


Validating Structural Predictions of Conjugated Macromolecules in Espaloma-Enabled Reproducible Workflows



Abstract
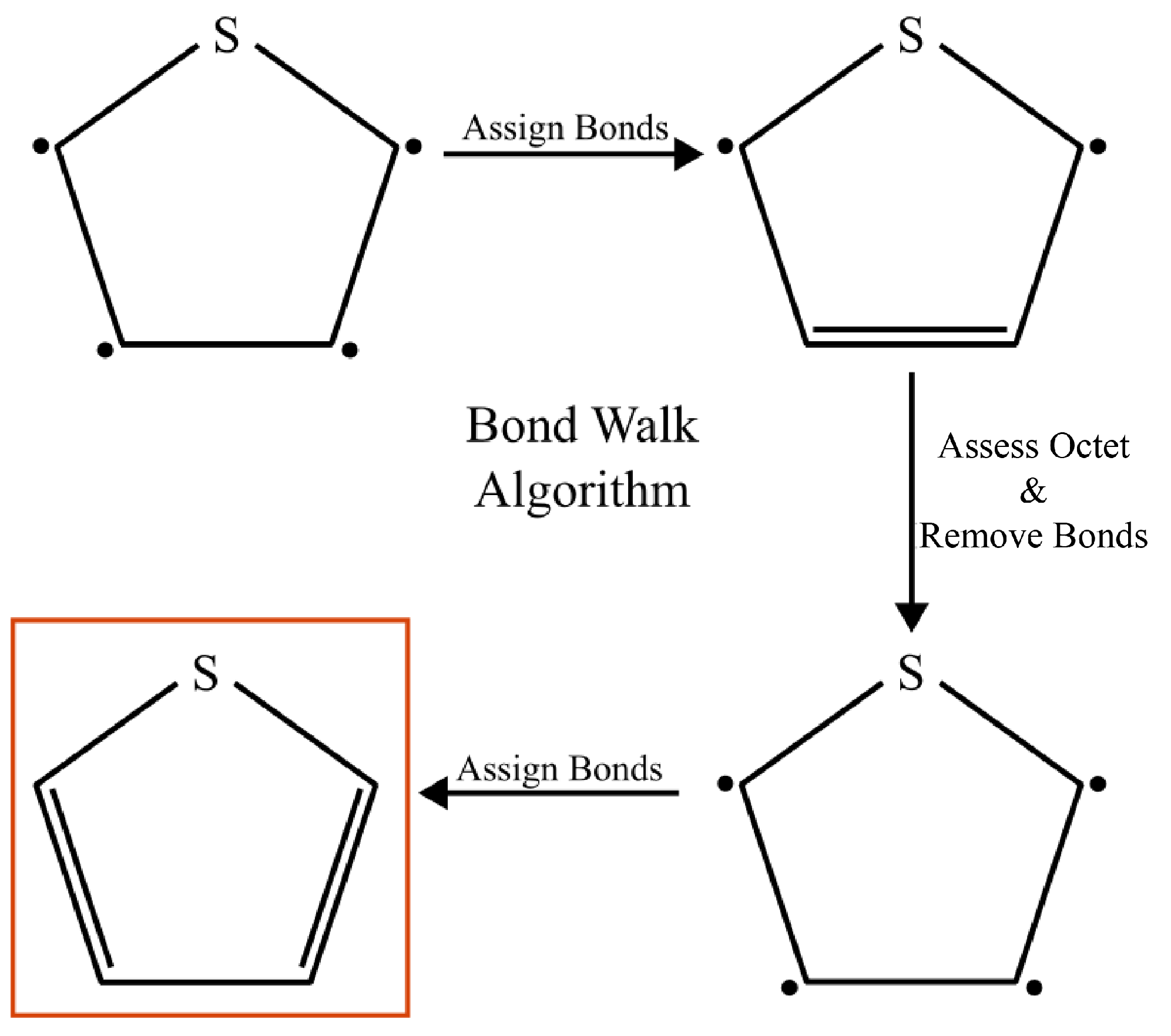
1. Introduction
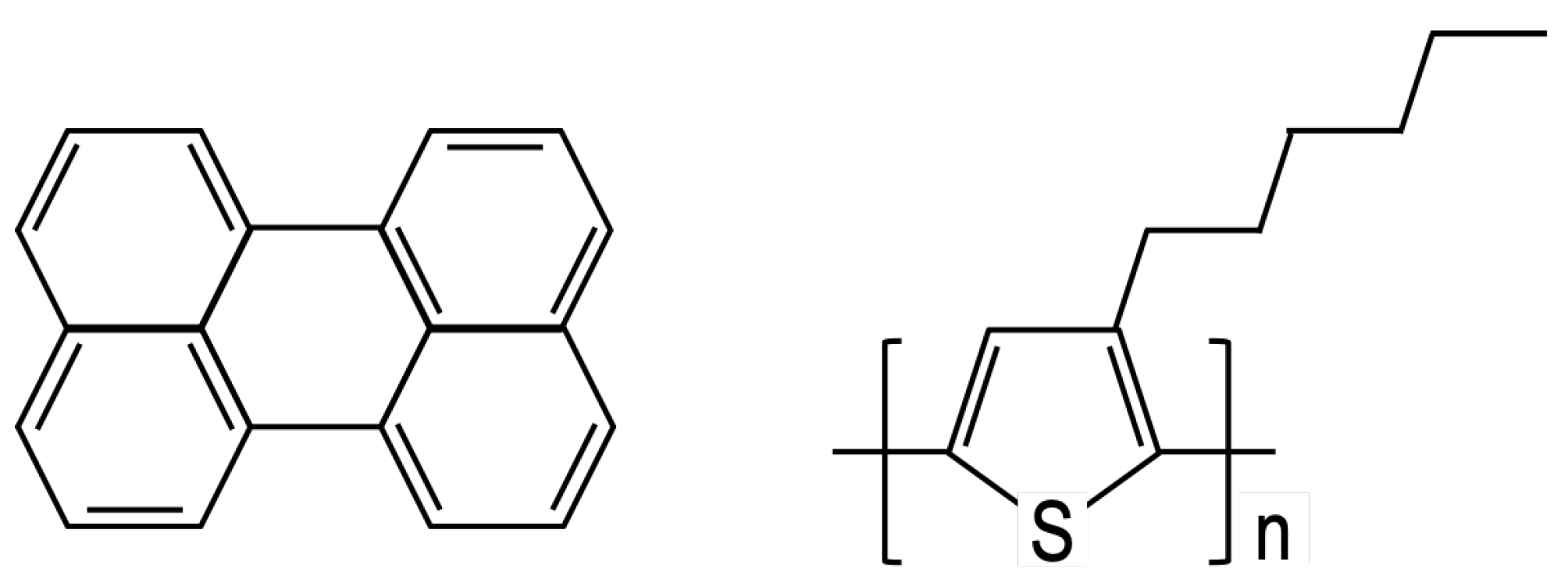
2. Model
3. Results and Discussion
3.1. Espaloma vs. OPLS-UA and GAFF
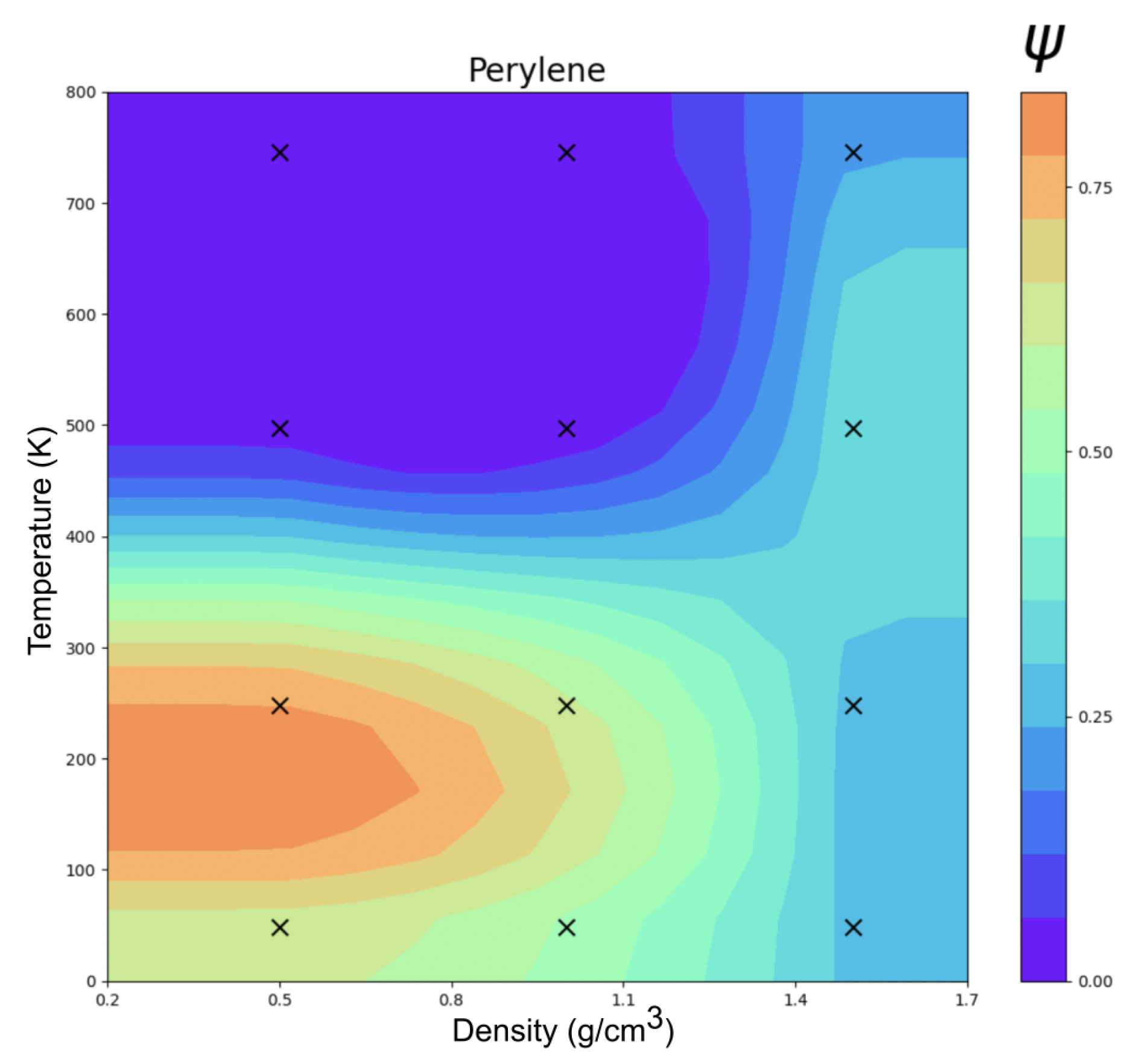
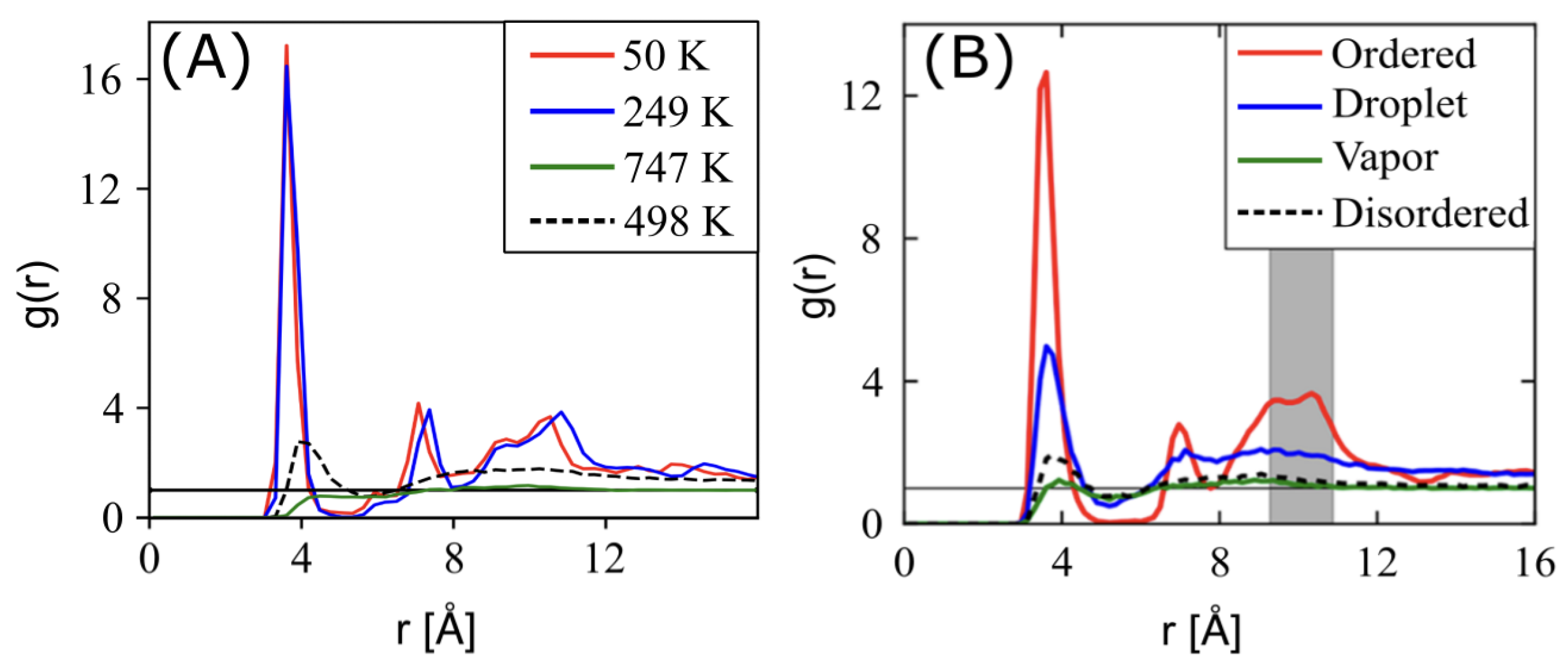
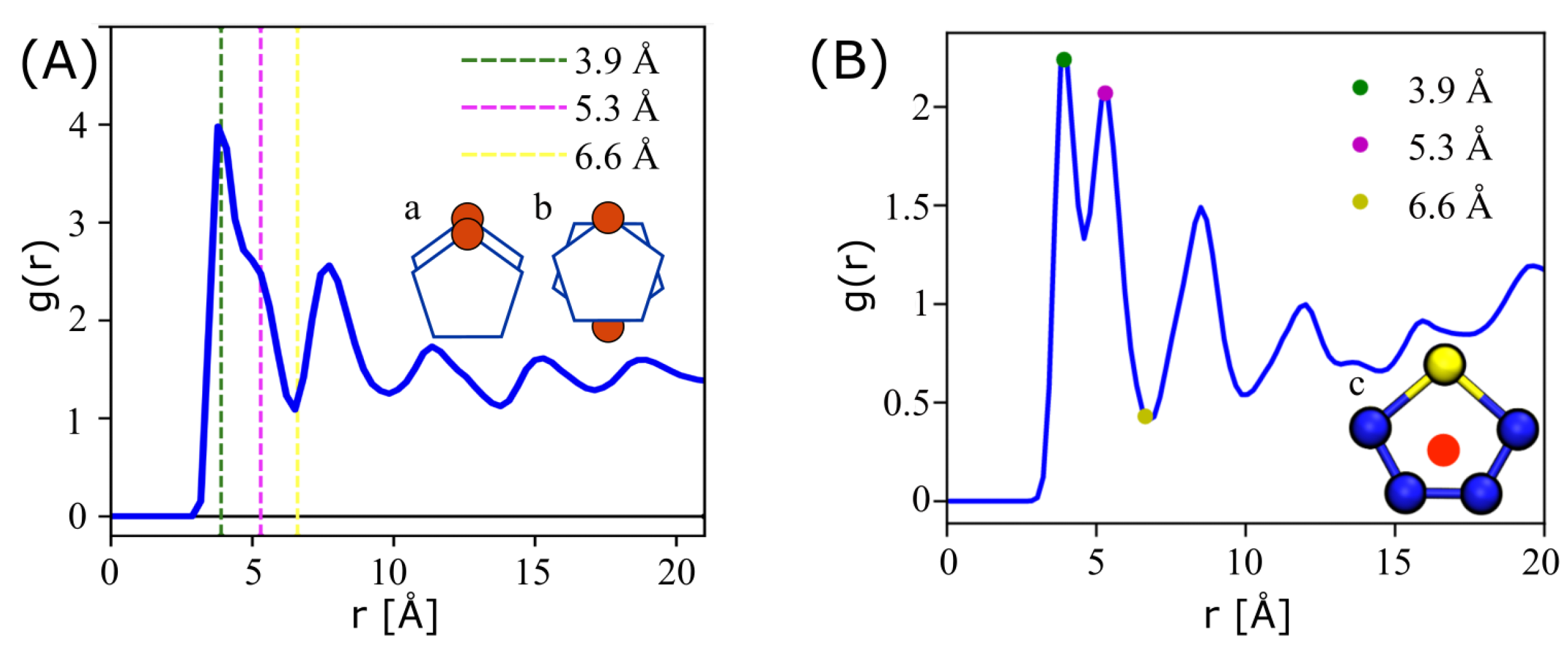
3.2. Perylene
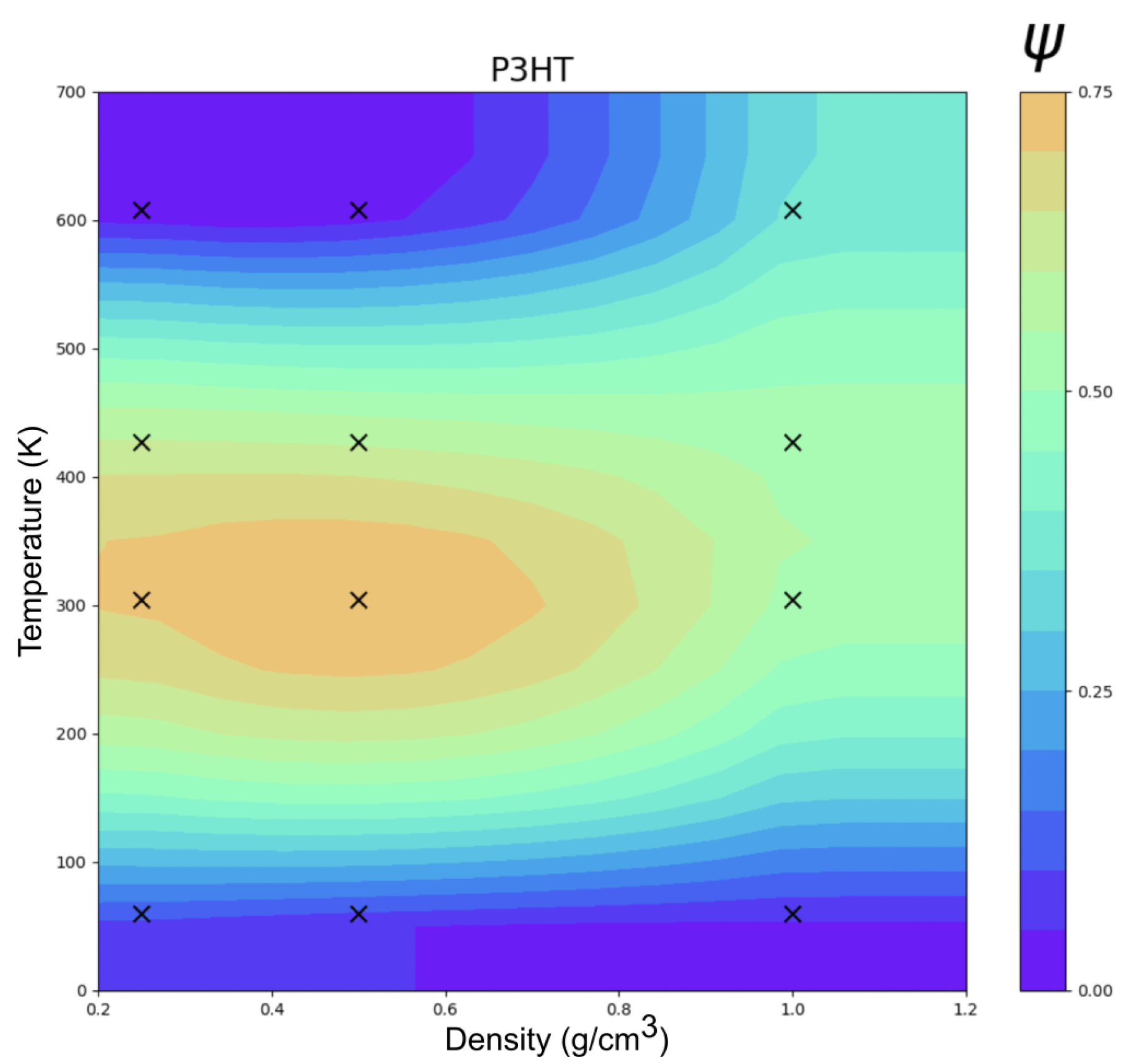
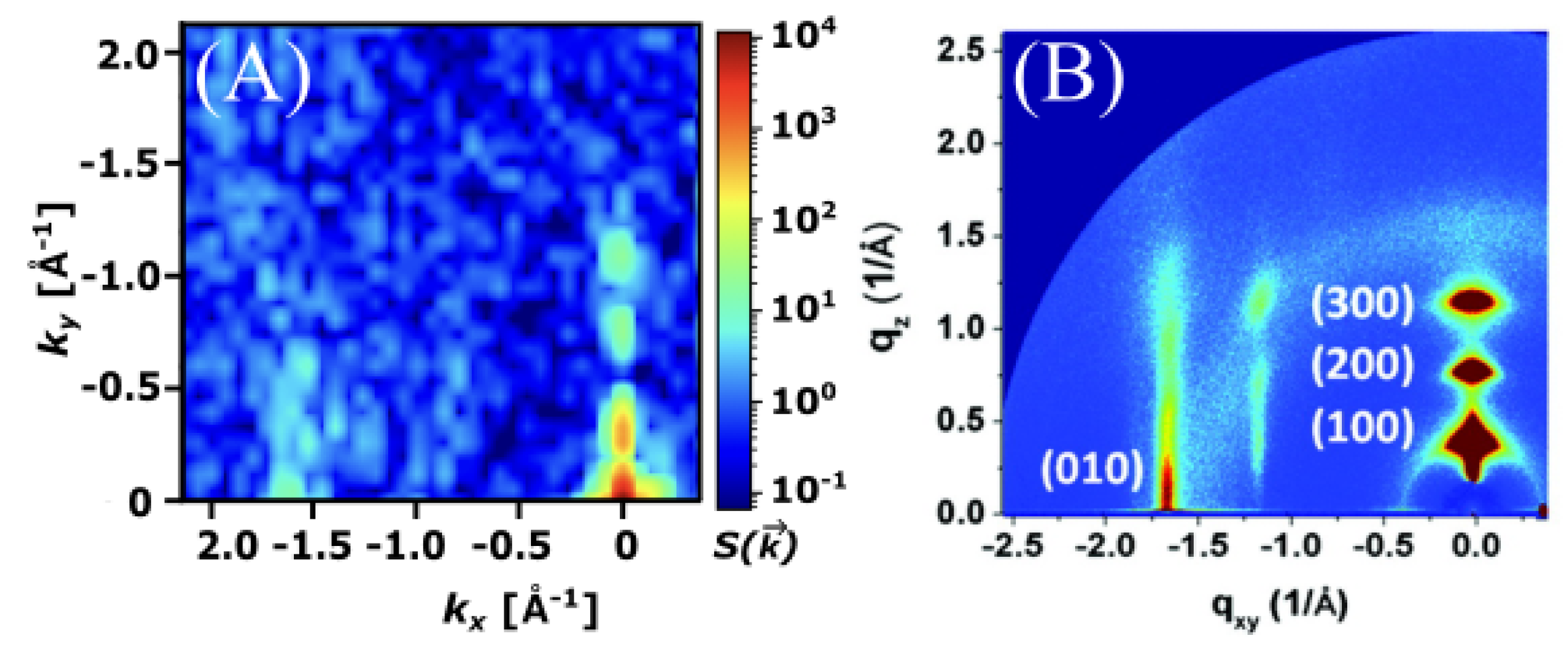
3.3. Poly-3-Hexylthiophene (P3HT)
4. Methods
4.1. Molecular Dynamics
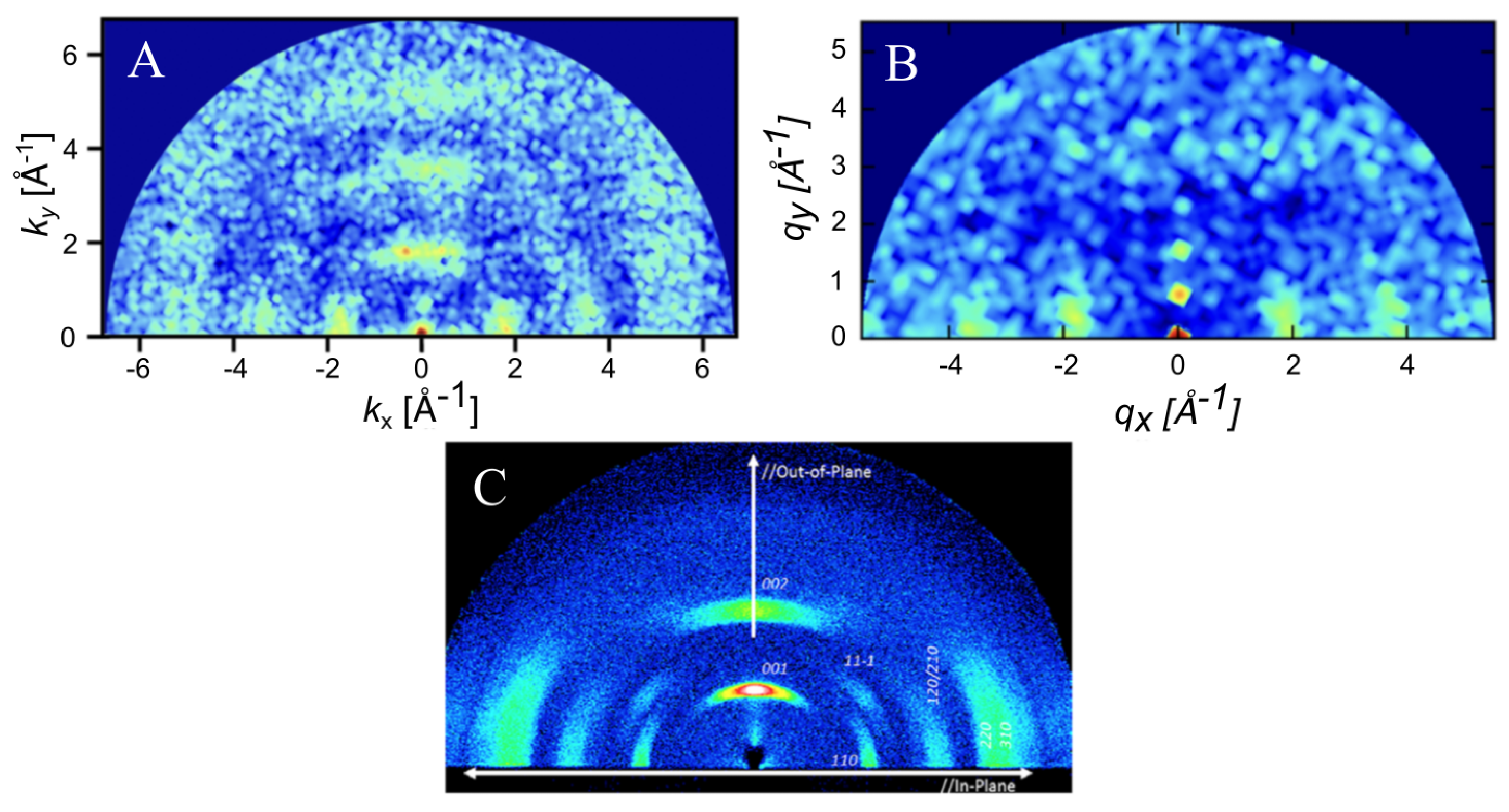
4.2. Morphology Characterization
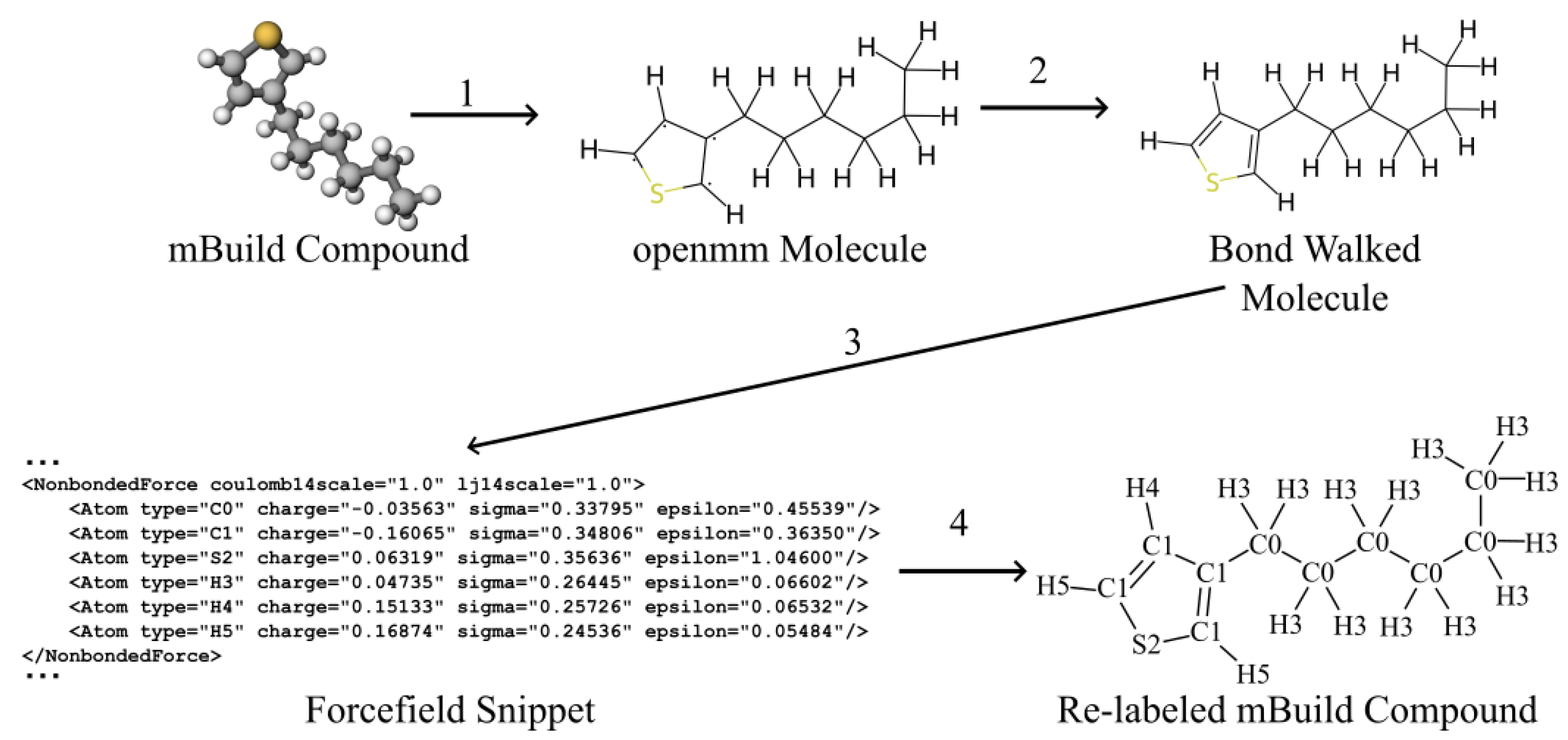
4.3. Integrating Espaloma Parameterization into MoSDeF
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Daglar, H.; Keskin, S. Recent advances, opportunities, and challenges in high-throughput computational screening of MOFs for gas separations. Coord. Chem. Rev. 2020, 422, 213470. [Google Scholar] [CrossRef]
- Liu, X.; Shi, D.; Zhou, S.; Liu, H.; Liu, H.; Yao, X. Molecular dynamics simulations and novel drug discovery. Expert Opin. Drug Discov. 2018, 13, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Li, X.; Lin, X. A review on applications of computational methods in drug screening and design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef]
- Afzal, M.A.F.; Browning, A.R.; Goldberg, A.; Halls, M.D.; Gavartin, J.L.; Morisato, T.; Hughes, T.F.; Giesen, D.J.; Goose, J.E. High-throughput molecular dynamics simulations and validation of thermophysical properties of polymers for various applications. ACS Appl. Polym. Mater. 2020, 3, 620–630. [Google Scholar] [CrossRef]
- Meier, M.A.; Hoogenboom, R.; Schubert, U.S. Combinatorial methods, automated synthesis and high-throughput screening in polymer research: The evolution continues. Macromol. Rapid Commun. 2004, 25, 21–33. [Google Scholar] [CrossRef]
- Quach, C.D.; Gilmer, J.B.; Pert, D.; Mason-Hogans, A.; Iacovella, C.R.; Cummings, P.T.; McCabe, C. High-throughput screening of tribological properties of monolayer films using molecular dynamics and machine learning. J. Chem. Phys. 2022, 156, 154902. [Google Scholar] [CrossRef]
- Wang, Y.; Fass, J.; Kaminow, B.; Herr, J.E.; Rufa, D.; Zhang, I.; Pulido, I.; Henry, M.; Chodera, J.D. End-to-End Differentiable Molecular Mechanics Force Field Construction. Chem. Sci. 2022, 13, 12016–12033. [Google Scholar] [CrossRef]
- Hopfinger, A.; Pearlstein, R. Molecular mechanics force-field parameterization procedures. J. Comput. Chem. 1984, 5, 486–499. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Guvench, O. Molecular mechanics. Curr. Pharm. Des. 2014, 20, 3281–3292. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef]
- Ghahremanpour, M.M.; Tirado-Rives, J.; Jorgensen, W.L. Refinement of the Optimized Potentials for Liquid Simulations Force Field for Thermodynamics and Dynamics of Liquid Alkanes. J. Phys. Chem. B 2022, 126, 5896–5907. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.W.; Gilmer, J.B.; Matsumoto, R.A.; Quach, C.D.; Shamaprasad, P.; Yang, A.H.; Iacovella, C.R.; McCabe, C.; Cummings, P.T. Towards molecular simulations that are transparent, reproducible, usable by others, and extensible (TRUE). Mol. Phys. 2020, 118, e1742938. [Google Scholar] [CrossRef] [PubMed]
- Jankowski, E.; Ellyson, N.; Fothergill, J.W.; Henry, M.M.; Leibowitz, M.H.; Miller, E.D.; Chesser, S.; Guevara, J.D.; Jones, C.D.; Klopfenstein, M.; et al. Perspective on coarse-graining, cognitive load, and materials simulation. Comput. Mater. Sci. 2020, 171, 109129. [Google Scholar] [CrossRef]
- Klein, C.; Summers, A.Z.; Thompson, M.W.; Gilmer, J.B.; McCabe, C.; Cummings, P.T.; Sallai, J.; Iacovella, C.R. Formalizing atom-typing and the dissemination of force fields with foyer. Comput. Mater. Sci. 2019, 167, 215–227. [Google Scholar] [CrossRef]
- Davel, C.M.; Bernat, T.; Jeffery, R.; Wagner, M.R.S. Parameterization of general organic polymers within the Open Force Field framework. J. Chem. Inf. Model. 2024, 64, 1290–1305. [Google Scholar] [CrossRef]
- Takaba, K.; Friedman, A.J.; Cavender, C.E.; Behara, P.K.; Pulido, I.; Henry, M.M.; MacDermott-Opeskin, H.; Iacovella, C.R.; Nagle, A.M.; Payne, A.M.; et al. Machine-learned molecular mechanics force fields from large-scale quantum chemical data. Chem. Sci. 2024, 15, 12861–12878. [Google Scholar] [CrossRef]
- Danielsen, S.P.O.; Bridges, C.R.; Segalman, R.A. Chain Stiffness of Donor–Acceptor Conjugated Polymers in Solution. Macromolecules 2022, 55, 437–449. [Google Scholar] [CrossRef]
- Miller, E.D.; Jones, M.L.; Jankowski, E. Enhanced Computational Sampling of Perylene and Perylothiophene Packing with Rigid-Body Models. ACS Omega 2017, 2, 353–362. [Google Scholar] [CrossRef]
- Miller, E.; Jones, M.; Henry, M.; Chery, P.; Miller, K.; Jankowski, E. Optimization and Validation of Efficient Models for Predicting Polythiophene Self-Assembly. Polymers 2018, 10, 1305. [Google Scholar] [CrossRef]
- Lennard-Jones, J. The electronic structure of some diatomic molecules. Trans. Faraday Soc. 1929, 25, 668–686. [Google Scholar] [CrossRef]
- Bhatta, R.S.; Yimer, Y.Y.; Perry, D.S.; Tsige, M. Improved Force Field for Molecular Modeling of Poly(3-hexylthiophene). J. Phys. Chem. B 2013, 117, 10035–10045. [Google Scholar] [CrossRef] [PubMed]
- Newbloom, G.M.; Weigandt, K.M.; Pozzo, D.C. Structure and property development of poly (3-hexylthiophene) organogels probed with combined rheology, conductivity and small angle neutron scattering. Soft Matter 2012, 8, 8854–8864. [Google Scholar] [CrossRef]
- Botoshansky, M.; Herbstein, F.H.; Kapon, M. Towards a complete description of a polymorphic crystal: The example of perylene: Redetermination of the structures of the (Z = 2 and 4) polymorphs. Helv. Chim. Acta 2003, 86, 1113–1128. [Google Scholar] [CrossRef]
- Ishii, A.; Miyasaka, T. A high voltage organic–inorganic hybrid photovoltaic cell sensitized with metal–ligand interfacial complexes. Chem. Commun. 2012, 48, 9900–9902. [Google Scholar] [CrossRef]
- Ishii, A.; Jena, A.K.; Miyasaka, T. Fully crystalline perovskite-perylene hybrid photovoltaic cell capable of 1.2 V output with a minimized voltage loss. APL Mater. 2014, 2, 091102. [Google Scholar] [CrossRef]
- Marsh, H.S.; Jankowski, E.; Jayaraman, A. Controlling the Morphology of Model Conjugated Thiophene Oligomers through Alkyl Side Chain Length, Placement, and Interactions. Macromolecules 2014, 47, 2736–2747. [Google Scholar] [CrossRef]
- Ko, S.; Hoke, E.T.; Pandey, L.; Hong, S.; Mondal, R.; Risko, C.; Yi, Y.; Noriega, R.; McGehee, M.D.; Brédas, J.L.; et al. Controlled Conjugated Backbone Twisting for an Increased Open-Circuit Voltage while Having a High Short-Circuit Current in Poly(hexylthiophene) Derivatives. J. Am. Chem. Soc. 2012, 134, 5222–5232. [Google Scholar] [CrossRef]
- Anderson, J.A.; Glaser, J.; Glotzer, S.C. HOOMD-blue: A Python package for high-performance molecular dynamics and hard particle Monte Carlo simulations. Comput. Mater. Sci. 2020, 173, 109363. [Google Scholar] [CrossRef]
- Cummings, P.T.; Iacovella, C.R.; Ledeczi, A.; Jankowski, E.; Jayaraman, A.; Palmer, J.C.; Maginn, E.J.; Glotzer, S.C.; Anderson, J.A.; Ilja Siepmann, J.; et al. Open-source molecular modeling software in chemical engineering focusing on the Molecular Simulation Design Framework. AIChE J. 2021, 67, e17206. [Google Scholar] [CrossRef]
- Adorf, C.S.; Dodd, P.M.; Ramasubramani, V.; Glotzer, S.C. Simple data and workflow management with the signac framework. Comput. Mater. Sci. 2018, 146, 220–229. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Albooyeh, M.; Jones, C.; Barrett, R.; Jankowski, E. FlowerMD: Flexible Library of Organic Workflows and Extensible Recipes for Molecular Dynamics. J. Open Source Softw. 2023, 8, 5989. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Cao, J.; Martyna, G.J. Adiabatic path integral molecular dynamics methods. II. Algorithms. J. Chem. Phys. 1996, 104, 2028–2035. [Google Scholar] [CrossRef]
- Ramasubramani, V.; Dice, B.D.; Harper, E.S.; Spellings, M.P.; Anderson, J.A.; Glotzer, S.C. freud: A Software Suite for High Throughput Analysis of Particle Simulation Data. Comput. Phys. Commun. 2020, 254, 107275. [Google Scholar] [CrossRef]
- Klein, C.; Sallai, J.; Jones, T.J.; Iacovella, C.R.; McCabe, C.; Cummings, P.T.; Snurr, R.Q.; Adjiman, C.S.; Kofke, D.A. A Hierarchical, Component Based Approach to Screening Properties of Soft Matter. In Foundations of Molecular Modeling and Simulation: Select Papers from FOMMS 2015; Molecular Modeling and Simulation; Springer: Singapore, 2016; pp. 79–92. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (Å) | (kJ/mol) | |||||
|---|---|---|---|---|---|---|
| C1 | C0 | S | C1 | C0 | S | |
| Espaloma | 3.481 | 3.380 | 3.564 | 0.3635 | 0.4554 | 1.046 |
| OPLS-UA | 3.436 | 3.905 | 3.436 | 0.4602 | 0.7113 | 1.339 |
| GAFF | 3.3997 | 3.3997 | 3.564 | 0.3598 | 0.4577 | 1.046 |
| P3HT | Perylene | |
|---|---|---|
| Temperature Range (K) | [60.4, 304.4, 427.7, 608.9] | [49.8, 248.8, 497.7, 746.5] |
| Density Range (g/cm3) | [0.25, 0.5, 1.0] | [0.5, 1.0, 1.5] |
| N | 100 | 250 |
| dt | 0.0003 | 0.0001 |
| M (amu) | 32.06 | 12.011 |
| (Å) | 3.56 | 3.40 |
| (kJ/mol) | 1.046 | 0.360 |
| Nmonomers | 15 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, M.E.; Jones, C.D.; Jankowski, E. Validating Structural Predictions of Conjugated Macromolecules in Espaloma-Enabled Reproducible Workflows. Int. J. Mol. Sci. 2025, 26, 478. https://doi.org/10.3390/ijms26020478
Paul ME, Jones CD, Jankowski E. Validating Structural Predictions of Conjugated Macromolecules in Espaloma-Enabled Reproducible Workflows. International Journal of Molecular Sciences. 2025; 26(2):478. https://doi.org/10.3390/ijms26020478
Chicago/Turabian StylePaul, Madilyn E., Chris D. Jones, and Eric Jankowski. 2025. "Validating Structural Predictions of Conjugated Macromolecules in Espaloma-Enabled Reproducible Workflows" International Journal of Molecular Sciences 26, no. 2: 478. https://doi.org/10.3390/ijms26020478
APA StylePaul, M. E., Jones, C. D., & Jankowski, E. (2025). Validating Structural Predictions of Conjugated Macromolecules in Espaloma-Enabled Reproducible Workflows. International Journal of Molecular Sciences, 26(2), 478. https://doi.org/10.3390/ijms26020478