
APOE Genotyping in Cognitive Disorders: Preliminary Observations from the Greek Population

 , ,
, ,  , ,
, ,  , , ,
, , ,  , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results
2.1. APOE and Age of Disease (Symptom) Onset
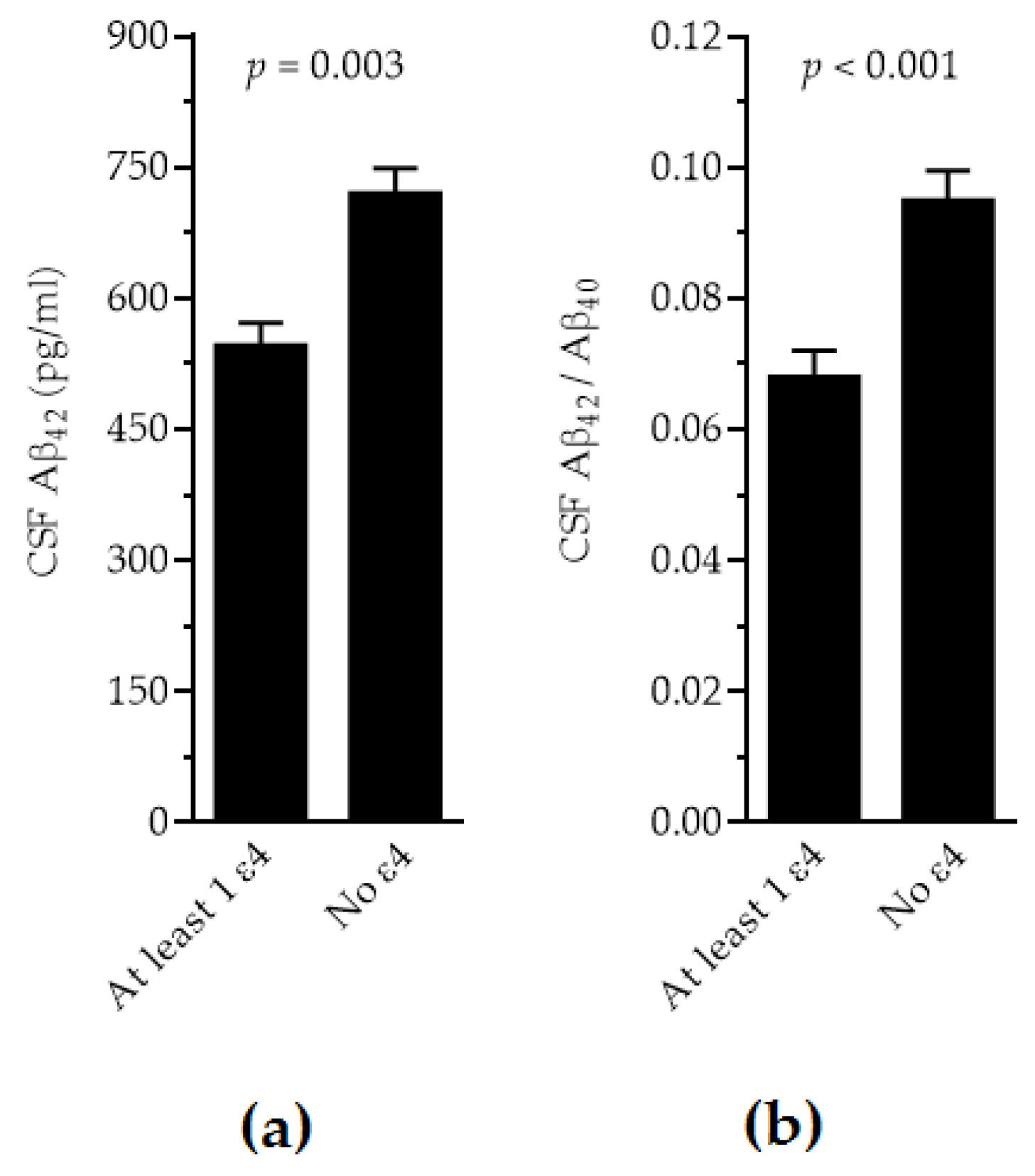
2.2. APOE and CSF Biomarkers
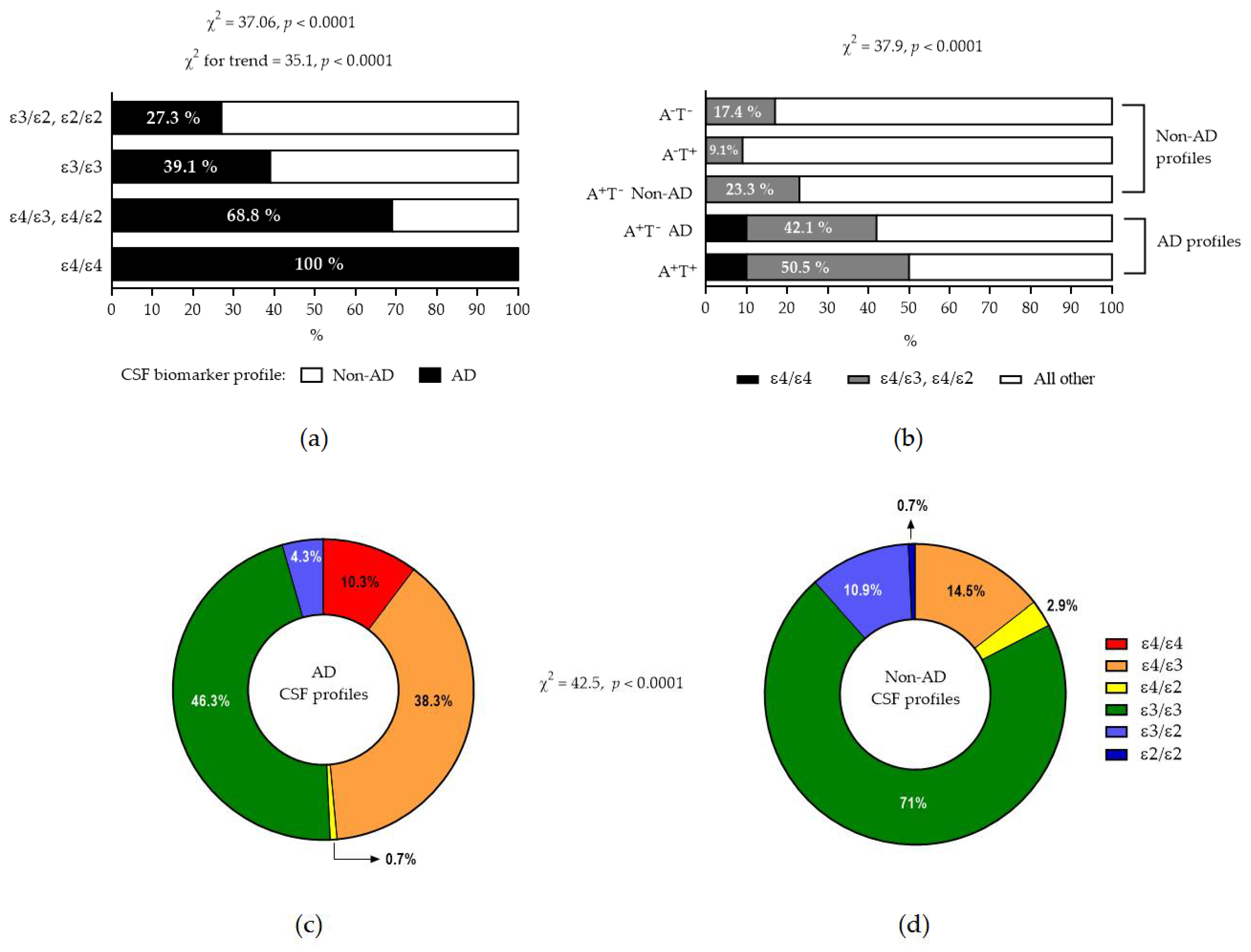
2.3. APOE and Biomarker Profiles
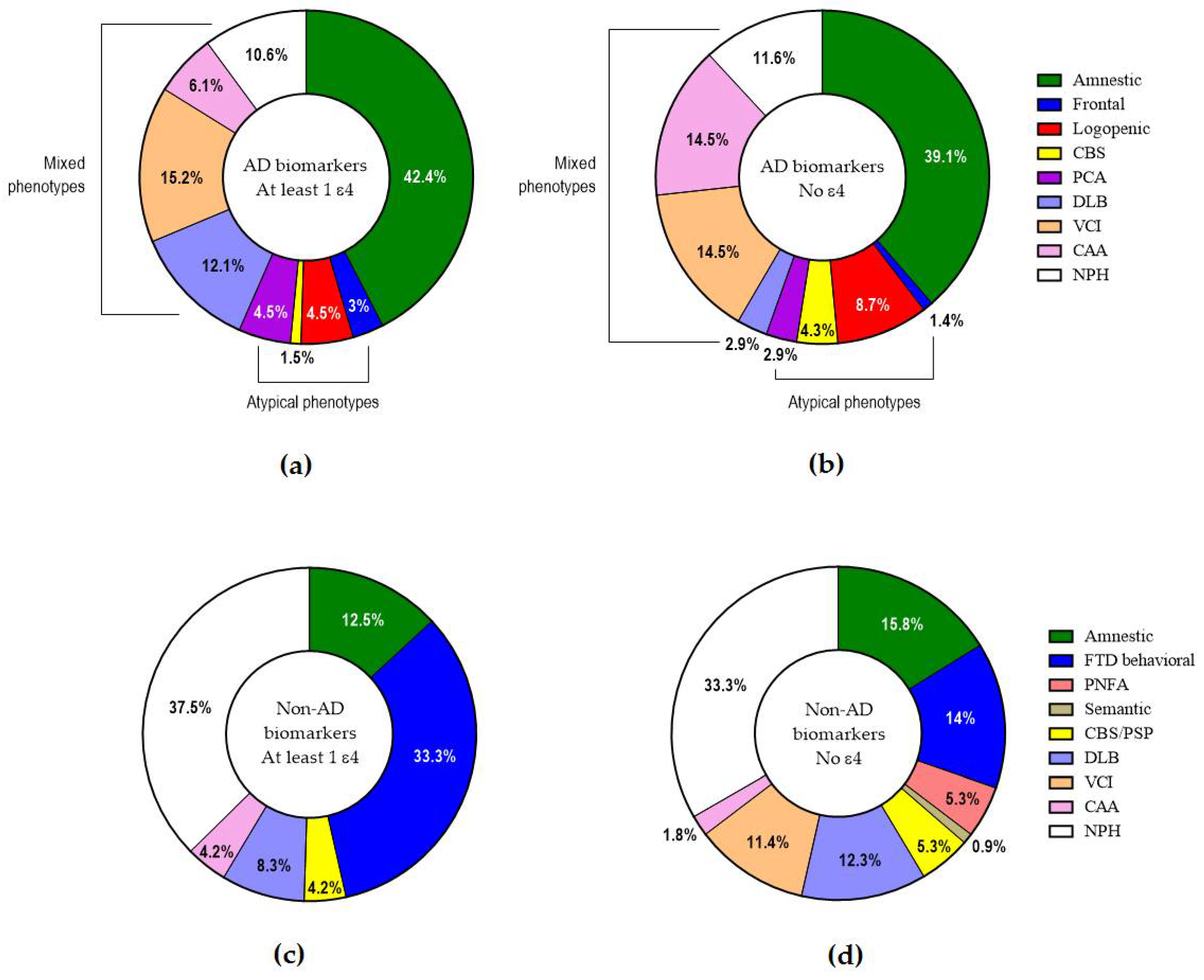
2.4. APOE and Clinical Phenotypes
3. Discussion
4. Materials and Methods
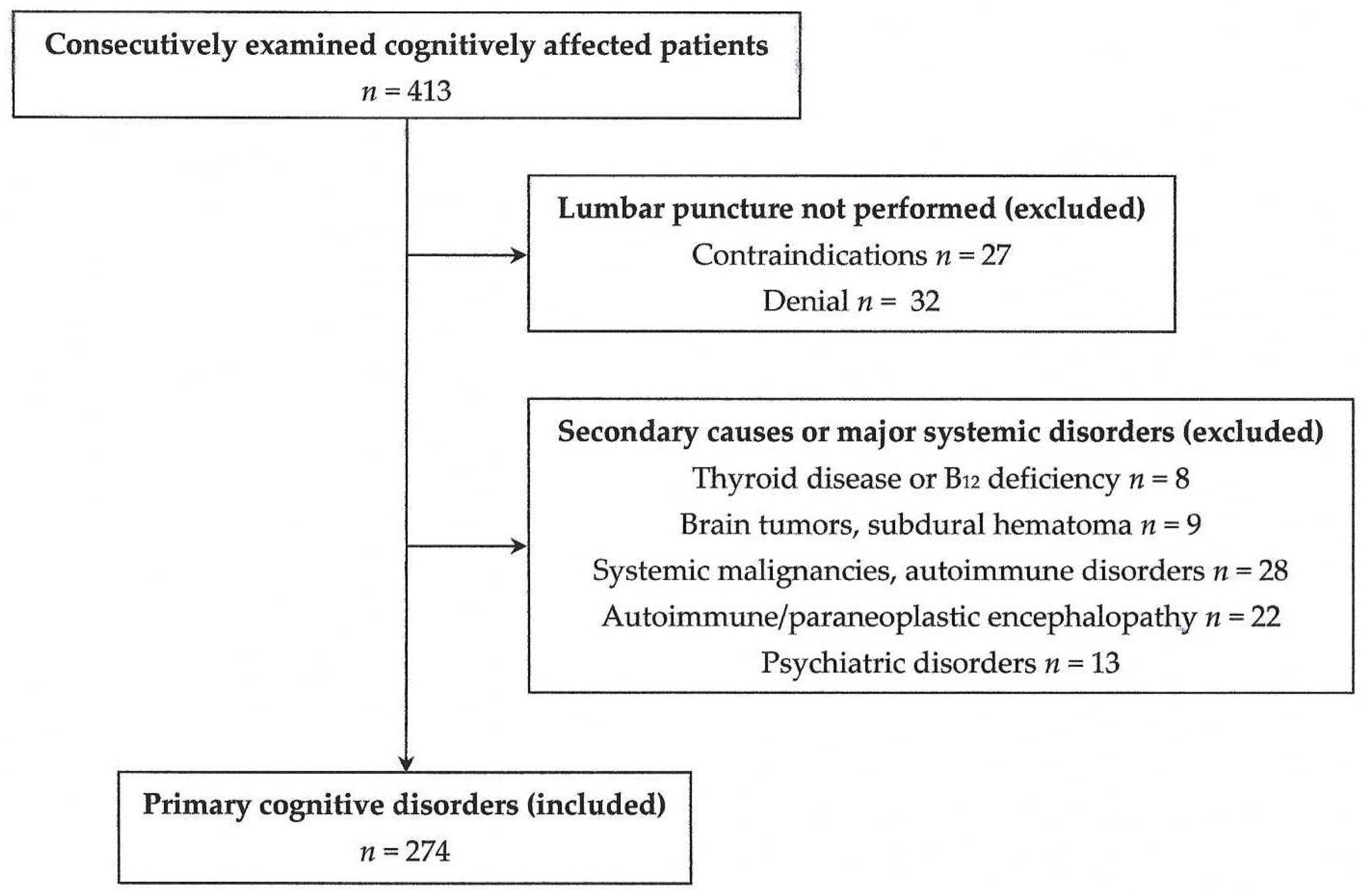
4.1. Study Design
4.2. Patient Clinical Approach
4.3. Lumbar Puncture and CSF Biomarker Assessments
4.4. CSF Biomarker Profiling
4.5. APOE Genotyping
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APOE | Apolipoprotein E |
| MMSE | Mini Mental State Examination |
| ACE-R | Addenbrooke’s Cognitive Examination—Revised version |
| MTA | Medial Temporal Atrophy score |
| ERICA | Entorhinal Cortex Atrophy score |
| ANOVA | Analysis of Variance |
| CBS | Corticobasal syndrome |
| FTD | Frontotemporal dementia |
| PSP | Progressive supranuclear palsy |
| PCA | Posterior cortical atrophy |
| DLB | Dementia with Lewy bodies |
| VCI | Vascular cognitive impairment |
| CAA | Cerebral amyloid angiopathy |
| NPH | Normal-pressure hydrocephalus |
References
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef]
- Dubois, B.; Villain, N.; Frisoni, G.B.; Rabinovici, G.D.; Sabbagh, M.; Cappa, S.; Bejanin, A.; Bombois, S.; Epelbaum, S.; Teichmann, M.; et al. Clinical diagnosis of Alzheimer’s disease: Recommendations of the International Working Group. Lancet Neurol. 2021, 20, 484–496. [Google Scholar] [CrossRef]
- De Strooper, B.; Karran, E. The cellular phase of Alzheimer’s disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Andrews, J.S.; Beach, T.G.; Buracchio, T.; Dunn, B.; Graf, A.; Hansson, O.; Ho, C.; Jagust, W.; McDade, E.; et al. Revised criteria for diagnosis and staging of Alzheimer’s disease: Alzheimer’s Association Workgroup. Alzheimers Dement. 2024, 20, 5143–5169. [Google Scholar] [CrossRef]
- Dubois, B.; Villain, N.; Schneider, L.; Fox, N.; Campbell, N.; Galasko, D.; Kivipelto, M.; Jessen, F.; Hanseeuw, B.; Boada, M.; et al. Alzheimer Disease as a Clinical-Biological Construct-An International Working Group Recommendation. JAMA Neurol. 2024, 81, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Frisoni, G.B.; Festari, C.; Massa, F.; Cotta Ramusino, M.; Orini, S.; Aarsland, D.; Agosta, F.; Babiloni, C.; Borroni, B.; Cappa, S.F.; et al. European intersocietal recommendations for the biomarker-based diagnosis of neurocognitive disorders. Lancet Neurol. 2024, 23, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Villain, N.; Planche, V.; Lilamand, M.; Cordonnier, C.; Soto-Martin, M.; Mollion, H.; Bombois, S.; Delrieu, J.; French Federation of Memory Clinics Work Group on Anti-Amyloid Immunotherapies. Lecanemab for early Alzheimer’s disease: Appropriate use recommendations from the French federation of memory clinics. J. Prev. Alzheimers Dis. 2025, 12, 100094. [Google Scholar] [CrossRef]
- Koutsodendris, N.; Nelson, M.R.; Rao, A.; Huang, Y. Apolipoprotein E and Alzheimer’s disease: Findings, hypotheses, and potential mechanisms. Annu. Rev. Pathol. 2022, 17, 73–99. [Google Scholar] [CrossRef]
- Papastefanopoulou, V.; Stanitsa, E.; Koros, C.; Simoudis, A.; Florou-Hatziyiannidou, C.; Beratis, I.; Antonelou, R.; Andronas, N.; Voskou, P.; Angelopoulou, E.; et al. APOE Allele Frequency in Southern Greece: Exploring the Role of Geographical Gradient in the Greek Population. Geriatrics 2022, 8, 1. [Google Scholar] [CrossRef]
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.J.; Engelborghs, S.; De Deyn, P.; Berr, C.; et al. APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol. Psychiatry 2011, 16, 903–907. [Google Scholar] [CrossRef]
- Hu, H.; Wan, S.; Hu, Y.; Wang, Q.; Li, H.; Zhang, N. Deciphering the role of APOE in cerebral amyloid angiopathy: From genetic insights to therapeutic horizons. Ann. Med. 2025, 57, 2445194. [Google Scholar] [CrossRef]
- Li, Z.; Shue, F.; Zhao, N.; Shinohara, M.; Bu, G. APOE2: Protective mechanism and therapeutic implications for Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 63. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.; Dadar, M.; Kamal, F.; Collins, D.L.; Alzheimer’s Disease Neuroimaging Initiative. Differences in Alzheimer’s Disease-Related Pathology Profiles across Apolipoprotein Groups. J. Gerontol. A Biol. Sci. Med. Sci. 2024, 79, 1–10 glad254. [Google Scholar]
- Fortea, J.; Pegueroles, J.; Alcolea, D.; Belbin, O.; Dols-Icardo, O.; Vaqué-Alcázar, L.; Videla, L.; Gispert, J.D.; Suárez-Calvet, M.; Johnson, S.C.; et al. APOE4 homozygozity represents a distinct genetic form of Alzheimer’s disease. Nat. Med. 2024, 30, 1284–1291. [Google Scholar] [CrossRef] [PubMed]
- Paraskevas, G.P.; Kapaki, E. Cerebrospinal Fluid Biomarkers for Alzheimer’s Disease in the Era of Disease-Modifying Treatments. Brain Sci. 2021, 11, 1258. [Google Scholar] [CrossRef]
- Paterson, R.W.; Toombs, J.; Slattery, C.F.; Nicholas, J.M.; Andreasson, U.; Magdalinou, N.K.; Blennow, K.; Warren, J.D.; Mummery, C.J.; Rossor, M.N.; et al. Dissecting IWG-2 typical and atypical Alzheimer’s disease: Insights from cerebrospinal fluid analysis. J. Neurol. 2015, 262, 2722–2730. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.X.X.; Graff-Radford, J.; Ahmed, S.; Chapleau, M.; Ossenkoppele, R.; Putcha, D.; Rabinovici, G.D.; Suarez-Gonzalez, A.; Schott, J.M.; Crutch, S.; et al. Diagnosis and Management of Posterior Cortical Atrophy. Curr. Treat. Options Neurol. 2023, 25, 23–43. [Google Scholar] [CrossRef]
- Dubois, B.; von Arnim, C.A.F.; Burnie, N.; Bozeat, S.; Cummings, J. Biomarkers in Alzheimer’s disease: Role in early and differential diagnosis and recognition of atypical variants. Alzheimers Res. Ther. 2023, 15, 175. [Google Scholar] [CrossRef]
- Tsantzali, I.; Athanasaki, A.; Boufidou, F.; Constantinides, V.C.; Stefanou, M.I.; Moschovos, C.; Zompola, C.; Paraskevas, S.G.; Bonakis, A.; Giannopoulos, S.; et al. Cerebrospinal Fluid Classical Biomarker Levels in Mixed vs. Pure A+T+ (A+T1+) Alzheimer’s Disease. Biomedicines 2024, 12, 2904. [Google Scholar] [CrossRef]
- Paraskevas, G.P.; Kasselimis, D.; Kourtidou, E.; Constantinides, V.; Bougea, A.; Potagas, C.; Evdokimidis, I.; Kapaki, E. Cerebrospinal Fluid Biomarkers as a Diagnostic Tool of the Underlying Pathology of Primary Progressive Aphasia. J. Alzheimers Dis. 2017, 55, 1453–31461. [Google Scholar] [CrossRef]
- Josephs, K.A.; Weigand, S.D.; Whitwell, J.L. Characterizing Amyloid-Positive Individuals With Normal Tau PET Levels After 5 Years: An ADNI Study. Neurology 2022, 98, e2282–e2292. [Google Scholar] [CrossRef] [PubMed]
- Prosser, L.; Sudre, C.H.; Oxtoby, N.P.; Young, A.L.; Malone, I.B.; Manning, E.N.; Pemberton, H.; Walsh, P.; Barkhof, F.; Biessels, G.J.; et al. Biomarker pathway heterogeneity of amyloid-positive individuals. Alzheimers Dement. 2024, 20, 8503–8515. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Katsumata, Y.; Wu, X.; Aung, K.Z.; Fardo, D.W.; Forrest, S.L.; Alzheimer’s Disease Genetics Consortium; Nelson, P.T. Amyloid-beta predominant Alzheimer’s disease neuropathologic change. Brain 2025, 148, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Schramm, C.; Charbonnier, C.; Zaréa, A.; Lacour, M.; Wallon, D.; CNRMAJ collaborators; Boland, A.; Deleuze, J.F.; Olaso, R.; ADES consortium; et al. Penetrance estimation of Alzheimer disease in SORL1 loss-of-function variant carriers using a family-based strategy and stratification by APOE genotypes. Genome Med. 2022, 14, 69. [Google Scholar] [CrossRef]
- Saddiki, H.; Fayosse, A.; Cognat, E.; Sabia, S.; Engelborghs, S.; Wallon, D.; Alexopoulos, P.; Blennow, K.; Zetterberg, H.; Parnetti, L.; et al. Age and the association between apolipoprotein E genotype and Alzheimer disease: A cerebrospinal fluid biomarker-based case-control study. PLoS Med. 2020, 17, e1003289. [Google Scholar] [CrossRef]
- Lozupone, M.; Leccisotti, I.; Mollica, A.; Berardino, G.; Moretti, M.C.; Altamura, M.; Bellomo, A.; Daniele, A.; Dibello, V.; Solfrizzi, V.; et al. Neuropsychiatric symptoms and apolipoprotein E genotypes in neurocognitive disorders. Neural Regen. Res. 2025, 21, 1528–1541. [Google Scholar] [CrossRef]
- Theodorou, A.; Tsantzali, I.; Kapaki, E.; Constantinides, V.C.; Voumvourakis, K.; Tsivgoulis, G.; Paraskevas, G.P. Cerebrospinal fluid biomarkers and apolipoprotein E genotype in cerebral amyloid angiopathy. A narrative review. Cereb. Circ. Cogn. Behav. 2021, 2, 100010. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.A.; Thorpe, R.J., Jr.; Odden, M.C. Age-dependent interactions of APOE isoform 4 and Alzheimer’s disease neuropathology: Findings from the NACC. Acta Neuropathol. Commun. 2025, 13, 102. [Google Scholar] [CrossRef]
- Hou, X.; Heckman, M.G.; Fiesel, F.C.; Koga, S.; Soto-Beasley, A.I.; Watzlawik, J.O.; Zhao, J.; Valentino, R.R.; Johnson, P.W.; White, L.J.; et al. Genome-wide association analysis identifies APOE as a mitophagy modifier in Lewy body disease. Alzheimers Dement. 2025, 21, e70198. [Google Scholar] [CrossRef]
- Fernández-Calle, R.; Konings, S.C.; Frontiñán-Rubio, J.; García-Revilla, J.; Camprubí-Ferrer, L.; Svensson, M.; Martinson, I.; Boza-Serrano, A.; Venero, J.L.; Nielsen, H.M.; et al. APOE in the bullseye of neurodegenerative diseases: Impact of the APOE genotype in Alzheimer’s disease pathology and brain diseases. Mol. Neurodegener. 2022, 17, 62. [Google Scholar] [CrossRef]
- Islam, S.; Noorani, A.; Sun, Y.; Michikawa, M.; Zou, K. Multi-functional role of apolipoprotein E in neurodegenerative diseases. Front. Aging Neurosci. 2025, 17, 1535280. [Google Scholar] [CrossRef]
- Grimaldi, L.; Bovi, E.; Formisano, R.; Sancesario, G. ApoE: The Non-Protagonist Actor in Neurological Diseases. Genes 2024, 15, 1397. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.Y.; Mormino, E.C. APOE genotype and early β-amyloid accumulation in older adults without dementia. Neurology 2017, 89, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Mattsson, N.; Ossenkoppele, R.; Smith, R.; Strandberg, O.; Ohlsson, T.; Jögi, J.; Palmqvist, S.; Stomrud, E.; Hansson, O. Greater tau load and reduced cortical thickness in APOE ε4-negative Alzheimer’s disease: A cohort study. Alzheimers Res. Ther. 2018, 10, 77. [Google Scholar] [CrossRef] [PubMed]
- Losinski, G.M.; Key, M.N.; Vidoni, E.D.; Clutton, J.; Morris, J.K.; Burns, J.M.; Watts, A. APOE4 and chronic health risk factors are associated with sex-specific preclinical Alzheimer’s disease neuroimaging biomarkers. Front. Glob. Womens Health 2025, 6, 1531062. [Google Scholar] [CrossRef]
- Gorno-Tempini, M.L.; Hillis, A.E.; Weintraub, S.; Kertesz, A.; Mendez, M.; Cappa, S.F.; Ogar, J.M.; Rohrer, J.D.; Black, S.; Boeve, B.F.; et al. Classification of primary progressive aphasia and its variants. Neurology 2011, 76, 1006–1014. [Google Scholar] [CrossRef]
- Crutch, S.J.; Schott, J.M.; Rabinovici, G.D.; Murray, M.; Snowden, J.S.; van der Flier, W.M.; Dickerson, B.C.; Vandenberghe, R.; Ahmed, S.; Bak, T.H.; et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 2017, 13, 870–884. [Google Scholar] [CrossRef]
- Sachdev, P.; Kalaria, R.; O’Brien, J.; Skoog, I.; Alladi, S.; Black, S.E.; Blacker, D.; Blazer, D.G.; Chen, C.; Chui, H.; et al. Diagnostic criteria for vascular cognitive disorders: A VASCOG statement. Alzheimer Dis. Assoc. Disord. 2014, 28, 206–218. [Google Scholar] [CrossRef]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; Halliday, G.; Taylor, J.P.; Weintraub, D.; Aarsland, D.; Galvin, J.; Attems, J.; Ballard, C.G.; et al. Diagnosis and management of dementia with Lewy bodies. Fourth consensus report of the DLB Consortium. Neurology 2017, 89, 88–100. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Rascovsky, K.; Hodges, J.R.; Knopman, D.; Mendez, M.F.; Kramer, J.H.; Neuhaus, J.; Van Swieten, J.C.; Seelaar, H.; Dopper, E.G.P.; Onyike, C.U.; et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011, 134, 2456–2477. [Google Scholar] [CrossRef]
- Höglinger, G.U.; Respondek, G.; Stamelou, M.; Kurz, C.; Josephs, K.A.; Lang, A.E.; Mollenhauer, B.; Müller, U.; Nilsson, C.; Whitwell, J.L.; et al. Clinical diagnosis of progressive supranuclear palsy: The movement disorder society criteria. Mov. Disord. 2017, 32, 853–864. [Google Scholar] [CrossRef]
- Armstrong, M.J.; Litvan, I.; Lang, A.E.; Bak, T.H.; Bhatia, K.P.; Borroni, B.; Boxer, A.L.; Dickson, D.W.; Grossman, M.; Hallett, M.; et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013, 80, 496–503. [Google Scholar] [CrossRef]
- Nakajima, M.; Yamada, S.; Miyajima, M.; Ishii, K.; Kuriyama, N.; Kazui, H.; Kanemoto, H.; Suehiro, T.; Yoshiyama, K.; Kameda, M.; et al. Guidelines for Management of Idiopathic Normal Pressure Hydrocephalus (Third Edition): Endorsed by the Japanese Society of Normal Pressure Hydrocephalus. Neurol. Med.-Chir. 2021, 61, 63–97. [Google Scholar] [CrossRef] [PubMed]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Mioshi, E.; Dawson, K.; Mitchell, J.; Arnold, R.; Hodges, J.R. The Addenbrooke’s Cognitive Examination Revised (ACE-R): A brief cognitive test battery for dementia screening. Int. J. Geriatr. Psychiatry 2006, 21, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Fountoulakis, K.N.; Tsolaki, M.; Chantzi, H.; Kazis, A. Mini Mental State Examination (MMSE): A validation study in Greece. Am. J. Alzheimers Dis. Other Demen. 2000, 15, 342–345. [Google Scholar] [CrossRef]
- Konstantinopoulou, E.; Kosmidis, M.H.; Ioannidis, P.; Kiosseoglou, G.; Karacostas, D.; Taskos, N. Adaptation of Addenbrooke’s Cognitive Examination-Revised for the Greek population. Eur. J. Neurol. 2011, 18, 442–447. [Google Scholar] [CrossRef]
- Scheltens, P.; Leys, D.; Barkhof, F.; Huglo, D.; Weinstein, H.C.; Vermersch, P.; Kuiper, M.; Steinling, M.; Wolters, E.C.; Valk, J. Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: Diagnostic value and neuropsychological correlates. J. Neurol. Neurosurg. Psychiatry 1992, 55, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Enkirch, S.J.; Traschütz, A.; Müller, A.; Widmann, C.N.; Gielen, G.H.; Heneka, M.T.; Jurcoane, A.; Schild, H.H.; Hattingen, E. The ERICA Score: An MR Imaging-based Visual Scoring System for the Assessment of Entorhinal Cortex Atrophy in Alzheimer Disease. Radiology 2018, 288, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Tsantzali, I.; Boufidou, F.; Sideri, E.; Mavromatos, A.; Papaioannou, M.G.; Foska, A.; Tollos, I.; Paraskevas, S.G.; Bonakis, A.; Voumvourakis, K.I.; et al. From Cerebrospinal Fluid Neurochemistry to Clinical Diagnosis of Alzheimer’s Disease in the Era of Anti-Amyloid Treatments. Report of Four Patients. Biomedicines 2021, 9, 1376. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Boufidou, F.; Stergiou, C.; Kuhle, J.; Willemse, E.; Palaiodimou, L.; Tsantzali, I.; Sideri, E.; Bonakis, A.; Giannopoulos, S.; et al. Plasma P-Tau181 for the Discrimination of Alzheimer’s Disease from Other Primary Dementing and/or Movement Disorders. Biomolecules 2022, 12, 1099. [Google Scholar] [CrossRef] [PubMed]
- University of Gothenburg, Institute of Neuroscience and Neurophysiology, The Alzheimer’s Association’s QC Program for CSF and Blood Biomarkers. Available online: https://www.gu.se/en/neuroscience-physiology/the-alzheimers-association-qc-program-for-csf-and-blood-biomarkers (accessed on 27 June 2025).
- INSTAND e.V. Gesellschaft zur Förderung der Qualitätssicherung in Medizinischen Laboratorien e. V. (Düsseldorf, Germany). Available online: https://www.instand-ev.de/en/ (accessed on 27 June 2025).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ε4/ε4 | ε4/ε3 | ε4/ε2 | ε3/ε3 | ε3/ε2 | ε2/ε2 | p Value | |
|---|---|---|---|---|---|---|---|
| n (%) | 14 (5.1%) | 72 (26.3%) | 5 (1.8%) | 161 (58.8%) | 21 (7.7%) | 1 (0.3%) | |
| Gender (M/F) | 5/9 | 26/46 | 1/4 | 67/94 | 8/13 | 1/0 | NS * |
| Age (y) | 68.6 ± 5.2 | 68.9 ± 8.4 | 66.0 ± 14.4 | 72.1 ± 8.9 a | 69.9 ± 7.4 | 83.0 | 0.044 † |
| Duration (y) | 3.2 ± 2.0 | 3.7 ± 3.7 | 5.5 ± 0.7 | 3.6 ± 3.4 | 3.9 ± 3.6 | 1.5 | NS † |
| Onset (y) | 65.4 ± 6.1 | 65.2 ± 8.3 | 60.4 ± 13.4 | 69.5 ± 8.3 b | 66.1. ± 8.7 | 81.5 | 0.0064 † |
| Education (y) | 11.0 ± 5.9 | 12.0 ± 4.3 | 7.5 ± 2.1 | 10.6 ± 5.0 | 9.1 ± 4.0 | 6.0 | NS † |
| MMSE score | 19.6 ± 5.8 21 (14–23) | 21.6 ± 7.3 24 (17–27) | 26.5 ± 3.5 27 (24–29) | 22.8 ± 5.8 24 (21–27) | 22.6 ± 5.4 24 (18–26) | 25 | NS † NS ‡ |
| ACE-R total score | 57.9 ± 18.4 | 65.0 ± 20.7 | 82.5 ± 3.5 | 66.1 ± 18.6 | 63.4 ± 20.8 | 65 | NS † |
| Attention-orientation | 13.0 ± 3.9 | 13.7 ± 4.7 | 16.1 ± 1.2 | 14.2 ± 3.9 | 14.6 ± 3.7 | 17 | NS † |
| Memory | 10.4 ± 4.9 | 14.2 ± 6.9 | 19.0 ± 4.2 | 14.8 ± 6.6 | 15.0 ± 7.0 | 19 | NS † |
| Fluency | 5.2 ± 3.5 | 5.9 ± 3.6 | 9.0 ±1.4 | 5.5 ± 3.3 | 5.8 ± 3.3 | 3 | NS † |
| Language | 17.7 ± 6.3 | 20.7 ± 5.0 | 24.0 ± 1.4 | 20.2 ± 4.9 | 17.5 ± 6.3 | 14 | NS † |
| Visuospatial | 11.7 ± 2.8 | 10.7 ± 3.9 | 13.5 ± 2.1 | 11.6 ± 3.1 | 9.9 ± 4.2 | 12 | NS † |
| MTA score | 3 (1.5–3) | 2.5 (1–3) | 0.5 (0–3) | 2 (1–3) | 2 (0.1–2.9) | 3 | NS ‡ |
| ERICA score | 2 (1.5–2) | 2 (0.5–2) | 1.5 (0–2) | 1 (1–2) c | 1 (0–1) c | 2 | 0.0016 ‡ |
| Aβ42 (pg/mL) | 457 ± 143 | 564 ± 249 | 767 ± 384 | 710 ± 379 | 788 ± 461 | 626 | 0.003 # |
| Aβ40 (pg/mL) | 7471 ± 2235 | 10077 ± 5482 | 9988 ± 2655 | 9171 ± 5238 | 9347 ± 3433 | 6852 | NS # |
| Aβ42/Aβ40 | 0.069 ± 0.019 | 0.069 ± 0.032 | 0.076 ± 0.032 | 0.093 ± 0.047 | 0.114 ± 0.050 | 0.091 | <0.001 # |
| τP181 (pg/mL) | 94.7 ± 47.3 86 (55–118) | 83.9 ± 50.9 72 (46–114) | 45.4 ± 20.1 39 (33–58) | 61.5 ± 41.1 49 (32–81) | 52.9 ± 23.2 48 (38–62) | 35 | NS # |
| τT (pg/mL) | 520 ± 247 500 (361–626) | 512 ± 391 419 (257–588) | 269 ± 83.2 246 (218–320) | 384 ± 285 298 (178–487) | 460 ± 269 357 (268–718) | 376 | NS # |
| τP181/Aβ42 | 0.21 ± 0.13 0.16 (0.12–0.26) | 0.18 ± 0.13 0.16 (0.08–0.24) | 0.07 ± 0.04 0.07 (0.04–0.10) | 0.13 ± 0.11 0.10 (0.05–0.17) | 0.12 ± 0.17 0.06 (0.05–0.10) | 0.06 | NS # |
| ε4/ε4 | ε4/ε3 | ε4/ε2 | ε3/ε3 | ε3/ε2 | ε2/ε2 | |
|---|---|---|---|---|---|---|
| n (%) | 14 (5.1%) | 72 (26.3%) | 5 (1.8%) | 161 (58.8%) | 21 (7.7%) | 1 (0.3%) |
| Total suggestive of AD pathology | 14 (100%) | 52 (72.2%) | 1 (20%) | 63 (39.1%) | 6 (28.6%) | 0 (0%) |
| A+T+ | 12 (85.7%) | 46 (63.9%) | 1 (20%) | 53 (32.9%) | 5 (23.8%) | 0 (0%) |
| A+T− with abnormal τP181/Aβ42 ratio | 2 (14.3%) | 6 (8.3%) | 0 (0%) | 10 (6.2%) | 1 (4.8%) | 0 (0%) |
| Total suggestive of non-AD pathology | 0 (0%) | 20 (27.8%) | 4 (80%) | 98 (60.9%) | 15 (71.4%) | 1 (100%) |
| A+T− with normal τP181/Aβ42 ratio | 0 (0%) | 6 (8.3%) | 1 (20%) | 21 (13.1%) | 2 (9.5%) | 0 (0%) |
| A−T+ | 0(0%) | 1 (1.4%) | 1 (20%) | 16 (9.9%) | 3 (14.3%) | 1 (100%) |
| A−T− | 0 (0%) | 13 (18.1%) | 2 (40%) | 61 (37.9%) | 10 (47.6%) | 0 (0%) |
| p Value a | Odds ratio b | Sensitivity b | Specificity b | PPV b | NPV b | |
|---|---|---|---|---|---|---|
| At least one ε4 allele 1 | <0.0001 | 5.87 (3.35–10.3) | 0.53 (0.44–0.62) | 0.84 (0.77–0.89) | 0.74 (0.63–0.82) | 0.68 (0.60–0.74) |
| ε4/ε3 and ε4/ε2 1 | <0.0001 | 4.61 (2.62–8.24) | 0.47 (0.38–0.57) | 0.84 (0.77–0.89) | 0.69 (0.52–0.79) | 0.68 (0.60–0.74) |
| ε4/ε4 1 | <0.0001 | 60.7 (3.56–1035) | 0.19 (0.11–0.30) | 1.00 (0.97–1.00) | 1.00 (0.77–1.00) | 0.68 (0.60–0.74) |
| ε4/ε4 2 | <0.0001 | 38.3 (2.26–649) | 0.11 (0.06–0.18) | 1.00 (0.98–1.00) | 1.00 (0.77–1.00) | 0.57 (0.51–0.63) |
| p Value | Odds Ratio | AUC | |
|---|---|---|---|
| First model: ε4 presence vs. ε4 absence | |||
| Model | <0.0001 | 0.75 (0.68–0.81) | |
| Education | NS | 0.97 (0.90–1.04) | |
| Sex | 0.03 | 2.04 (1.20–3.45) | |
| At least one ε4 allele | <0.0001 | 6.31 (3.12–12.76) | |
| Second model: others vs. ε3/ε3 as reference | |||
| Model | <0.0001 | 0.76 (0.69–0.82) | |
| Education | NS | 0.96 ((0.89–1.03) | |
| Sex | 0.04 | 1.9 (1.03–3.88) | |
| ε4/ε3 and ε4/ε2 | 0.0001 | 4.46 (2.15–9.25) | |
| ε4/ε4 | 0.05 | 37.7 (2.1–501) | |
| ε3/ε2 and ε2/ε2 | NS | 0.37 (0.09–1.03) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Athanasaki, A.; Tsantzali, I.; Kroupis, C.; Theodorou, A.; Boufidou, F.; Constantinides, V.C.; Tzartos, J.S.; Tzartos, S.J.; Velonakis, G.; Zompola, C.; et al. APOE Genotyping in Cognitive Disorders: Preliminary Observations from the Greek Population. Int. J. Mol. Sci. 2025, 26, 7410. https://doi.org/10.3390/ijms26157410
Athanasaki A, Tsantzali I, Kroupis C, Theodorou A, Boufidou F, Constantinides VC, Tzartos JS, Tzartos SJ, Velonakis G, Zompola C, et al. APOE Genotyping in Cognitive Disorders: Preliminary Observations from the Greek Population. International Journal of Molecular Sciences. 2025; 26(15):7410. https://doi.org/10.3390/ijms26157410
Chicago/Turabian StyleAthanasaki, Athanasia, Ioanna Tsantzali, Christos Kroupis, Aikaterini Theodorou, Fotini Boufidou, Vasilios C. Constantinides, John S. Tzartos, Socrates J. Tzartos, Georgios Velonakis, Christina Zompola, and et al. 2025. "APOE Genotyping in Cognitive Disorders: Preliminary Observations from the Greek Population" International Journal of Molecular Sciences 26, no. 15: 7410. https://doi.org/10.3390/ijms26157410
APA StyleAthanasaki, A., Tsantzali, I., Kroupis, C., Theodorou, A., Boufidou, F., Constantinides, V. C., Tzartos, J. S., Tzartos, S. J., Velonakis, G., Zompola, C., Michalopoulou, A., Paraskevas, P. G., Bonakis, A., Giannopoulos, S., Moutsatsou, P., Tsivgoulis, G., Kapaki, E., & Paraskevas, G. P. (2025). APOE Genotyping in Cognitive Disorders: Preliminary Observations from the Greek Population. International Journal of Molecular Sciences, 26(15), 7410. https://doi.org/10.3390/ijms26157410