
MYC Overexpression Enhances Sensitivity to MEK Inhibition in Head and Neck Squamous Cell Carcinoma


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
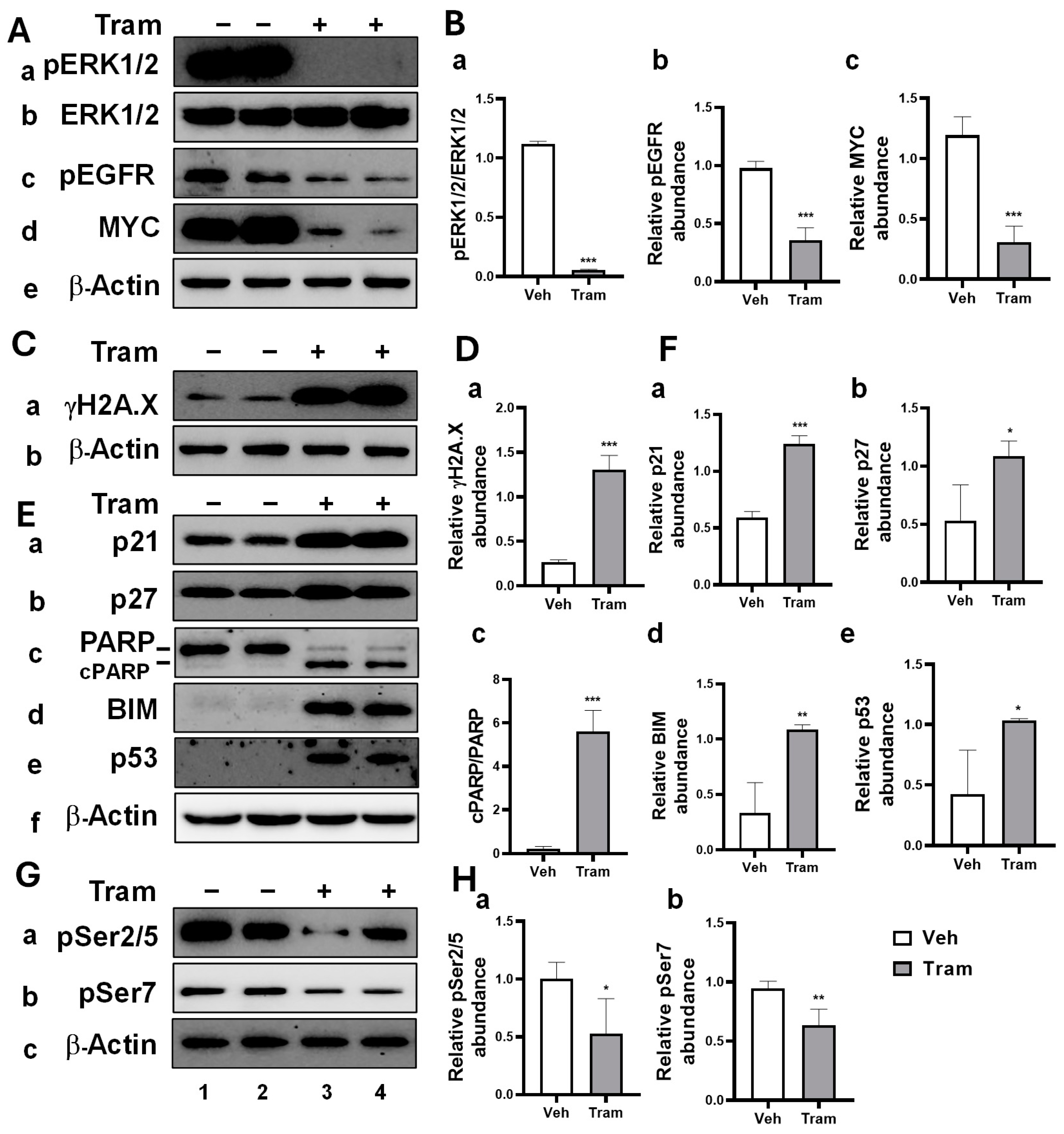
2.1. Trametinib-Induced Multiple Anti-Cancer Effects in HNSCC Cells Through MEK Inhibition
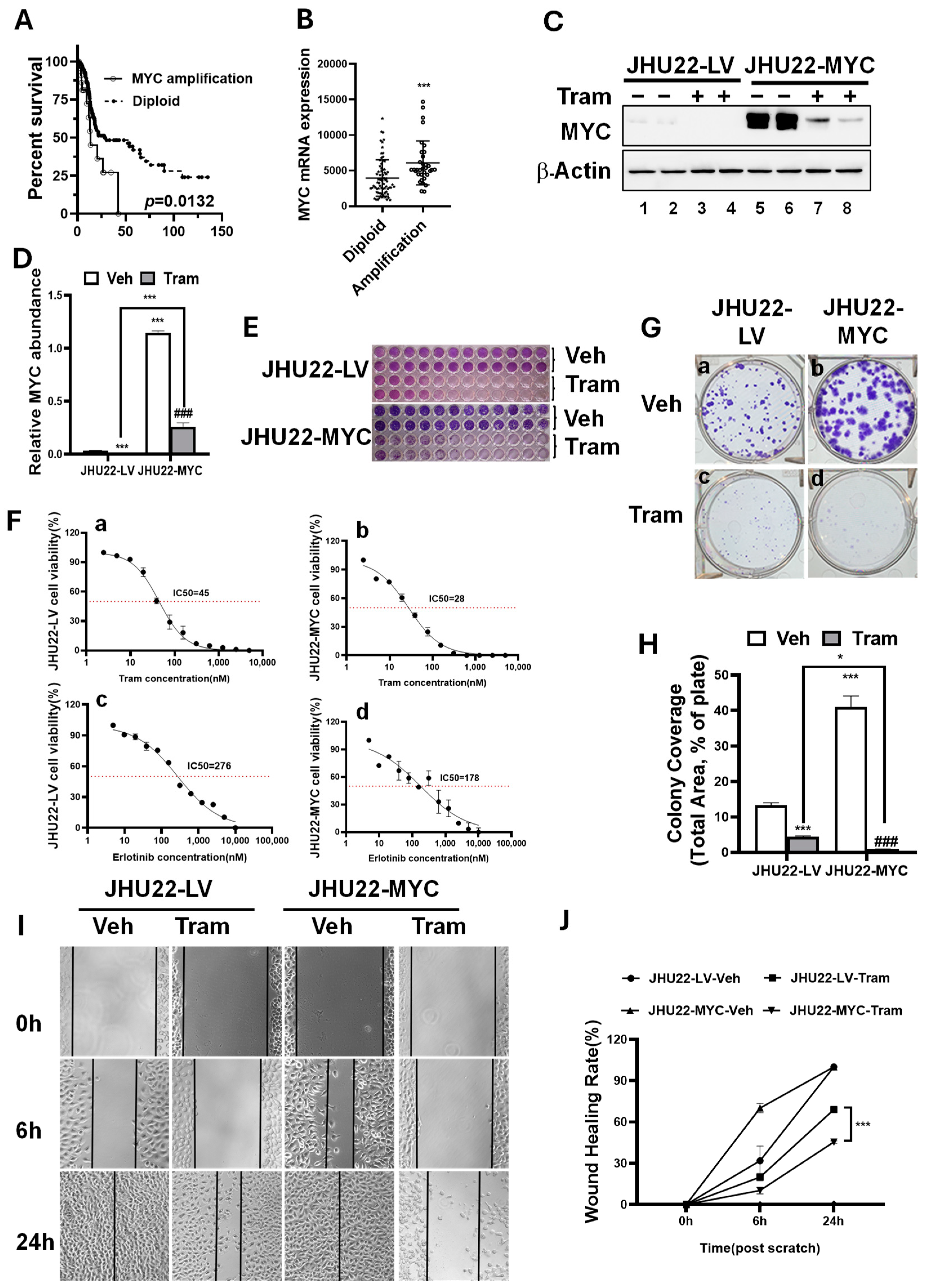
2.2. MYC Overexpression Renders HNSCC Cells More Sensitive to Trametinib
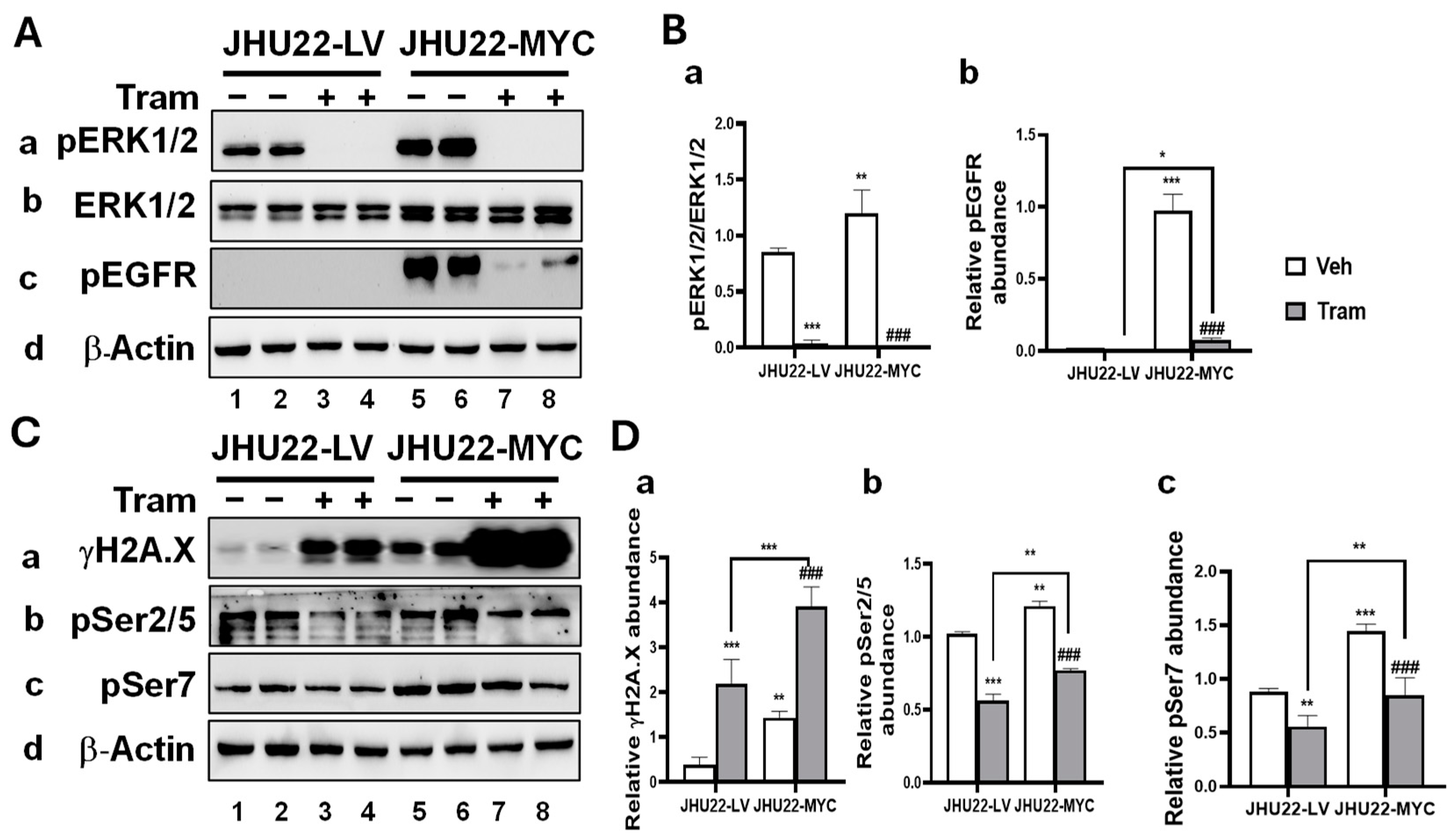
2.3. MYC Overexpression Alters the Response of HNSCC Cells to Trametinib
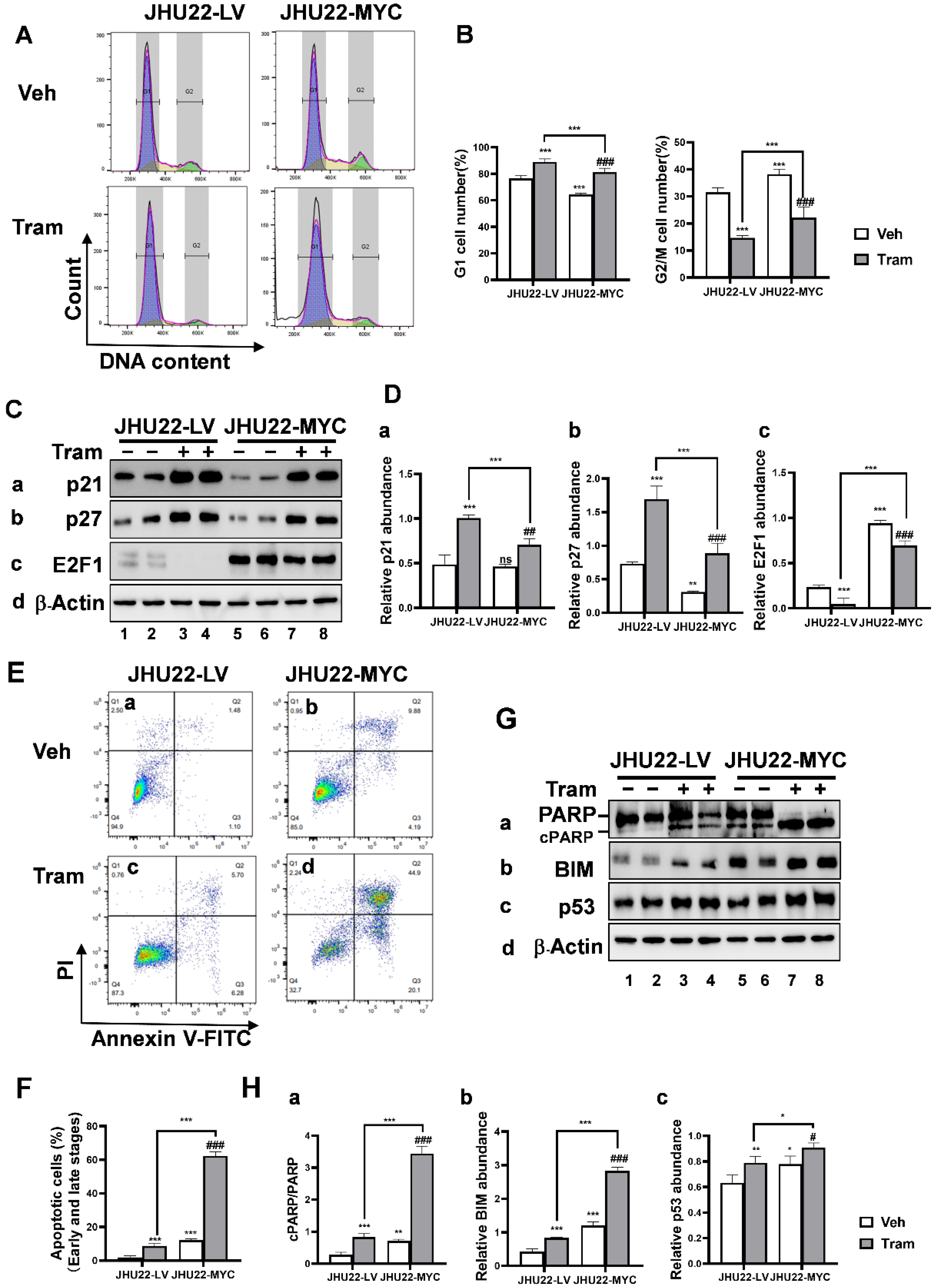
2.4. MYC Overexpression Promotes Cell Cycle Progression and Apoptosis in Responses to Trametinib in JHU22 HNSCC Cells
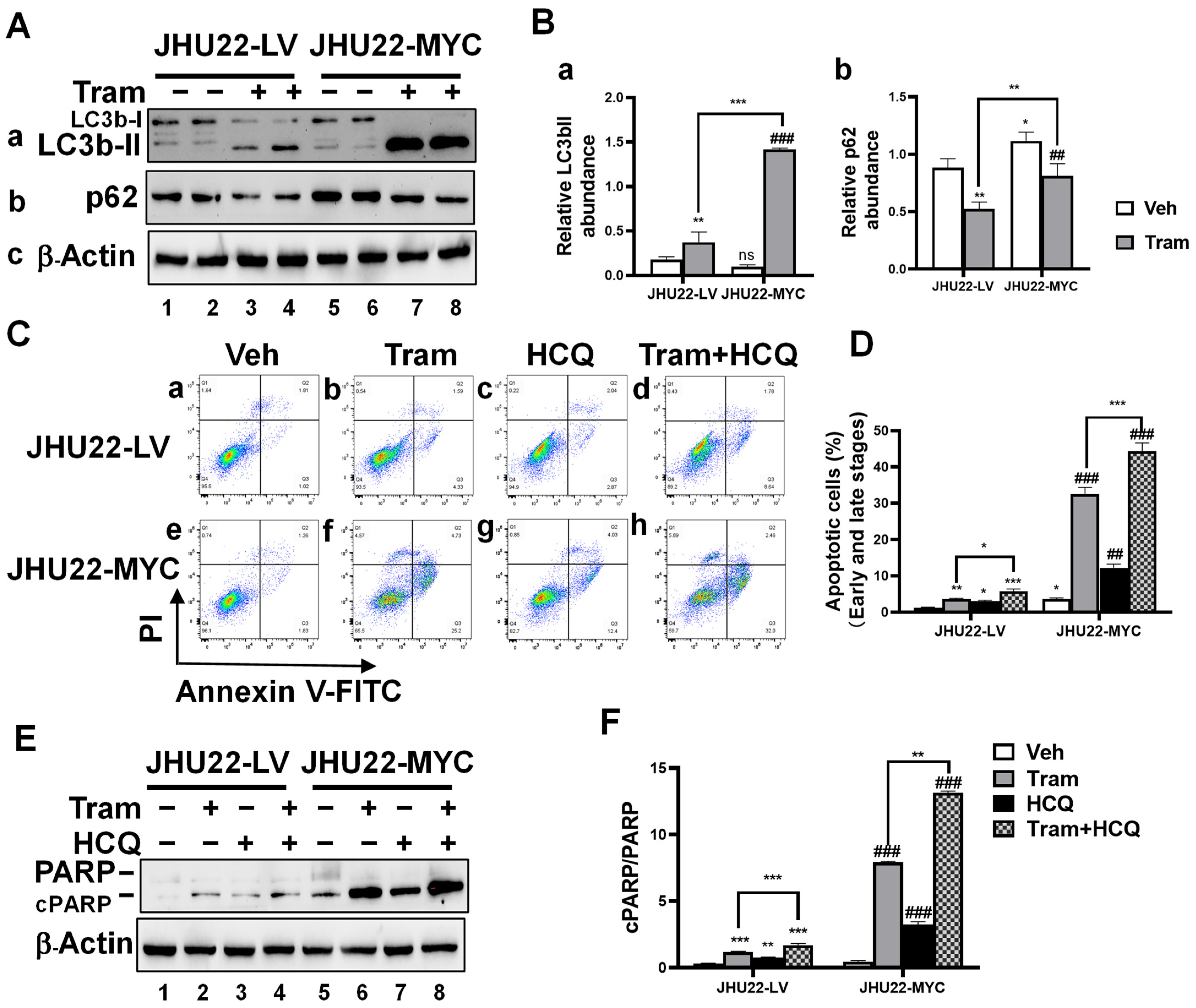
2.5. Inhibition of Trametinib-Induced Autophagy Enhances Apoptotic Cell Death in HNSCC Cells
2.6. Impact of Trametinib on Xenograft Tumors Overexpressing MYC in HNSCC
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Exogenous Overexpression of MYC in the JHU22 HNSCC Cells
4.3. Cell Viability Assay
4.4. Colony Formation Assay
4.5. Wound Healing Assay
4.6. Flow Cytometric Analysis of Apoptosis and Cell Cycle
4.7. Western Blot and Antibodies
4.8. Xenograft Tumor Models in Athymic Nude Mice
4.9. Immunohistochemical (IHC) Analysis
4.10. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Canning, M.; Guo, G.; Yu, M.; Myint, C.; Groves, M.W.; Byrd, J.K.; Cui, Y. Heterogeneity of the Head and Neck Squamous Cell Carcinoma Immune Landscape and Its Impact on Immunotherapy. Front. Cell Dev. Biol. 2019, 7, 52. [Google Scholar] [CrossRef] [PubMed]
- Ferris, R.L.; Blumenschein, G., Jr.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef] [PubMed]
- Ragin, C.C.; Langevin, S.M.; Marzouk, M.; Grandis, J.; Taioli, E. Determinants of head and neck cancer survival by race. Head Neck 2011, 33, 1092–1098. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Bonner, J.A.; Harari, P.M.; Giralt, J.; Azarnia, N.; Shin, D.M.; Cohen, R.B.; Jones, C.U.; Sur, R.; Raben, D.; Jassem, J.; et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N. Engl. J. Med. 2006, 354, 567–578. [Google Scholar] [CrossRef]
- Byeon, H.K.; Ku, M.; Yang, J. Beyond EGFR inhibition: Multilateral combat strategies to stop the progression of head and neck cancer. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Vermorken, J.B.; Mesia, R.; Rivera, F.; Remenar, E.; Kawecki, A.; Rottey, S.; Erfan, J.; Zabolotnyy, D.; Kienzer, H.R.; Cupissol, D.; et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N. Engl. J. Med. 2008, 359, 1116–1127. [Google Scholar] [CrossRef]
- Ngan, H.L.; Law, C.H.; Choi, Y.C.Y.; Chan, J.Y.; Lui, V.W.Y. Precision drugging of the MAPK pathway in head and neck cancer. NPJ Genom. Med. 2022, 7, 20. [Google Scholar] [CrossRef]
- Ngan, H.L.; Liu, Y.; Fong, A.Y.; Poon, P.H.Y.; Yeung, C.K.; Chan, S.S.M.; Lau, A.; Piao, W.; Li, H.; Tse, J.S.W.; et al. MAPK pathway mutations in head and neck cancer affect immune microenvironments and ErbB3 signaling. Life Sci. Alliance 2020, 3, 545. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, A.G.; Bleam, M.R.; Groy, A.; Moss, K.G.; Minthorn, E.A.; Kulkarni, S.G. GSK1120212 (JTP-74057) Is an Inhibitor of MEK Activity and Activation with Favorable Pharmacokinetic Properties for Sustained In Vivo Pathway Inhibition. Clin. Cancer Res. 2011, 17, 989–1000, Erratum in Clin. Cancer Res. 2012, 18, 2413. [Google Scholar] [CrossRef]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, A.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- King, J.W.; Nathan, P.D. Role of the MEK inhibitor trametinib in the treatment of metastatic melanoma. Future Oncol. 2014, 10, 1559–1570. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas, N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015, 517, 576–582. [Google Scholar] [CrossRef]
- Uppaluri, R.; Winkler, A.E.; Lin, T.; Law, J.H.; Haughey, B.H.; Nussenbaum, B.; Paniello, R.C.; Rich, J.T.; Diaz, J.A.; Michel, L.P.; et al. Biomarker and Tumor Responses of Oral Cavity Squamous Cell Carcinoma to Trametinib: A Phase II Neoadjuvant Window-of-Opportunity Clinical Trial. Clin Cancer Res 2017, 23, 2186–2194. [Google Scholar] [CrossRef]
- Xie, G.; Zhu, A.; Gu, X. Mitogen-activated protein kinase inhibition-induced modulation of epidermal growth factor receptor signaling in human head and neck squamous cell carcinoma. Head Neck 2021, 43, 1721–1729. [Google Scholar] [CrossRef]
- Kress, T.R.; Sabo, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef]
- Dalla-Favera, R.; Martinotti, S.; Gallo, R.C.; Erikson, J.; Croce, C.M. Translocation and rearrangements of the c-myc oncogene locus in human undifferentiated B-cell lymphomas. Science 1983, 219, 963–967. [Google Scholar] [CrossRef]
- Gurel, B.; Iwata, T.; Koh, C.M.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear MYC protein overexpression is an early alteration in human prostate carcinogenesis. Mod. Pathol. 2008, 21, 1156–1167. [Google Scholar] [CrossRef]
- Jain, S.; Wang, X.; Chang, C.C.; Ibarra-Drendall, C.; Wang, H.; Zhang, Q.; Brady, S.W.; Li, P.; Zhao, H.; Dobbs, J.; et al. Src Inhibition Blocks c-Myc Translation and Glucose Metabolism to Prevent the Development of Breast Cancer. Cancer Res. 2015, 75, 4863–4875. [Google Scholar] [CrossRef] [PubMed]
- Porter, M.J.; Field, J.K.; Leung, S.F.; Lo, D.; Lee, J.C.; Spandidos, D.A.; van Hasselt, C.A. The detection of the c-myc and ras oncogenes in nasopharyngeal carcinoma by immunohistochemistry. Acta Oto-Laryngol. 1994, 114, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Qin, Z.; Mao, Y.; Zhang, W.; Wang, Y.; Jia, L.; Peng, X. Therapeutic Targeting of MYC in Head and Neck Squamous Cell Carcinoma. Oncoimmunology 2022, 11, 2130583. [Google Scholar] [CrossRef]
- Liu, W.; Ding, Z.; Tao, Y.; Liu, S.; Jiang, M.; Yi, F.; Wang, Z.; Han, Y.; Zong, H.; Li, D.; et al. A positive feedback loop between PFKP and c-Myc drives head and neck squamous cell carcinoma progression. Mol. Cancer 2024, 23, 141. [Google Scholar] [CrossRef] [PubMed]
- Cyberski, T.F.; Singh, A.; Korzinkin, M.; Mishra, V.; Pun, F.; Shen, L.; Wing, C.; Cheng, X.; Baird, B.; Miao, Y.; et al. Acquired resistance to immunotherapy and chemoradiation in MYC amplified head and neck cancer. NPJ Precis. Oncol. 2024, 8, 114. [Google Scholar] [CrossRef]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. gammaH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Bresciani, A.; Spiezia, M.C.; Boggio, R.; Cariulo, C.; Nordheim, A.; Altobelli, R.; Kuhlbrodt, K.; Dominguez, C.; Munoz-Sanjuan, I.; Wityak, J.; et al. Quantifying autophagy using novel LC3B and p62 TR-FRET assays. PLoS ONE 2018, 13, e0194423. [Google Scholar] [CrossRef]
- Kalyankrishna, S.; Grandis, J.R. Epidermal growth factor receptor biology in head and neck cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 2666–2672. [Google Scholar] [CrossRef]
- Affolter, A.; Muller, M.F.; Sommer, K.; Stenzinger, A.; Zaoui, K.; Lorenz, K.; Wolf, T.; Sharma, S.; Wolf, J.; Perner, S.; et al. Targeting irradiation-induced mitogen-activated protein kinase activation in vitro and in an ex vivo model for human head and neck cancer. Head Neck 2016, 38 (Suppl. 1), E2049–E2061. [Google Scholar] [CrossRef] [PubMed]
- Horn, D.; Hess, J.; Freier, K.; Hoffmann, J.; Freudlsperger, C. Targeting EGFR-PI3K-AKT-mTOR signaling enhances radiosensitivity in head and neck squamous cell carcinoma. Expert Opin. Ther. Targets 2015, 19, 795–805. [Google Scholar] [CrossRef]
- Li, Q.; Tie, Y.; Alu, A.; Ma, X.; Shi, H. Targeted therapy for head and neck cancer: Signaling pathways and clinical studies. Signal Transduct. Target. Ther. 2023, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Shan, L.; Liu, Y.; Wu, T.C.; Gu, X. Antitumor Efficacy of EGFR-Targeted Recombinant Immunotoxin in Human Head and Neck Squamous Cell Carcinoma. Biology 2022, 11, 486. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Shan, L.; Yang, C.; Liu, Y.; Pang, X.; Teng, S.; Wu, T.C.; Gu, X. Recombinant immunotoxin induces tumor intrinsic STING signaling against head and neck squamous cell carcinoma. Sci. Rep. 2023, 13, 18476. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Goto, Y.; Allevato, M.M.; Wu, V.H.; Saddawi-Konefka, R.; Gilardi, M.; Alvarado, D.; Yung, B.S.; O’Farrell, A.; Molinolo, A.A.; et al. Disruption of the HER3-PI3K-mTOR oncogenic signaling axis and PD-1 blockade as a multimodal precision immunotherapy in head and neck cancer. Nat. Commun. 2021, 12, 2383. [Google Scholar] [CrossRef]
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942. [Google Scholar] [CrossRef]
- Rong, C.; Muller, M.F.; Xiang, F.; Jensen, A.; Weichert, W.; Major, G.; Plinkert, P.K.; Hess, J.; Affolter, A. Adaptive ERK signalling activation in response to therapy and in silico prognostic evaluation of EGFR-MAPK in HNSCC. Br. J. Cancer 2020, 123, 288–297. [Google Scholar] [CrossRef]
- Meyer, N.; Penn, L.Z. Reflecting on 25 years with MYC. Nat. Rev. Cancer 2008, 8, 976–990. [Google Scholar] [CrossRef]
- Luscher, B.; Larsson, L.G. The basic region/helix-loop-helix/leucine zipper domain of Myc proto-oncoproteins: Function and regulation. Oncogene 1999, 18, 2955–2966. [Google Scholar] [CrossRef]
- Lee, K.M.; Lin, C.C.; Servetto, A.; Bae, J.; Kandagatla, V.; Ye, D.; Kim, G.; Sudhan, D.R.; Mendiratta, S.; Gonzalez Ericsson, P.I.; et al. Epigenetic Repression of STING by MYC Promotes Immune Evasion and Resistance to Immune Checkpoint Inhibitors in Triple-Negative Breast Cancer. Cancer Immunol. Res. 2022, 10, 829–843. [Google Scholar] [CrossRef] [PubMed]
- Silvis, M.R.; Silva, D.; Rohweder, R.; Schuman, S.; Gudipaty, S.; Truong, A.; Yap, J.; Affolter, K.; McMahon, M.; Kinsey, C. MYC-mediated resistance to trametinib and HCQ in PDAC is overcome by CDK4/6 and lysosomal inhibition. J. Exp. Med. 2023, 220, 1524. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.Y.; Xiao, Y.; Wei, J.L.; Xu, X.E.; Jin, X.; Hu, X.; Li, D.Q.; Jiang, Y.Z.; Shao, Z.M. MYC suppresses STING-dependent innate immunity by transcriptionally upregulating DNMT1 in triple-negative breast cancer. J. Immunother. Cancer 2021, 9, 2528. [Google Scholar] [CrossRef] [PubMed]
- Goga, A.; Yang, D.; Tward, A.D.; Morgan, D.O.; Bishop, J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007, 13, 820–827. [Google Scholar] [CrossRef]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schuttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef]
- Yang, D.; Liu, H.; Goga, A.; Kim, S.; Yuneva, M.; Bishop, J.M. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proc. Natl. Acad. Sci. USA 2010, 107, 13836–13841. [Google Scholar] [CrossRef]
- Frenzel, A.; Zirath, H.; Vita, M.; Albihn, A.; Henriksson, M.A. Identification of cytotoxic drugs that selectively target tumor cells with MYC overexpression. PLoS ONE 2011, 6, e27988. [Google Scholar] [CrossRef]
- Dumontet, C.; Jordan, M.A. Microtubule-binding agents: A dynamic field of cancer therapeutics. Nat. Rev. Drug Discov. 2010, 9, 790–803. [Google Scholar] [CrossRef]
- Biederstadt, A.; Hassan, Z.; Schneeweis, C.; Schick, M.; Schneider, L.; Muckenhuber, A.; Hong, Y.; Siegers, G.; Nilsson, L.; Wirth, M.; et al. SUMO pathway inhibition targets an aggressive pancreatic cancer subtype. Gut 2020, 69, 1472–1482. [Google Scholar] [CrossRef]
- Tang, L.; Zhang, H.; Zhou, F.; Wei, Q.; Du, M.; Wu, J.; Li, C.; Luo, W.; Zhou, J.; Wang, X.; et al. Targeting autophagy overcomes cancer-intrinsic resistance to CAR-T immunotherapy in B-cell malignancies. Cancer Commun. 2024, 44, 408–432. [Google Scholar] [CrossRef]
- Grbovic-Huezo, O.; Pitter, K.L.; Lecomte, N.; Saglimbeni, J.; Askan, G.; Holm, M.; Melchor, J.P.; Chandwani, R.; Joshi, S.; Haglund, C.; et al. Unbiased in vivo preclinical evaluation of anticancer drugs identifies effective therapy for the treatment of pancreatic adenocarcinoma. Proc. Natl. Acad. Sci. USA 2020, 117, 30670–30678. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, V.; Lan, T.; Wang, W.; Kong, J.; Lopes, E.C.; Wang, J.; Khayati, K.; Raju, A.; Rangel, M.; Lopez, E.; et al. Inhibition of autophagy and MEK promotes ferroptosis in Lkb1-deficient Kras-driven lung tumors. Cell Death Discov. 2023, 14, 61. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; You, X.; Xu, L.; Wang, L.; Chen, Y. PAQR3 suppresses the growth of non-small cell lung cancer cells via modulation of EGFR-mediated autophagy. Autophagy 2020, 16, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Y.; Jiao, L.; Lin, C.; Lu, C.; Zhang, K.; Hu, C.; Ye, J.; Zhang, D.; Wu, H.; et al. Protective autophagy decreases osimertinib cytotoxicity through regulation of stem cell-like properties in lung cancer. Cancer Lett. 2019, 452, 191–202. [Google Scholar] [CrossRef]
- Lu, C.; Yu, R.; Zhang, C.; Lin, C.; Dou, Y.; Wu, D.; Pan, Y.; Peng, T.; Tang, H.; Han, R.; et al. Protective autophagy decreases lorlatinib cytotoxicity through Foxo3a-dependent inhibition of apoptosis in NSCLC. Cell Death Discov. 2022, 8, 221. [Google Scholar] [CrossRef]
- Li, N.; Zuo, R.; He, Y.; Gong, W.; Wang, Y.; Chen, L.; Luo, Y.; Zhang, C.; Liu, Z.; Chen, P.; et al. PD-L1 induces autophagy and primary resistance to EGFR-TKIs in EGFR-mutant lung adenocarcinoma via the MAPK signaling pathway. Cell Death Discov. 2024, 15, 555. [Google Scholar] [CrossRef]
- Zaryouh, H.; Van Loenhout, J.; Peeters, M.; Vermorken, J.B.; Lardon, F.; Wouters, A. Co-Targeting the EGFR and PI3K/Akt Pathway to Overcome Therapeutic Resistance in Head and Neck Squamous Cell Carcinoma: What about Autophagy? Cancers 2022, 14, 6128. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Pang, X.; Teng, S.; Wilson, S.; Gu, X.; Xie, G. MYC Overexpression Enhances Sensitivity to MEK Inhibition in Head and Neck Squamous Cell Carcinoma. Int. J. Mol. Sci. 2025, 26, 588. https://doi.org/10.3390/ijms26020588
Yang C, Pang X, Teng S, Wilson S, Gu X, Xie G. MYC Overexpression Enhances Sensitivity to MEK Inhibition in Head and Neck Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2025; 26(2):588. https://doi.org/10.3390/ijms26020588
Chicago/Turabian StyleYang, Cuicui, Xiaowu Pang, Shaolei Teng, Shamel Wilson, Xinbin Gu, and Guiqin Xie. 2025. "MYC Overexpression Enhances Sensitivity to MEK Inhibition in Head and Neck Squamous Cell Carcinoma" International Journal of Molecular Sciences 26, no. 2: 588. https://doi.org/10.3390/ijms26020588
APA StyleYang, C., Pang, X., Teng, S., Wilson, S., Gu, X., & Xie, G. (2025). MYC Overexpression Enhances Sensitivity to MEK Inhibition in Head and Neck Squamous Cell Carcinoma. International Journal of Molecular Sciences, 26(2), 588. https://doi.org/10.3390/ijms26020588