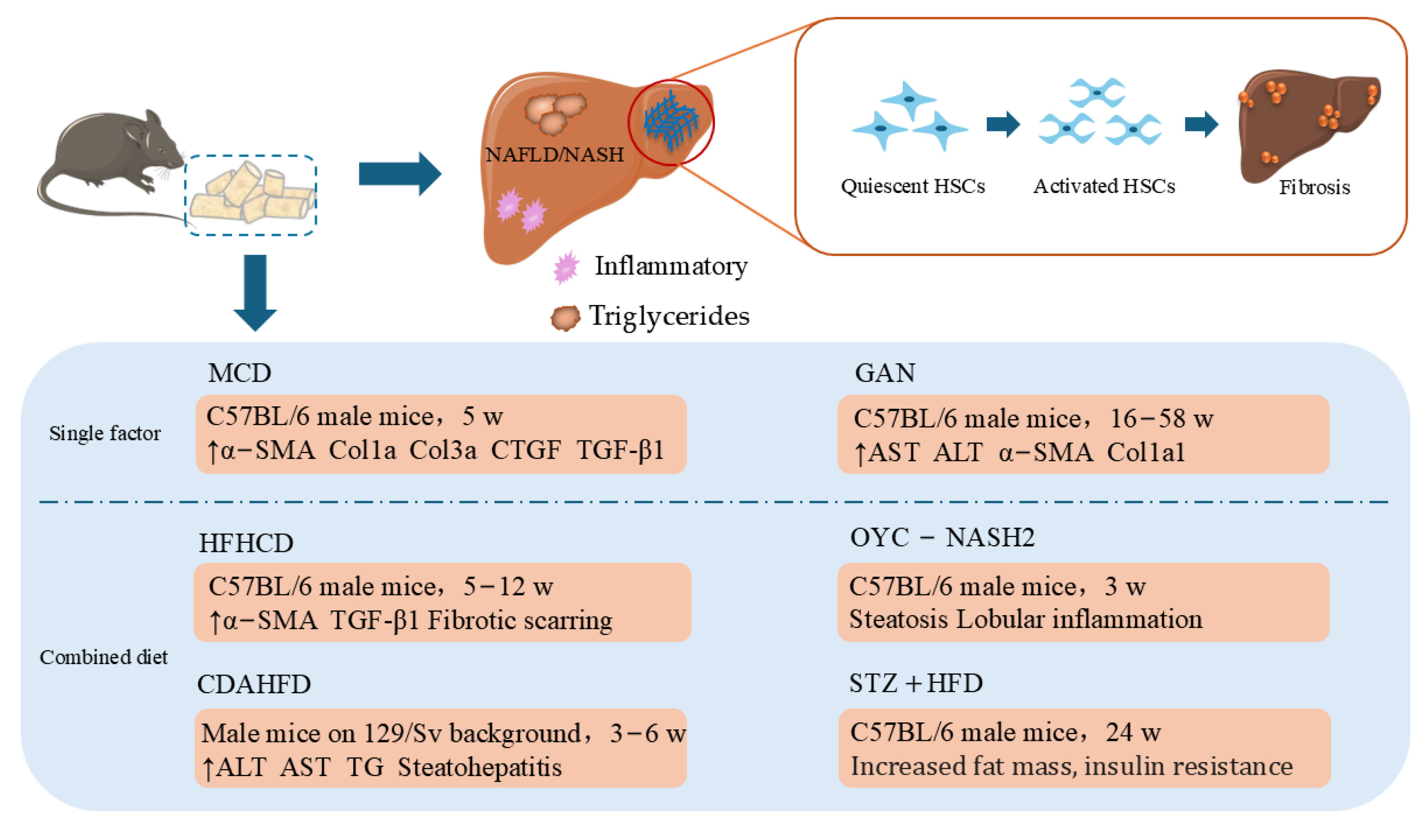
2.4. HF Animal Models Induced by Specific Diets
2.4.1. MCD Diet
2.4.2. Gubra–Amylin (GAN)-Induced NASH
2.4.3. Combined Diet
2.5. Genetically Modified HF Animal Models
3. In Vitro HF Models
3.1. Primary HSCs
3.2. HSCs Lines
3.3. Three-Dimensional In Vitro HF Model
3.3.1. Three-Dimensional Coculture Models
3.3.2. Three-Dimensional Organoid Models
3.3.3. Three-Dimensional-Bioprinted Liver Tissues
3.3.4. Precision-Cut Liver Slice Culture
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Mohammed, O.S.; Attia, H.G.; Mohamed, B.; Elbaset, M.A.; Fayed, H.M. Current investigations for liver fibrosis treatment: Between repurposing the FDA-approved drugs and the other emerging approaches. J. Pharm. Pharm. Sci. 2023, 26, 11808. [Google Scholar] [CrossRef] [PubMed]
- Horn, P.; Tacke, F. Metabolic reprogramming in liver fibrosis. Cell Metab. 2024, 36, 1439–1455. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Li, Z.J.; Gou, H.Z.; Song, X.J.; Zhang, L. The gut microbiota-bile acid axis: A potential therapeutic target for liver fibrosis. Front. Cell. Infect. Microbiol. 2022, 12, 945368. [Google Scholar] [CrossRef] [PubMed]
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef]
- Aydın, M.M.; Akçalı, K.C. Liver fibrosis. Turk. J. Gastroenterol. 2018, 29, 14–21. [Google Scholar] [CrossRef]
- Sun, M.; Kisseleva, T. Reversibility of liver fibrosis. Clin. Res. Hepatol. Gastroenterol. 2015, 39, S60–S63. [Google Scholar] [CrossRef]
- Caligiuri, A.; Gentilini, A.; Pastore, M.; Gitto, S.; Marra, F.J.C. Cellular and molecular mechanisms underlying liver fibrosis regression. Cells 2021, 10, 2759. [Google Scholar] [CrossRef]
- Arab, J.P.; Addolorato, G.; Mathurin, P.; Thursz, M.R. Alcohol-Associated Liver Disease: Integrated Management With Alcohol Use Disorder. Clin. Gastroenterol. Hepatol. 2023, 21, 2124–2134. [Google Scholar] [CrossRef]
- Buchanan, R.; Sinclair, J.M. Alcohol use disorder and the liver. Addiction 2021, 116, 1270–1278. [Google Scholar] [CrossRef]
- Minea, H.; Singeap, A.-M.; Sfarti, C.V.; Girleanu, I.; Chiriac, S.; Muzica, C.; Cuciureanu, T.; Petrea, O.C.; Huiban, L.; Zenovia, S. The Impact of Alcohol Consumption Pattern on Liver Fibrosis in Asymptomatic Patients. J. Clin. Med. 2023, 12, 7405. [Google Scholar] [CrossRef]
- Mackowiak, B.; Fu, Y.; Maccioni, L.; Gao, B. Alcohol-associated liver disease. J. Clin. Investig. 2024, 134, e176345. [Google Scholar] [CrossRef] [PubMed]
- Hyun, J.; Han, J.; Lee, C.; Yoon, M.; Jung, Y. Pathophysiological Aspects of Alcohol Metabolism in the Liver. Int. J. Mol. Sci. 2021, 22, 5717. [Google Scholar] [CrossRef] [PubMed]
- Blomdahl, J.; Nasr, P.; Ekstedt, M.; Kechagias, S. Moderate alcohol consumption is associated with advanced fibrosis in non-alcoholic fatty liver disease and shows a synergistic effect with type 2 diabetes mellitus. Metabolism 2021, 115, 154439. [Google Scholar] [CrossRef]
- Sayed, E.A.; Badr, G.; Hassan, K.A.-H.; Waly, H.; Ozdemir, B.; Mahmoud, M.H.; Alamery, S. Induction of liver fibrosis by CCl4 mediates pathological alterations in the spleen and lymph nodes: The potential therapeutic role of propolis. Saudi J. Biol. Sci. 2021, 28, 1272–1282. [Google Scholar] [CrossRef]
- Mohi-Ud-Din, R.; Mir, R.H.; Sawhney, G.; Dar, M.A.; Bhat, Z.A. Possible pathways of hepatotoxicity caused by chemical agents. Curr. Drug Metab. 2019, 20, 867–879. [Google Scholar] [CrossRef]
- Unsal, V.; Cicek, M.; Sabancilar, İ. Toxicity of carbon tetrachloride, free radicals and role of antioxidants. Rev. Environ. Health. 2021, 36, 279–295. [Google Scholar] [CrossRef]
- Scholten, D.; Trebicka, J.; Liedtke, C.; Weiskirchen, R. The carbon tetrachloride model in mice. Lab. Anim. 2015, 49, 4–11. [Google Scholar] [CrossRef]
- Zhao, Z.; Dong, H.; Li, B.; Shen, B.; Guo, Y.; Gu, T.; Qu, Y.; Cai, X.; Lu, L. Hydroxynitone suppresses hepatic stellate cell activation by inhibiting TGF-β1 phosphorylation to alleviate CCl 4-induced liver fibrosis in rats. Nan Fang Yi Ke Da Xue Xue Bao 2022, 42, 1511–1516. [Google Scholar]
- Dai, Z.; Song, G.; Balakrishnan, A.; Yang, T.; Yuan, Q.; Möbus, S.; Weiss, A.C.; Bentler, M.; Zhu, J.; Jiang, X.; et al. Growth differentiation factor 11 attenuates liver fibrosis via expansion of liver progenitor cells. Gut 2020, 69, 1104–1115. [Google Scholar] [CrossRef]
- Dong, S.; Chen, Q.L.; Song, Y.N.; Sun, Y.; Wei, B.; Li, X.Y.; Hu, Y.Y.; Liu, P.; Su, S.B. Mechanisms of CCl4-induced liver fibrosis with combined transcriptomic and proteomic analysis. J. Toxicol. Sci. 2016, 41, 561–572. [Google Scholar] [CrossRef]
- McGill, M.R.; Jaeschke, H. Animal models of drug-induced liver injury. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Fitzhugh, O.G.; Nelson, A.A. Liver Tumors in Rats Fed Thiourea or Thioacetamide. Science 1948, 108, 626–628. [Google Scholar] [CrossRef] [PubMed]
- Elnfarawy, A.A.; Nashy, A.E.; Abozaid, A.M.; Komber, I.F.; Elweshahy, R.H.; Abdelrahman, R.S. Vinpocetine attenuates thioacetamide-induced liver fibrosis in rats. Hum. Exp. Toxicol. 2021, 40, 355–368. [Google Scholar] [CrossRef]
- Ghanim, A.M.H.; Younis, N.S.; Metwaly, H.A. Vanillin augments liver regeneration effectively in Thioacetamide induced liver fibrosis rat model. Life Sci. 2021, 286, 120036. [Google Scholar] [CrossRef]
- Salem, G.E.M.; Azzam, S.M.; Nasser, M.A.F.; El Malah, T.; Abd El-Latief, H.M.; Khan, R.H.; Chavanich, S.; Anwar, H.M. Bacterial protease alleviate chronic liver fibrosis induced by thioacetamide through suppression of hepatic stellate cells consequently decrease its proliferative index. Int. J. Biol. Macromol. 2023, 239, 124243. [Google Scholar] [CrossRef]
- Enciso, N.; Amiel, J.; Fabián-Domínguez, F.; Pando, J.; Rojas, N.; Cisneros-Huamaní, C.; Nava, E.; Enciso, J. Model of Liver Fibrosis Induction by Thioacetamide in Rats for Regenerative Therapy Studies. Anal. Cell. Pathol. 2022, 2022, 2841894. [Google Scholar] [CrossRef]
- Stenbäck, F.; Ala-Kokko, L.; Ryhänen, L. Morphological, immunohistochemical and ultrastructural changes in dimethylnitrosamine [correction of dimenthylnitrosamine] induced liver injury. Effect of malotilate. Histol. Histopathol. 1989, 4, 95–104. [Google Scholar]
- Chooi, K.F.; Kuppan Rajendran, D.B.; Phang, S.S.; Toh, H.H. The Dimethylnitrosamine Induced Liver Fibrosis Model in the Rat. J. Vis. Exp. 2016, 112, e54208. [Google Scholar] [CrossRef]
- George, J.; Chandrakasan, G. Molecular characteristics of dimethylnitrosamine induced fibrotic liver collagen. Biochim. Biophys. Acta 1996, 1292, 215–222. [Google Scholar] [CrossRef]
- George, J.; Tsuchishima, M.; Tsutsumi, M. Molecular mechanisms in the pathogenesis of N-nitrosodimethylamine induced hepatic fibrosis. Cell Death Dis. 2019, 10, 18. [Google Scholar] [CrossRef]
- George, J.; Tsuchishima, M.; Tsutsumi, M. Metabolism of N-nitrosodimethylamine, methylation of macromolecules, and development of hepatic fibrosis in rodent models. J. Mol. Med. 2020, 98, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Kim, H.G.; Wang, J.H.; Choi, M.K.; Han, J.M.; Lee, J.S.; Son, C.G. Comparison of TGF-β, PDGF, and CTGF in hepatic fibrosis models using DMN, CCl4, and TAA. Drug Chem. Toxicol. 2016, 39, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Fortea, J.I.; Fernández-Mena, C.; Puerto, M.; Ripoll, C.; Almagro, J.; Bañares, J.; Bellón, J.M.; Bañares, R.; Vaquero, J. Comparison of Two Protocols of Carbon Tetrachloride-Induced Cirrhosis in Rats—Improving Yield and Reproducibility. Sci. Rep. 2018, 8, 9163. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wang, X.; Xing, W.; Li, F.; Liang, M.; Li, K.; He, Y.; Wang, J. An update on animal models of liver fibrosis. Front. Med. 2023, 10, 1160053. [Google Scholar] [CrossRef]
- Czaja, A.J. Global Disparities and Their Implications in the Occurrence and Outcome of Autoimmune Hepatitis. Dig. Dis. Sci. 2017, 62, 2277–2292. [Google Scholar] [CrossRef]
- Llanwarne, F.; Helmby, H. Granuloma formation and tissue pathology in Schistosoma japonicum versus Schistosoma mansoni infections. Parasite Immunol. 2021, 43, e12778. [Google Scholar] [CrossRef]
- Li, T.Y.; Yang, Y.; Zhou, G.; Tu, Z.K. Immune suppression in chronic hepatitis B infection associated liver disease: A review. World J. Gastroenterol. 2019, 25, 3527–3537. [Google Scholar] [CrossRef]
- Ye, L.; Yu, H.; Li, C.; Hirsch, M.L.; Zhang, L.; Samulski, R.J.; Li, W.; Liu, Z. Adeno-Associated Virus Vector Mediated Delivery of the HBV Genome Induces Chronic Hepatitis B Virus Infection and Liver Fibrosis in Mice. PLoS ONE 2015, 10, e0130052. [Google Scholar] [CrossRef]
- Kan, F.; Ye, L.; Yan, T.; Cao, J.; Zheng, J.; Li, W. Proteomic and transcriptomic studies of HBV-associated liver fibrosis of an AAV-HBV-infected mouse model. BMC Genom. 2017, 18, 641. [Google Scholar] [CrossRef]
- Li, H.C.; Yang, C.H.; Lo, S.Y. Roles of microRNAs in Hepatitis C Virus Replication and Pathogenesis. Viruses 2022, 14, 1776. [Google Scholar] [CrossRef]
- Shi, L.; Zhou, L.; Han, M.; Zhang, Y.; Zhang, Y.; Yuan, X.X.; Lu, H.P.; Wang, Y.; Yang, X.L.; Liu, C.; et al. Calcitriol attenuates liver fibrosis through hepatitis C virus nonstructural protein 3-transactivated protein 1-mediated TGF β1/Smad3 and NF-κB signaling pathways. World J. Gastroenterol. 2023, 29, 2798–2817. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Zhu, C.; Liao, F.; Zhao, L.; Shen, L.; Jiang, W. Reciprocal induction of hepatitis C virus replication and stimulation of hepatic profibrogenic cytokine release and cellular viability by YKL-40. Ann. Transl. Med. 2021, 9, 1649. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Duan, X.; Zhu, C.; Liu, X.; Jeyarajan, A.J.; Xu, M.; Tu, Z.; Sheng, Q.; Chen, D.; Zhu, C.; et al. Hepatitis B and Hepatitis C Virus Infection Promote Liver Fibrogenesis through a TGF-β1-Induced OCT4/Nanog Pathway. J. Immunol. 2022, 208, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayanan, B.; Davenport, M. Biliary atresia: A comprehensive review. J. Autoimmun. 2016, 73, 1–9. [Google Scholar] [CrossRef]
- Mohanty, S.K.; Donnelly, B.; Temple, H.; Mowery, S.; Poling, H.M.; Meller, J.; Malik, A.; McNeal, M.; Tiao, G. Rhesus rotavirus receptor-binding site affects high mobility group box 1 release, altering the pathogenesis of experimental biliary atresia. Hepatol. Commun. 2022, 6, 2702–2714. [Google Scholar] [CrossRef]
- Mohanty, S.K.; Donnelly, B.; Temple, H.; Tiao, G.M. A Rotavirus-Induced Mouse Model to Study Biliary Atresia and Neonatal Cholestasis. Methods Mol. Biol. 2019, 1981, 259–271. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, R.; Lin, Z.; Fu, M.; Chen, Y. A Mouse Model of Chronic Liver Fibrosis for the Study of Biliary Atresia. J. Vis. Exp. 2023, 192, e65044. [Google Scholar] [CrossRef]
- Zhao, J.; Jeong, H.; Yang, D.; Tian, W.; Kim, J.W.; Woong Lim, C.; Kim, B. Toll-like receptor-7 signaling in Kupffer cells exacerbates concanavalin A-induced liver injury in mice. Int. Immunopharmacol. 2023, 119, 110238. [Google Scholar] [CrossRef]
- Heymann, F.; Hamesch, K.; Weiskirchen, R.; Tacke, F. The concanavalin A model of acute hepatitis in mice. Lab. Anim. 2015, 49, 12–20. [Google Scholar] [CrossRef]
- Yan, F.; Cheng, D.; Wang, H.; Gao, M.; Zhang, J.; Cheng, H.; Wang, C.; Zhang, H.; Xiong, H. Corilagin Ameliorates Con A-Induced Hepatic Injury by Restricting M1 Macrophage Polarization. Front. Immunol. 2021, 12, 807509. [Google Scholar] [CrossRef]
- Ding, X.; Fan, S. Purple sweet potato polysaccharide ameliorates concanavalin A-induced hepatic injury by inhibiting inflammation and oxidative stress. Phytomedicine 2024, 129, 155652. [Google Scholar] [CrossRef] [PubMed]
- Salah, M.M.; Ashour, A.A.; Abdelghany, T.M.; Abdel-Aziz, A.H.; Salama, S.A. Pirfenidone alleviates concanavalin A-induced liver fibrosis in mice. Life Sci. 2019, 239, 116982. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Jin, H.; Wang, Y.; Dai, M.; Liu, S.; Tan, Y.; Liu, H.; Lu, Z. Depletion of serotonin relieves concanavalin A-induced liver fibrosis in mice by inhibiting inflammation, oxidative stress, and TGF-β1/Smads signaling pathway. Toxicol. Lett. 2021, 340, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Cai, P.; Liu, S.; Piao, X.; Hou, N.; Gobert, G.N.; McManus, D.P.; Chen, Q. Comprehensive Transcriptome Analysis of Sex-Biased Expressed Genes Reveals Discrete Biological and Physiological Features of Male and Female Schistosoma japonicum. PLoS Negl. Trop. Dis. 2016, 10, e0004684. [Google Scholar] [CrossRef]
- Wang, X.; Yang, Y.; Ren, D.; Xia, Y.; He, W.; Wu, Q.; Zhang, J.; Liu, M.; Du, Y.; Ren, C.; et al. JQ1, a bromodomain inhibitor, suppresses Th17 effectors by blocking p300-mediated acetylation of RORγt. Br. J. Pharmacol. 2020, 177, 2959–2973. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Li, H.; Jiang, J.; Guo, C.; Zhou, C.; Zhou, Z.; Ming, Y. Single-cell RNA sequencing to dissect the immunological network of liver fibrosis in Schistosoma japonicum-infected mice. Front. Immunol. 2022, 13, 980872. [Google Scholar] [CrossRef]
- Ding, H.; Yang, X.; Tian, J.; Wang, X.; Ji, Y.; El-Ashram, S.; Ren, C.; Shen, J.; Liu, M. JQ-1 ameliorates schistosomiasis liver fibrosis by suppressing JAK2 and STAT3 activation. Biomed. Pharmacother. 2021, 144, 112281. [Google Scholar] [CrossRef]
- Ye, Z.; Huang, S.; Zhang, Y.; Mei, X.; Zheng, H.; Li, M.; Chen, J.; Lu, F. Galectins, Eosinophiles, and Macrophages May Contribute to Schistosoma japonicum Egg-Induced Immunopathology in a Mouse Model. Front. Immunol. 2020, 11, 146. [Google Scholar] [CrossRef]
- Qi, X.; Pu, Y.; Chen, F.; Dong, L.; Ma, Y.; Wang, J.; Yin, G.; Lu, D.; Chen, X.; Zhu, J.; et al. Schistosome egg antigen stimulates the secretion of miR-33-carrying extracellular vesicles from macrophages to promote hepatic stellate cell activation and liver fibrosis in schistosomiasis. PLoS Negl. Trop. Dis. 2023, 17, e0011385. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, L.; Liang, Y.; Lu, L. Pathology and molecular mechanisms of Schistosoma japonicum-associated liver fibrosis. Front. Cell. Infect. Microbiol. 2022, 12, 1035765. [Google Scholar] [CrossRef]
- Sun, W.Y.; Gu, Y.J.; Li, X.R.; Sun, J.C.; Du, J.J.; Chen, J.Y.; Ma, Y.; Wang, Q.T.; Wei, W. β-arrestin2 deficiency protects against hepatic fibrosis in mice and prevents synthesis of extracellular matrix. Cell Death Dis. 2020, 11, 389. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.; Wang, X.; Xie, J.; Yi, X.; Liu, F. Deep sequencing analysis of microRNA expression in porcine serum-induced hepatic fibrosis rats. Ann. Hepatol. 2014, 13, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Huang, Q.; Wei, J.; Lv, S.; Chen, Y.; Liang, C.; Wei, L.; Lu, Z.; Lin, X. Gypsophila elegans isoorientin-2″-O-α-l-arabinopyranosyl ameliorates porcine serum-induced immune liver fibrosis by inhibiting NF-κB signaling pathway and suppressing HSC activation. Int. Immunopharmacol. 2018, 54, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Tag, C.G.; Sauer-Lehnen, S.; Weiskirchen, S.; Borkham-Kamphorst, E.; Tolba, R.H.; Tacke, F.; Weiskirchen, R. Bile duct ligation in mice: Induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J. Vis. Exp. 2015, 96, e52438. [Google Scholar] [CrossRef]
- Al-Najjar, A.H.; Ayob, A.R.; Awad, A.S. Role of Lactoferrin in Treatment of Bile Duct Ligation-Induced Hepatic Fibrosis in Rats: Impact on Inflammation and TGF-β1/Smad2/α SMA Signaling Pathway. J. Clin. Exp. Hepatol. 2023, 13, 428–436. [Google Scholar] [CrossRef]
- Liu, J.Y.; Cai, Y.Y.; Ding, Z.Y.; Zhou, Z.Y.; Lv, M.; Liu, H.; Zheng, L.Y.; Li, L.; Luo, Y.H.; Xiao, E.H. Characterizing Fibrosis and Inflammation in a Partial Bile Duct Ligation Mouse Model by Multiparametric Magnetic Resonance Imaging. J. Magn. Reson. Imaging 2022, 55, 1864–1874. [Google Scholar] [CrossRef]
- Li, J.; Zou, B.; Yeo, Y.H.; Feng, Y.; Xie, X.; Lee, D.H.; Fujii, H.; Wu, Y.; Kam, L.Y.; Ji, F.; et al. Prevalence, incidence, and outcome of non-alcoholic fatty liver disease in Asia, 1999–2019: A systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2019, 4, 389–398. [Google Scholar] [CrossRef]
- Lee, E.; Korf, H.; Vidal-Puig, A. An adipocentric perspective on the development and progression of non-alcoholic fatty liver disease. J. Hepatol. 2023, 78, 1048–1062. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Haczeyni, F.; Yeh, M.M.; Ioannou, G.N.; Leclercq, I.A.; Goldin, R.; Dan, Y.Y.; Yu, J.; Teoh, N.C.; Farrell, G.C. Mouse models of non-alcoholic steatohepatitis: A reflection on recent literature. J. Gastroenterol. Hepatol. 2018, 33, 1312–1320. [Google Scholar] [CrossRef]
- Alshawsh, M.A.; Alsalahi, A.; Alshehade, S.A.; Saghir, S.A.M.; Ahmeda, A.F.; Al Zarzour, R.H.; Mahmoud, A.M. A Comparison of the Gene Expression Profiles of Non-Alcoholic Fatty Liver Disease between Animal Models of a High-Fat Diet and Methionine-Choline-Deficient Diet. Molecules 2022, 27, 858. [Google Scholar] [CrossRef] [PubMed]
- György, P.; Goldblatt, H. Observations on the conditions of dietary hepatic injury (necrosis, cirrhosis) in rats. J. Exp. Med. 1942, 75, 355–368. [Google Scholar] [CrossRef]
- Rinella, M.E.; Green, R.M. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J. Hepatol. 2004, 40, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Zheng, Y.; Ying, H.; Ma, H.; Li, L.; Zhao, Y. Alanyl-Glutamine Protects Mice against Methionine- and Choline-Deficient-Diet-Induced Steatohepatitis and Fibrosis by Modulating Oxidative Stress and Inflammation. Nutrients 2022, 14, 3796. [Google Scholar] [CrossRef] [PubMed]
- Aljobaily, N.; Krutsinger, K.; Viereckl, M.J.; Joly, R.; Menlove, B.; Cone, B.; Suppes, A.; Han, Y. Low-Dose Administration of Cannabigerol Attenuates Inflammation and Fibrosis Associated with Methionine/Choline Deficient Diet-Induced NASH Model via Modulation of Cannabinoid Receptor. Nutrients 2022, 15, 178. [Google Scholar] [CrossRef]
- Zhen, Q.; Liang, Q.; Wang, H.; Zheng, Y.; Lu, Z.; Bian, C.; Zhao, X.; Guo, X. Theabrownin ameliorates liver inflammation, oxidative stress, and fibrosis in MCD diet-fed C57BL/6J mice. Front. Endocrinol. 2023, 14, 1118925. [Google Scholar] [CrossRef]
- Hansen, H.H.; Ægidius, H.M.; Oró, D.; Evers, S.S.; Heebøll, S.; Eriksen, P.L.; Thomsen, K.L.; Bengtsson, A.; Veidal, S.S.; Feigh, M.; et al. Human translatability of the GAN diet-induced obese mouse model of non-alcoholic steatohepatitis. BMC Gastroenterol. 2020, 20, 210. [Google Scholar] [CrossRef]
- Møllerhøj, M.B.; Veidal, S.S.; Thrane, K.T.; Oró, D.; Overgaard, A.; Salinas, C.G.; Madsen, M.R.; Pfisterer, L.; Vyberg, M.; Simon, E.; et al. Hepatoprotective effects of semaglutide, lanifibranor and dietary intervention in the GAN diet-induced obese and biopsy-confirmed mouse model of NASH. Clin. Transl. Sci. 2022, 15, 1167–1186. [Google Scholar] [CrossRef]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef]
- Ichimura-Shimizu, M.; Watanabe, S.; Kashirajima, Y.; Nagatomo, A.; Wada, H.; Tsuneyama, K.; Omagari, K. Dietary Cholic Acid Exacerbates Liver Fibrosis in NASH Model of Sprague-Dawley Rats Fed a High-Fat and High-Cholesterol Diet. Int. J. Mol. Sci. 2022, 23, 9268. [Google Scholar] [CrossRef]
- Fukuda, A.; Sasao, M.; Asakawa, E.; Narita, S.; Hisano, M.; Suruga, K.; Ichimura, M.; Tsuneyama, K.; Tanaka, K.; Omagari, K. Dietary fat, cholesterol, and cholic acid affect the histopathologic severity of nonalcoholic steatohepatitis in Sprague-Dawley rats. Pathol. Res. Pract. 2019, 215, 152599. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.; Hameed, H.; Dimanche-Boitrel, M.T.; Piquet-Pellorce, C.; Samson, M.; Le Seyec, J. Switching to Regular Diet Partially Resolves Liver Fibrosis Induced by High-Fat, High-Cholesterol Diet in Mice. Nutrients 2022, 14, 386. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Dai, M.; Wang, Y.; Yan, Z.; Mao, S.; Liu, A.; Lu, C. A CDAHFD-induced mouse model mimicking human NASH in the metabolism of hepatic phosphatidylcholines and acyl carnitines. Food Funct. 2024, 15, 2982–2995. [Google Scholar] [CrossRef]
- Zheng, Q.; Kawaguchi, M.; Mikami, H.; Diao, P.; Zhang, X.; Zhang, Z.; Nakajima, T.; Iwadare, T.; Kimura, T.; Nakayama, J.; et al. Establishment of Novel Mouse Model of Dietary NASH Rapidly Progressing into Liver Cirrhosis and Tumors. Cancers 2023, 15, 3744. [Google Scholar] [CrossRef]
- Liao, Y.J.; Wang, Y.H.; Wu, C.Y.; Hsu, F.Y.; Chien, C.Y.; Lee, Y.C. Ketogenic Diet Enhances the Cholesterol Accumulation in Liver and Augments the Severity of CCl(4) and TAA-Induced Liver Fibrosis in Mice. Int. J. Mol. Sci. 2021, 22, 2934. [Google Scholar] [CrossRef]
- Jeong, B.K.; Choi, W.I.; Choi, W.; Moon, J.; Lee, W.H.; Choi, C.; Choi, I.Y.; Lee, S.H.; Kim, J.K.; Ju, Y.S.; et al. A male mouse model for metabolic dysfunction-associated steatotic liver disease and hepatocellular carcinoma. Nat. Commun. 2024, 15, 6506. [Google Scholar] [CrossRef]
- Yanguas, S.C.; Cogliati, B.; Willebrords, J.; Maes, M.; Colle, I.; van den Bossche, B.; de Oliveira, C.; Andraus, W.; Alves, V.A.F.; Leclercq, I.; et al. Experimental models of liver fibrosis. Arch. Toxicol. 2016, 90, 1025–1048. [Google Scholar] [CrossRef]
- Yang, J.; Gong, Y.; Xu, W.; Li, L.; Shi, Z.; Wang, Q.; He, Y.; Zhang, C.; Luo, C.; Fang, Z.; et al. Smad3 gene C-terminal phosphorylation site mutation exacerbates CCl4-induced hepatic fibrogenesis by promoting pSmad2L/C-mediated signaling transduction. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2021, 394, 1779–1786. [Google Scholar] [CrossRef]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β mediated SMAD signaling for the prevention of fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef]
- Lu, Z.; Li, Y.; Li, A.-J.; Syn, W.-K.; Wank, S.A.; Lopes-Virella, M.F.; Huang, Y. Loss of GPR40 in LDL receptor-deficient mice exacerbates high-fat diet-induced hyperlipidemia and nonalcoholic steatohepatitis. PLoS ONE 2022, 17, e0277251. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic β cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Phung, H.H.; Lee, C.H. Mouse models of nonalcoholic steatohepatitis and their application to new drug development. Arch. Pharm. Res. 2022, 45, 761–794. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Puche, J.E.; Saiman, Y.; Friedman, S.L. Hepatic stellate cells and liver fibrosis. Compr. Physiol. 2013, 3, 1473–1492. [Google Scholar]
- Khomich, O.; Ivanov, A.V.; Bartosch, B. Metabolic Hallmarks of Hepatic Stellate Cells in Liver Fibrosis. Cells 2019, 9, 24. [Google Scholar] [CrossRef]
- Gutiérrez-Ruiz, M.C.; Gómez-Quiroz, L.E. Liver fibrosis: Searching for cell model answers. Liver Int. 2007, 27, 434–439. [Google Scholar] [CrossRef]
- Lai, X.; Li, C.; Xiang, C.; Pan, Z.; Zhang, K.; Wang, L.; Xie, B.; Cao, J.; Shi, J.; Deng, J.; et al. Generation of functionally competent hepatic stellate cells from human stem cells to model liver fibrosis in vitro. Stem Cell Rep. 2022, 17, 2531–2547. [Google Scholar] [CrossRef]
- Xu, L.; Hui, A.Y.; Albanis, E.; Arthur, M.J.; O’Byrne, S.M.; Blaner, W.S.; Mukherjee, P.; Friedman, S.L.; Eng, F.J. Human hepatic stellate cell lines, LX-1 and LX-2: New tools for analysis of hepatic fibrosis. Gut 2005, 54, 142–151. [Google Scholar] [CrossRef]
- Meurer, S.K.; Alsamman, M.; Sahin, H.; Wasmuth, H.E.; Kisseleva, T.; Brenner, D.A.; Trautwein, C.; Weiskirchen, R.; Scholten, D. Overexpression of endoglin modulates TGF-β1-signalling pathways in a novel immortalized mouse hepatic stellate cell line. PLoS ONE 2013, 8, e56116. [Google Scholar] [CrossRef] [PubMed]
- Fausther, M.; Goree, J.R.; Lavoie, É.G.; Graham, A.L.; Sévigny, J.; Dranoff, J.A. Establishment and characterization of rat portal myofibroblast cell lines. PLoS ONE 2015, 10, e0121161. [Google Scholar] [CrossRef] [PubMed]
- Giraudi, P.J.; Becerra, V.J.; Marin, V.; Chavez-Tapia, N.C.; Tiribelli, C.; Rosso, N. The importance of the interaction between hepatocyte and hepatic stellate cells in fibrogenesis induced by fatty accumulation. Exp. Mol. Pathol. 2015, 98, 85–92. [Google Scholar] [CrossRef]
- Nieto, N. Oxidative-stress and IL-6 mediate the fibrogenic effects of [corrected] Kupffer cells on stellate cells. Hepatology 2006, 44, 1487–1501. [Google Scholar] [CrossRef] [PubMed]
- Wirz, W.; Antoine, M.; Tag, C.G.; Gressner, A.M.; Korff, T.; Hellerbrand, C.; Kiefer, P. Hepatic stellate cells display a functional vascular smooth muscle cell phenotype in a three-dimensional co-culture model with endothelial cells. Differ. Res. Biol. Divers. 2008, 76, 784–794. [Google Scholar] [CrossRef]
- Mannaerts, I.; Eysackers, N.; Anne van Os, E.; Verhulst, S.; Roosens, T.; Smout, A.; Hierlemann, A.; Frey, O.; Leite, S.B.; van Grunsven, L.A. The fibrotic response of primary liver spheroids recapitulates in vivo hepatic stellate cell activation. Biomaterials 2020, 261, 120335. [Google Scholar] [CrossRef]
- Zahmatkesh, E.; Othman, A.; Braun, B.; Aspera, R.; Ruoß, M.; Piryaei, A.; Vosough, M.; Nüssler, A. In vitro modeling of liver fibrosis in 3D microtissues using scalable micropatterning system. Arch. Toxicol. 2022, 96, 1799–1813. [Google Scholar] [CrossRef]
- Lee, H.J.; Mun, S.J.; Jung, C.R.; Kang, H.M.; Kwon, J.E.; Ryu, J.S.; Ahn, H.S.; Kwon, O.S.; Ahn, J.; Moon, K.S.; et al. In vitro modeling of liver fibrosis with 3D co-culture system using a novel human hepatic stellate cell line. Biotechnol. Bioeng. 2023, 120, 1241–1253. [Google Scholar] [CrossRef]
- Kanebratt, K.P.; Janefeldt, A.; Vilén, L.; Vildhede, A.; Samuelsson, K.; Milton, L.; Björkbom, A.; Persson, M.; Leandersson, C.; Andersson, T.B.; et al. Primary Human Hepatocyte Spheroid Model as a 3D In Vitro Platform for Metabolism Studies. J. Pharm. Sci. 2021, 110, 422–431. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Artegiani, B.; Clevers, H. Use and application of 3D-organoid technology. Hum. Mol. Genet. 2018, 27, R99–R107. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Gehart, H.; Artegiani, B.; LÖpez-Iglesias, C.; Dekkers, F.; Basak, O.; van Es, J.; Chuva de Sousa Lopes, S.M.; Begthel, H.; Korving, J.; et al. Long-Term Expansion of Functional Mouse and Human Hepatocytes as 3D Organoids. Cell 2018, 175, 1591–1606.e1519. [Google Scholar] [CrossRef] [PubMed]
- Shiota, J.; Samuelson, L.C.; Razumilava, N. Hepatobiliary Organoids and Their Applications for Studies of Liver Health and Disease: Are We There Yet? Hepatology 2021, 74, 2251–2263. [Google Scholar] [CrossRef]
- Mun, S.J.; Ryu, J.S.; Lee, M.O.; Son, Y.S.; Oh, S.J.; Cho, H.S.; Son, M.Y.; Kim, D.S.; Kim, S.J.; Yoo, H.J.; et al. Generation of expandable human pluripotent stem cell-derived hepatocyte-like liver organoids. J. Hepatol. 2019, 71, 970–985. [Google Scholar] [CrossRef]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e376. [Google Scholar] [CrossRef]
- Matai, I.; Kaur, G.; Seyedsalehi, A.; McClinton, A.; Laurencin, C.T. Progress in 3D bioprinting technology for tissue/organ regenerative engineering. Biomaterials 2020, 226, 119536. [Google Scholar] [CrossRef]
- Nguyen, D.G.; Funk, J.; Robbins, J.B.; Crogan-Grundy, C.; Presnell, S.C.; Singer, T.; Roth, A.B. Bioprinted 3D Primary Liver Tissues Allow Assessment of Organ-Level Response to Clinical Drug Induced Toxicity In Vitro. PLoS ONE 2016, 11, e0158674. [Google Scholar] [CrossRef]
- Ma, X.; Yu, C.; Wang, P.; Xu, W.; Wan, X.; Lai, C.S.E.; Liu, J.; Koroleva-Maharajh, A.; Chen, S. Rapid 3D bioprinting of decellularized extracellular matrix with regionally varied mechanical properties and biomimetic microarchitecture. Biomaterials 2018, 185, 310–321. [Google Scholar] [CrossRef]
- Norona, L.M.; Nguyen, D.G.; Gerber, D.A.; Presnell, S.C.; LeCluyse, E.L. Editor’s Highlight: Modeling Compound-Induced Fibrogenesis In Vitro Using Three-Dimensional Bioprinted Human Liver Tissues. Toxicol. Sci. 2016, 154, 354–367. [Google Scholar] [CrossRef]
- Carvalho, A.M.; Bansal, R.; Barrias, C.C.; Sarmento, B. The Material World of 3D-Bioprinted and Microfluidic-Chip Models of Human Liver Fibrosis. Adv. Mater. 2024, 36, e2307673. [Google Scholar] [CrossRef] [PubMed]
- Palma, E.; Doornebal, E.J.; Chokshi, S. Precision-cut liver slices: A versatile tool to advance liver research. Hepatol. Int. 2019, 13, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Dewyse, L.; Reynaert, H.; van Grunsven, L.A. Best Practices and Progress in Precision-Cut Liver Slice Cultures. Int. J. Mol. Sci. 2021, 22, 7137. [Google Scholar] [CrossRef] [PubMed]
- Rastovic, U.; Bozzano, S.F.; Riva, A.; Simoni-Nieves, A.; Harris, N.; Miquel, R.; Lackner, C.; Zen, Y.; Zamalloa, A.; Menon, K.; et al. Human Precision-Cut Liver Slices: A Potential Platform to Study Alcohol-Related Liver Disease. Int. J. Mol. Sci. 2023, 25, 150. [Google Scholar] [CrossRef] [PubMed]
- Dewyse, L.; De Smet, V.; Verhulst, S.; Eysackers, N.; Kunda, R.; Messaoudi, N.; Reynaert, H.; van Grunsven, L.A. Improved Precision-Cut Liver Slice Cultures for Testing Drug-Induced Liver Fibrosis. Front. Med. 2022, 9, 862185. [Google Scholar] [CrossRef]
- Lee, Y.S.; Seki, E. In Vivo and in Vitro Models to Study Liver Fibrosis: Mechanisms and Limitations. Cell. Mol. Gastroenterol. Hepatol. 2023, 16, 355–367. [Google Scholar] [CrossRef]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef]

{kind=link}
| Inducements | Species | Methods | Duration | Characteristics | Reference |
|---|---|---|---|---|---|
| Ethanol | / | Mixed with water or liquid diets, Intragastric infusion | / | ROS ↑, HSCs ↑ | [9,10,11,12] |
| CCl4 | Male SD rats | IP injection, Subcutaneous injection (50%, made with olive oil, 2 mL/kg) | 6–12 w | Kupffer cells ↑, HSCs ↑, ECM ↑ | [15,16,17,18,19] |
| TAA | Male SD rats | IP injection (200 mg/kg, 3 times/week) | 6–8 w | MDA ↑, SOD ↑, NO ↑, ALT ↑, AST ↑, ALP ↑, Nrf2 ↓ | [23,24,25,26] |
| DMN | Male Wistar rats | IP injection(10 mg/mL, 10 mg/kg) | 4 w | Kupffer cells ↑, ECM ↑, III collagen ↑ | [28,29,30,31] |
| HBV | C57BL/6 mice | Tail vein injection | 4–5 w | Ground glass-like hepatocytes ↑, collagen deposition ↑ | [37,38,39] |
| HCV | C57BL/6 mice | Tail vein | / | TGF-β ↑, HSCs ↑, TNF-α, ROS-MAPK ↑ | [40,41,42,43] |
| RRV | BALB/c mice | IP injection (20 μL, titer 1.5 × 106 PFU/mL) | 2–6 w | ALT ↑ALP ↑, TBIL ↑, DBIL ↑, IBIL ↑ | [44,45,46,47] |
| ConA | Wild-type male C57BL/6 mice | Intravenous injection (10–20 mg/kg) | 4–8 w | T-cell mitosis ↑, II and IV collagens ↑, α-SMA mRNA, HP, TGF-β ↑, TNF-α ↑, TIMP-1 ↑, MMP-2 ↓ | [48,49,50,51,52,53] |
| Schistosoma | Female C57BL/6 mice | Percutaneous infection (S. japonicum-infected Oncomelania hupensis snails) | 4–9 w, 8 w (severe) | EVs ↑miR-1 ↑, SOCS3 ↓, HSCs ↑, TGF-β3 ↑ | [55,56,57,58,59] |
| PS | Male SD rats | IP injection (0.5 mL, twice weekly) | 12–16 w | HSCs ↑ECM ↑, β-arrestin2 ↑, TGF-β ↑, TβRⅢ ↓, collagen I and collagen III ↑ | [61,62,63] |
| BDL | Male C57BL/6 wild-type mice | Double ligation of the common bile duct | 3–4 w | TGF-β1/Smad2/α-SMA ↑, TNF-α ↑, IL-1β ↑ | [64,65,66] |
| MCD | C57BL/6 male mice | Feeding (methionine- and choline-sufficient diet) | 5 w | MDA ↑SOD ↓, GPX ↓, α-SMA ↑, Col1a ↑, Col3a ↑, CTGF ↑, TGF-β1 ↑ | [72,73,74,75,76] |
| GAN | C57BL/6 J mice | Feeding (4.49 kcal/g, 40 kcal-% fat, 22% fructose, 10% sucrose, 2% cholesterol) | 16–58 w | AST ↑, ALT ↑, α-SMA ↑, Col1a1 ↑ | [77,78] |
| HFHCD | C57BL/6J male mice | Feeding (HFD supplemented with cholesterol and containing a high percentage of saturated fatty acids) | 5–12 w | Fibrotic scarring ↑ | [82] |
| CDAHFD | Male mice on 129/Sv background | Feeding (18% protein, 36% carbohydrate, 46% fat) | 3–6 w | TG ↑, Steatohepatitis | [83] |
| OYC-NASH2 | C57BL/6J male mice | Feeding (choline-deficient and methionine-restricted HFD) | 3 w | Steatosis ↑, TGF-β1 ↑, IL-1β ↑ | [84] |
| STZ + HFD | C57BL/6J male mice | IP injection (low-dose STZ) + Feeding(HFD) | 24 w | Fat mass ↑ | [86] |
| pSmad3C | C57BL/6J mice (WT, genotype: pSmad3C+/+) | Genetically modified mice | 6 w | TGF-β, Smad3 ↑, TGF-β1 ↑, pSmad2C ↑, pSmad2L ↑, PAI-1 ↑ | [87,88,89] |
| GPR40 knockout | LDLR-deficient mice on a C57BL/6 background | Genetically modified mice | 16 w | Mediate FFA ↑, Glucose and insulin secretion ↑ | [87,90,91] |
| Models | Inducers | Duration (Fibrosis) | Characteristics | Reference |
|---|---|---|---|---|
| Primary HSCs | TGF-β1, IL-1, TNF | / | TNF ↑, vitamin A ↓, PPARγ ↓, ECM ↑, α-SMA ↑ | [94,95,96,97] |
| HSCs lines | SV40 T antigen, Fetal bovine serum | / | βPDGF-R ↑, Ob-RL ↑, DDR2 ↑, MMP-2 ↑, TIMP-2 ↑, MT1-MMP ↑, α-SMA ↑ | [98,99,100,101,102,103] |
| HEP–HSCs 3D coculture | 2 mM APAP | 72 h | Acta2 ↑, Col3a1 ↑, Col1a1 ↑, Lox ↑, TGF-β1 ↑ | [105] |
| HepaRG–HUVEC 3D coculture | LX-2 cells, TGFβ-1 | / | TGF-β/SMAD, EMT, Acta2 ↑, Col1a1 ↑, MMP2 ↑, TIMP1 ↑ | [106] |
| LSC-1–HepaRG 3D coculture | SV-40, hTERT, Fibrinogen–thrombin | 3 w | Col4A3 ↑, LAMb1 ↑, CYP2B6 ↑, 3A4 ↑, Type IV collagen ↑, α-SMA ↑, Col1A1 ↑ | [107] |
| 3D organoid | HLO+ FFA | 1 w | VIM ↑, α-SMA ↑, P3NP ↑ | [108,109,110,111,112] |
| 3D-bioprinted liver tissues | TGF-β1, MTX, TAA | 2 w | ACTA2 ↑, COL1A1 ↑, Collagen I ↑, Collagen IV ↑, α-SMA ↑ | [115,116,117,118,119] |
| PCLS | TGF-β1, APAP | 48 h | Col1a1 ↑, Col5a2 ↑, Lox ↑, TIMP-1, TGF-β1, COL1A1, and PDGFRB ↑ | [120,121,122,123] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, Y.; Zhang, Z.; Adiham, A.; Li, H.; Gu, J.; Gong, P. In Vivo and In Vitro Models of Hepatic Fibrosis for Pharmacodynamic Evaluation and Pathology Exploration. Int. J. Mol. Sci. 2025, 26, 696. https://doi.org/10.3390/ijms26020696
Hu Y, Zhang Z, Adiham A, Li H, Gu J, Gong P. In Vivo and In Vitro Models of Hepatic Fibrosis for Pharmacodynamic Evaluation and Pathology Exploration. International Journal of Molecular Sciences. 2025; 26(2):696. https://doi.org/10.3390/ijms26020696
Chicago/Turabian StyleHu, Yanting, Zhongrui Zhang, Akida Adiham, Hong Li, Jian Gu, and Puyang Gong. 2025. "In Vivo and In Vitro Models of Hepatic Fibrosis for Pharmacodynamic Evaluation and Pathology Exploration" International Journal of Molecular Sciences 26, no. 2: 696. https://doi.org/10.3390/ijms26020696
APA StyleHu, Y., Zhang, Z., Adiham, A., Li, H., Gu, J., & Gong, P. (2025). In Vivo and In Vitro Models of Hepatic Fibrosis for Pharmacodynamic Evaluation and Pathology Exploration. International Journal of Molecular Sciences, 26(2), 696. https://doi.org/10.3390/ijms26020696