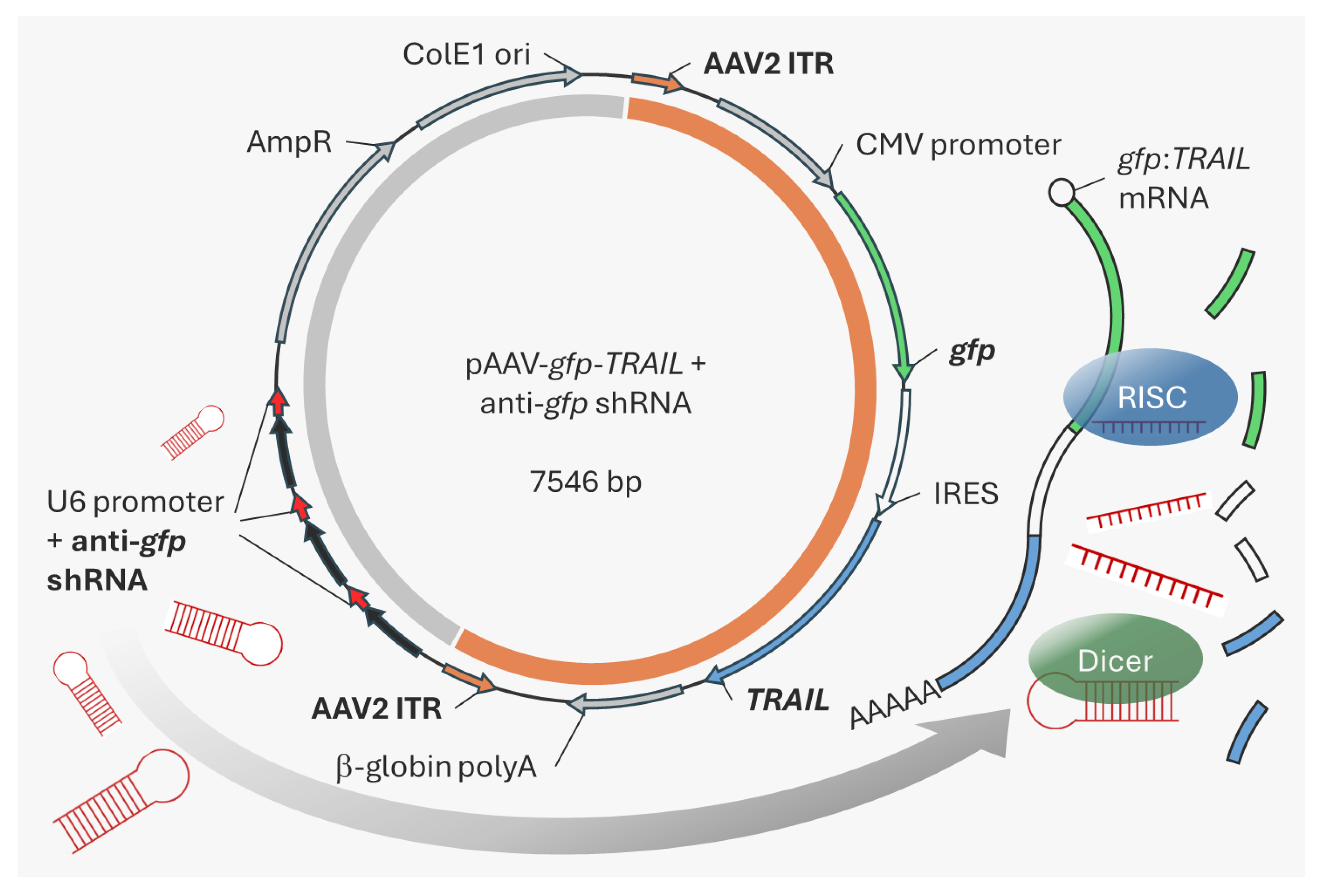
1. Introduction
Genetic diseases caused by missing or defective genes can be treated by transferring a wild-type copy of the affected gene into a patient’s cells. Because these defects may only be relevant in certain specialised cells, targeting gene therapy vectors to the correct cells is important. rAAV is one of the most popular vectors to achieve this due to its versatile range of target cells and low immunogenicity [
1,
2]. rAAV-based gene therapies consist of a therapeutic transgene flanked by inverted terminal repeats (ITRs) enclosed in a capsid shell, which determines the serotype and target cell range. Currently, the U.S. Food and Drug administration has approved 5 rAAV-based gene therapy drugs, with a further 136 undergoing clinical trials [
3].
Besides delivering replacement genes, an alternative use for rAAV in cancer treatment is to deliver toxic or pro-apoptotic genes to tumor cells. One such transgene is TNF-related apoptosis-inducing ligand (
TRAIL), which induces cell death by binding to the DR4 (TRAIL-R1) or DR5 (TRAIL-R2) death receptors [
4,
5]. rAAV-
TRAIL has been used successfully to induce tumor regression in mouse models [
6,
7,
8,
9].
Production of rAAV-
TRAIL (and rAAV in general) is challenging and expensive. rAAVs are commonly produced by transiently transfecting HEK293F cells with 3 plasmids encoding the ITR-flanked therapeutic transgene, the rAAV replication and capsid genes (
rep and
cap) and Adenovirus-derived helper genes, respectively. Following transfection, Rep binds the ITR region to initiate replication and packaging of the transgene into capsids produced from the
cap gene [
10]. While this approach is necessary to minimize the chance of recombination events leading to the production of wild-type, replication-competent rAAVs, the resulting yields are often low. Higher rAAV production yields would have positive economic implications and thus increase access to medication.
A further issue is that, unless under the control of a highly tissue-specific promoter sequence, the therapeutic transgene may be expressed in the producer cell line while the rAAV is being manufactured. This is likely to reduce yields as the cell expends resources to express the transgene instead of synthesizing rAAVs. Notably, if the transgene product is toxic to the producer cell, rAAV production may be highly compromised with a consequentially negative impact on process yield.
Tissue-specific promoters can be used to prevent undesirable transgene expression in producer cells [
11]. However, ‘leaky’ expression of a toxic transgene can still occur [
12,
13]. Furthermore, the maximum transgene packaging capacity of rAAV is relatively small at 4.7 kb and this may limit options in promoter choice.
Several groups have reported different approaches to overcoming this challenge, including the application of RNA interference (RNAi) for gene silencing. Mediators of RNAi include microRNAs (miRNAs) and small interfering RNAs (siRNAs). The main difference is that miRNAs can regulate multiple genes, while siRNAs have a single specific target. A further category, short hairpin RNAs (shRNAs) are single-stranded RNAs with self-complementary ends; a hairpin loop at the center allows the molecule to fold back on itself into a double-stranded RNA. All types of RNAi make use of the RNA-induced silencing complex (RISC); double-stranded RNAs are processed by the endo-ribonuclease Dicer into siRNAs, then formed into a complex with RISC which degrades mRNAs complementary to the siRNA, resulting in gene silencing [
14,
15].
Taking advantage of the cell type-specific nature of miRNA expression, Blahetek et al. added miRNA binding sites (MBS) in the 3′ untranslated region (UTR) of the therapeutic transgene. The particular miRNA was chosen due to it being highly expressed in HEK293 cells, thus silencing the transgene mRNA during rAAV production, but not in the patient target tissue [
16]. While effective, this approach does result in extra genetic material being included in the gene therapy being administered which may not be desirable. Riboswitch-mediated attenuation of transgene expression during the production stage was also shown to greatly improve the manufacturability of rAAVs containing the pro-apoptotic gene,
BAX. By placing a guanine-responsive riboswitch sequence in the 3′ UTR of the transgene, the addition of the inducer to the production culture caused destabilization of the mRNA and thus limited Bax protein production. In this case the inclusion of the riboswitch sequence in the transgene did not interfere with expression in the target cells in mice [
17].
In this study, we focused on
TRAIL as the model oncolytic gene and show that transgene expression during rAAV production in HEK293F cells reduced cell viability and thus rAAV yields. To address this problem, a transgene-specific shRNA cassette was inserted into the backbone of the rAAV genome-encoding plasmid. Following triple transfection in HEK293F cells, shRNA expression targets the
TRAIL-containing transcript and leads to improved rAAV production [
18]. Importantly, by placing the shRNA cassette in the plasmid backbone, we avoid introducing extraneous genetic material into the resulting rAAV product.
3. Discussion
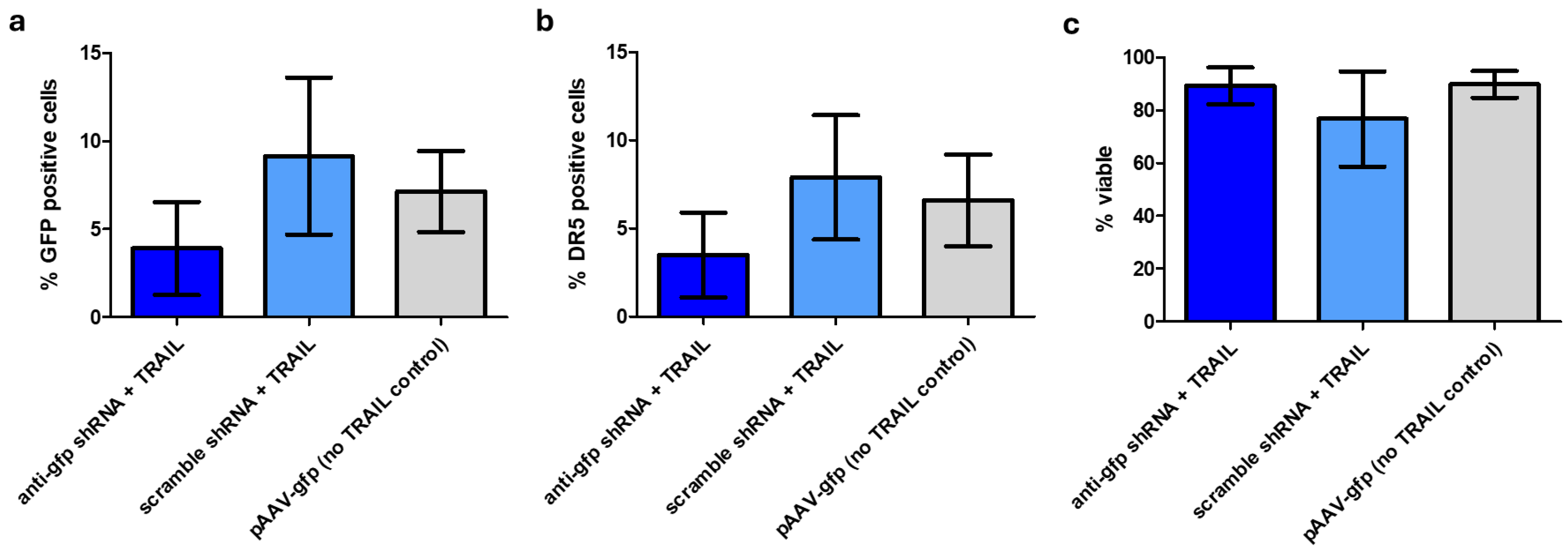
Oncolytic rAAVs are a promising new tool in cancer treatment, however production of rAAV in general is expensive based on current production yields, particularly to meet the demands of high-dose or common disease indications. This work demonstrated that expression of the cytotoxic gene, TRAIL, in HEK293F cells during rAAV production leads to reduced viability and increased apoptosis most likely via the DR5 receptor. Subsequently, these conditions lead to reduced yields of rAAV-TRAIL. A shRNA-based approach was used to inhibit TRAIL expression, which improved rAAV-TRAIL yields. Although expression of the anti-gfp shRNA was not sufficient to fully restore rAAV-TRAIL yields to the level of a non-TRAIL rAAV control, increasing the number of shRNA cassette repeats might offer further improvement. It might also be possible to further suppress transgene expression by using multiple shRNAs, targeting different regions of the same mRNA. However, the RNAi processing machinery of the cell may be the limiting factor here. Furthermore, optimization of the shRNA sequence could be undertaken for each individual target transgene to maximize the effect. Alternatively, expression of other elements in the apoptotic pathway could be silenced, e.g., through use of a DR5 antagonist.
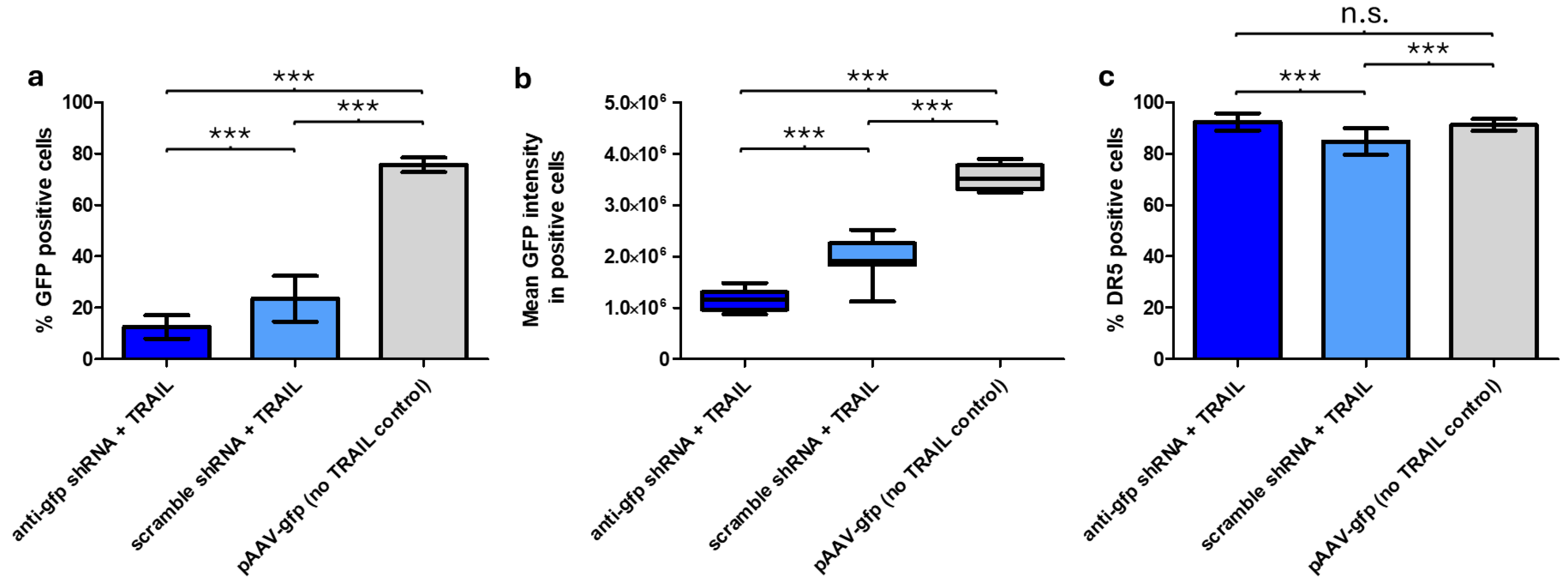
GFP expression was impacted by both TRAIL and the anti-
gfp shRNA; in the presence of TRAIL and the absence of the anti-
gfp shRNA, cells showed reduced viability, which most likely led to reduced GFP expression. When TRAIL and the anti-
gfp shRNA were both present, viability was restored, though not to non-TRAIL control levels. Furthermore, TRAIL-containing samples showed reduced GFP intensity in GFP-positive cells compared to the non-TRAIL control (
Figure 3b). This may be the result of
gfp being in a transcriptional fusion with
TRAIL, which could lead to lower GFP translation. As a result, in samples with TRAIL and the anti-
gfp shRNA, reduced viability, shRNA silencing and the transcriptional fusion with TRAIL all contributed to lower GFP expression. This would also explain the strong differences observed in the percentage of GFP positive cells, when comparing the anti-
gfp shRNA + TRAIL sample to the non-TRAIL control (
Figure 3a). A control experiment excluded the possibility of these differences being caused by varying transfection efficiency (
Figure S2).
In a similar approach, expression of the toxic transgene
G protein-coupled receptor 78 (GPR78) during AAV production was blocked by co-transfecting plasmids containing anti-
GPR78 shRNAs. AAV yields were comparable to a non-GPR78 control. However, this approach relies on transfection of a further plasmid, which would reduce the overall efficiency [
22].
A further option was explored in which toxic transgene expression was reduced by inserting miRNA target sites or riboswitches in the 3′ untranslated region of the transgene [
16,
17]. However, inserting such sequences anywhere between the ITRs further reduces the already limited packaging capacity of rAAV (~4.7 kb). Indeed, partial packaging, where some capsids contain less than the full transgene sequence, is a recognized ‘impurity’ in existing rAAV manufacturing processes, and becomes more pronounced as the maximum sequence capacity limit is approached [
23]. Additionally, using these sequences in rAAV drug production would lead to them being present in the final product administered to patients, which may not be desirable.
While rAAV-
TRAIL successfully transduced CHO-K1 cells, no effect on DR5 expression or apoptosis was observed. CHO-K1 was chosen as the target cell line as it has been reported to be more susceptible to transduction by rAAV5, however low levels of DR5 expression mean it would potentially not be as susceptible to TRAIL signaling [
21].
Overall, the system described here provides a useful in vitro screening tool to produce and test rAAVs carrying various toxic transgenes. An important benefit is that the toxic transgene can be easily exchanged without the need to re-design the anti-shRNA cassette. Once this has been established for a pro-apoptotic gene of interest, the logical next step would be to design the shRNA to target the toxic transgene directly, for the manufacturing process, and to remove the gfp gene.
Future applications of shRNA in rAAV manufacturing could extend to regulating other cellular pathways that interfere with rAAV production. For example, over-expression of a long non-coding RNA involved in regulating the ATP-binding cassette cellular transporter pathway resulted in increased production of rAAVs [
24]. As mentioned earlier, rather than using an artificial shRNA, Blahetek et al. added artificial miRNA binding sites to the 3′ UTR of the cytotoxic transgene, which only interacted with an endogenous miRNA that is only highly expressed in HEK293 cells but not in the patient target tissue [
16]. While useful in improving manufacturing characteristics, it does result in extra genetic material being included in the gene therapy going into the patient. Another option might include transient delivery of short double-stranded siRNAs targeting the cytotoxic transgene, along with the three AAV plasmids. These would suppress transgene expression in the HEK293 cells but not form any part of the AAV produced.
The approach described here is the first to use shRNAs to reduce toxic transgene expression during rAAV production, without the need for another plasmid or insertion of additional sequences that would be packaged into the final rAAV product.
4. Materials and Methods
4.1. TRAIL Vector Construction
Plasmids were acquired from Addgene (Watertown, MA, USA): pAdDeltaF6 (plasmid # 112867), pAAV2/5 (plasmid # 104964), and pAAV-gfp (plasmid # 32395). The TRAIL gene (derived from pEGFP-TRAIL, plasmid # 10953) was inserted downstream of gfp and an internal ribosome entry site. The anti-gfp shRNA and scramble control shRNA cassettes were inserted into the backbone of the pAAV-gfp plasmid, along with flanking SanDI sites. The resulting vector and PCR-amplified shRNA cassettes were digested with SanDI (Thermo Fisher Scientific, Dublin, Ireland), purified and ligated in a 1:6 vector:insert molar ratio. The resulting colonies were screened for the number of shRNA cassette inserts.
4.2. Cell Culture
HEK293F cells (Thermo Fisher Scientific, Waltham, MA, USA, Cat. R79007) were suspension-cultured in BalanCD HEK (Cat. 91165-1L, FUJIFILM Irvine Scientific, Wicklow, Ireland) chemically defined, serum-free medium supplemented with 4 mM L-Glutamine (Thermo Fisher Scientific, Dublin, Ireland). CHO-K1 cells were suspension cultured in CHO-SFM with anti-clumping agent (Thermo Fisher Scientific, Dublin, Ireland). All cells were cultured in 125 mL Erlenmeyer flasks (Thermo Fisher Scientific, Dublin, Ireland) at 37 °C, 5% CO2, 125 rpm agitation, 25 mm throw in a humidified atmosphere.
4.3. Triple Transfection and rAAV Purification
HEK293F cells were seeded in 4.5 mL BalanCD HEK293 + 4 mM L-Glutamine at 1.11 × 106 cells/mL in 50 mL bioreactor tubes, incubated overnight at 37 °C, 8% CO2, 125 rpm agitation in a humidified atmosphere. On the following day, cells were triple transfected with 3 µg/mL of pAAV-gfp-TRAIL, pAAV2/5, and pAdDeltaF6 (molar ratio 1:1:1) and 1:2 ratio of Polyethylenimine MAX (“PEI-MAX”, Polysciences, Warrington, PA, USA). The plasmids and PEI-MAX reagent were initially diluted in two separate tubes containing 250 µL of medium. The contents of the two tubes were then mixed and incubated for 15 min at room temperature before adding to the cells. Transfections were performed in biological triplicates. For negative controls to be used in qPCR, pAAV-gfp-TRAIL was transfected in the same concentration used in triple transfection, but without the other plasmids. For general negative controls, samples were mock-transfected by adding 500 µL of fresh cell culture medium. 72 h post-transfection, cell viability was measured by trypan blue staining on the LUNA-II Automated Cell Counter (Logos Biosystems, Villeneuve d’Ascq, France). 4 mL of each culture were collected and centrifuged at 16,000× g, 20 min, 4 °C. The supernatants were aliquoted and stored at −80 °C for further analysis, while the cell pellets were resuspended in 1 mL of medium. Cell pellets were processed by 3 liquid nitrogen/37 °C water bath freeze/thaw cycles. After the final thaw, MgCl2 and Benzonase (Merck, Darmstadt, Germany) were added to the samples to a final concentration of 2 mM and 100 U/mL, respectively. Samples were incubated at 37 °C for 1 h. Following incubation, samples were centrifuged at 16,000× g, 20 min, 4 °C. Supernatants were aspirated and stored at −80 °C until further analysis.
4.4. qPCR
rAAV viral genome titers were determined for supernatant and cell pellet samples by qPCR. First, 10 µL of each sample was treated with 1.25 U/µL Benzonase (Merck, Darmstadt, Germany), followed by 6 U/mL proteinase K (Thermo Fisher Scientific, Dublin, Ireland) digestion. Two dilutions of the digested sample (1:100 and 1:1000) were used for qPCR, performed using Fast SYBR Green Master Mix (Applied Biosystems, Carlsbad, CA, USA) and primers specific to the rAAV2 ITRs flanking the gfp expression cassette in the pAAV-gfp-TRAIL plasmid. qPCR was performed using a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific, Dublin, Ireland), and the viral genome titers were calculated using the QuantStudio design and analysis software (version 1.5.1) based on the generated standard curve.
4.5. ELISA
ELISA measurements were performed using a rAAV5 kit (Progen, Heidelberg, Germany), according to the manufacturer’s instructions. Results were obtained using a Biotek Synergy H1 plate reader at 450 and 650 nm wavelengths (Agilent, Santa Clara, CA, USA).
4.6. Transduction
For transduction experiments, 1 × 105 CHO-K1 cells were seeded into each well of a 24-well plate and grown in DMEM-F12 + 10% fetal bovine serum (Thermo Fisher Scientific, Dublin, Ireland) for 24 h, before adding 22,000 vg/cell, then incubating a further 72 h in a static incubator before harvesting. To recover cells which were attached to the culture ware surface, 200 µL of 0.25% trypsin-EDTA (Thermo Fisher Scientific, Dublin, Ireland) was added into each well and incubated for 3 min at 37 °C. Cells recovered in Trypsin-EDTA were combined with harvested suspension cells. After washing with phosphate buffered saline (Thermo Fisher Scientific, Dublin, Ireland), cells were stained and examined by flow cytometry.
4.7. Flow Cytometry
Samples were fixed in 1 mL of 4% v/v formaldehyde/Phosphate buffered saline (PBS) solution for 15 min, then centrifuged at 300× g, 5 min, 20 °C. The supernatant was discarded, and the cell pellet resuspended in 1 mL PBS. This wash step was repeated two times. Samples were loaded onto a 96-well U-bottom plate and analyzed by FACS Accuri C6 (BD, Franklin Lakes, NJ, USA). For staining with PE anti-DR5 antibody (Abcam, Cambridge, U.K., RRID: AB_879820), 1 mL of cells were harvested, centrifuged at 300× g, 5 min, 20 °C and resuspended in ice-cold PBS, 10% Fetal calf serum, 0.1% sodium azide solution (Thermo Fisher Scientific, Dublin, Ireland). 2 µL of antibody was added to the cells and incubated for 30 min at room temperature in the dark. Following centrifugation at 300× g, 5 min, 20 °C, cells were resuspended in 100 µL 4% formaldehyde and incubated at room temperature for 15 min. Finally, centrifugation was performed at 300× g, 5 min, 20 °C and cells were resuspended in 1 mL ice-cold PBS. The final wash step was repeated twice, followed by analysis using FACS Accuri C6 as described above.
4.8. Statistical Analysis
Statistical analysis was performed using Prism (Graphpad, Boston, MA, USA) and Student’s two sample unpaired t-test with a significance level p < 0.05.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}