

Computational Design of Potentially Multifunctional Antimicrobial Peptide Candidates via a Hybrid Generative Model

 , and
, and
Abstract
1. Introduction
2. Results and Discussion
2.1. Implementation Details
2.1.1. Data Gathering and Preprocessing
2.1.2. GAN Training and Evaluation
2.1.3. Training and Evaluation of the Multifunction Predictor
2.2. Experimental Results
2.2.1. Comparison of AMP Identification Models
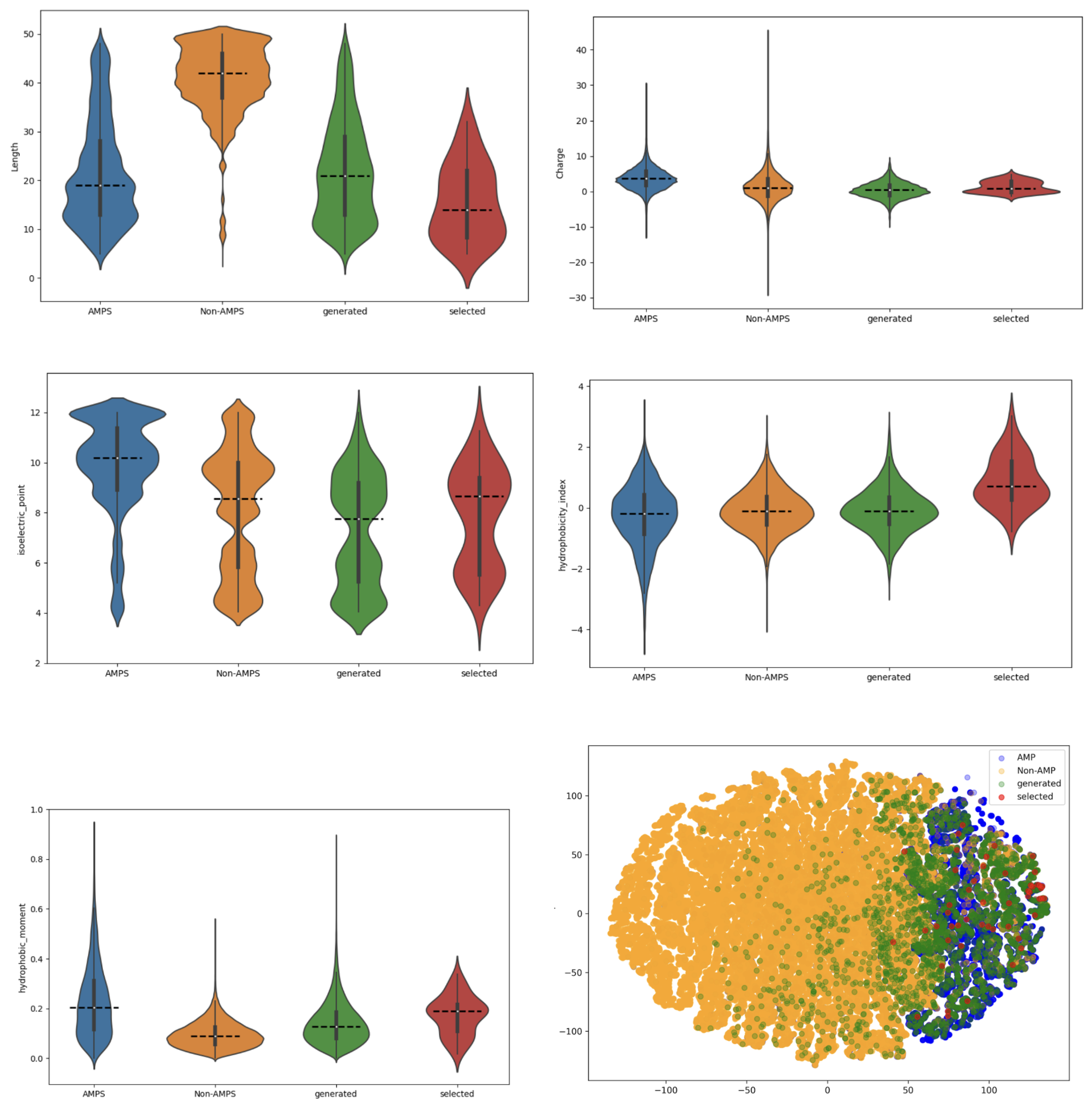
2.2.2. Analysis of the Quality and Multifunctionality of the Generated AMP Sequences
Multifunction Predictions
Physicochemical Properties
2.2.3. Ranking Generated AMP Sequences Based on Structural Confidence
3. Materials and Methods
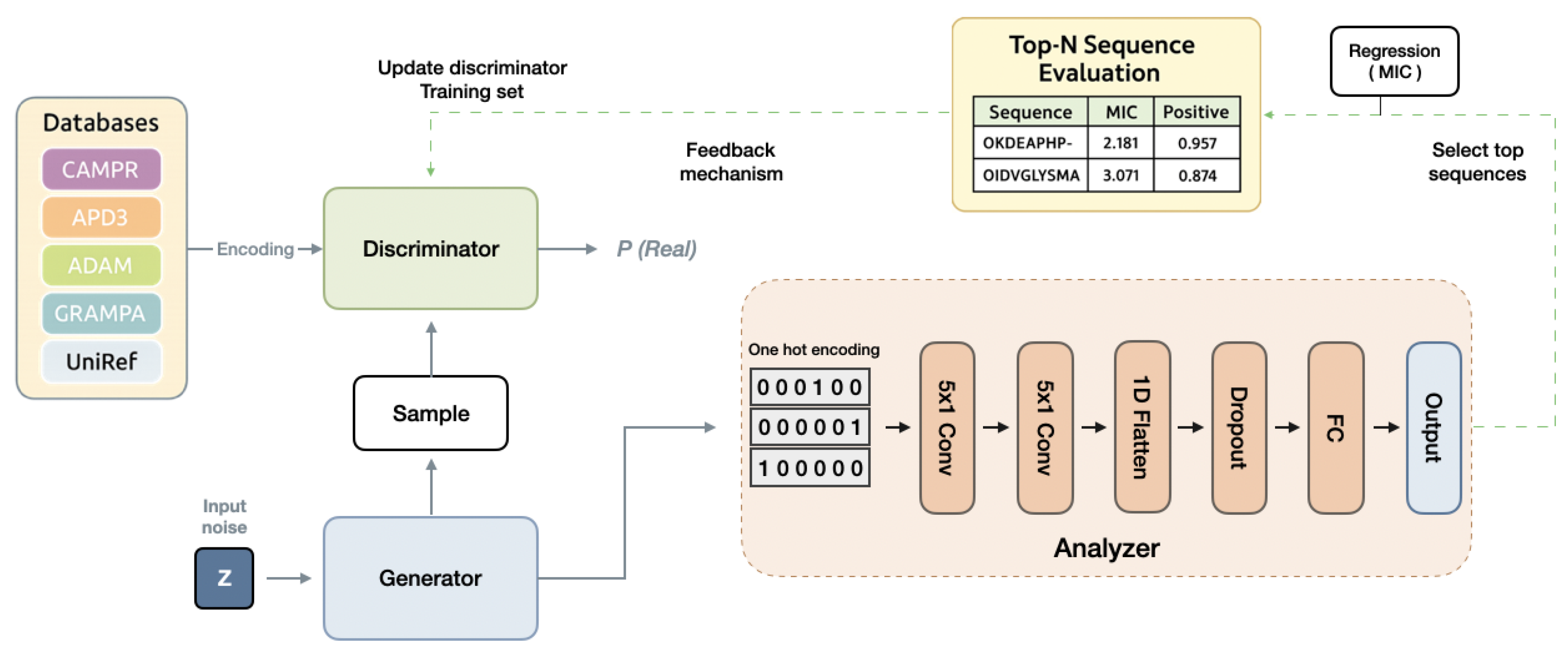
3.1. Overall Framework of FBGAN-Based Model
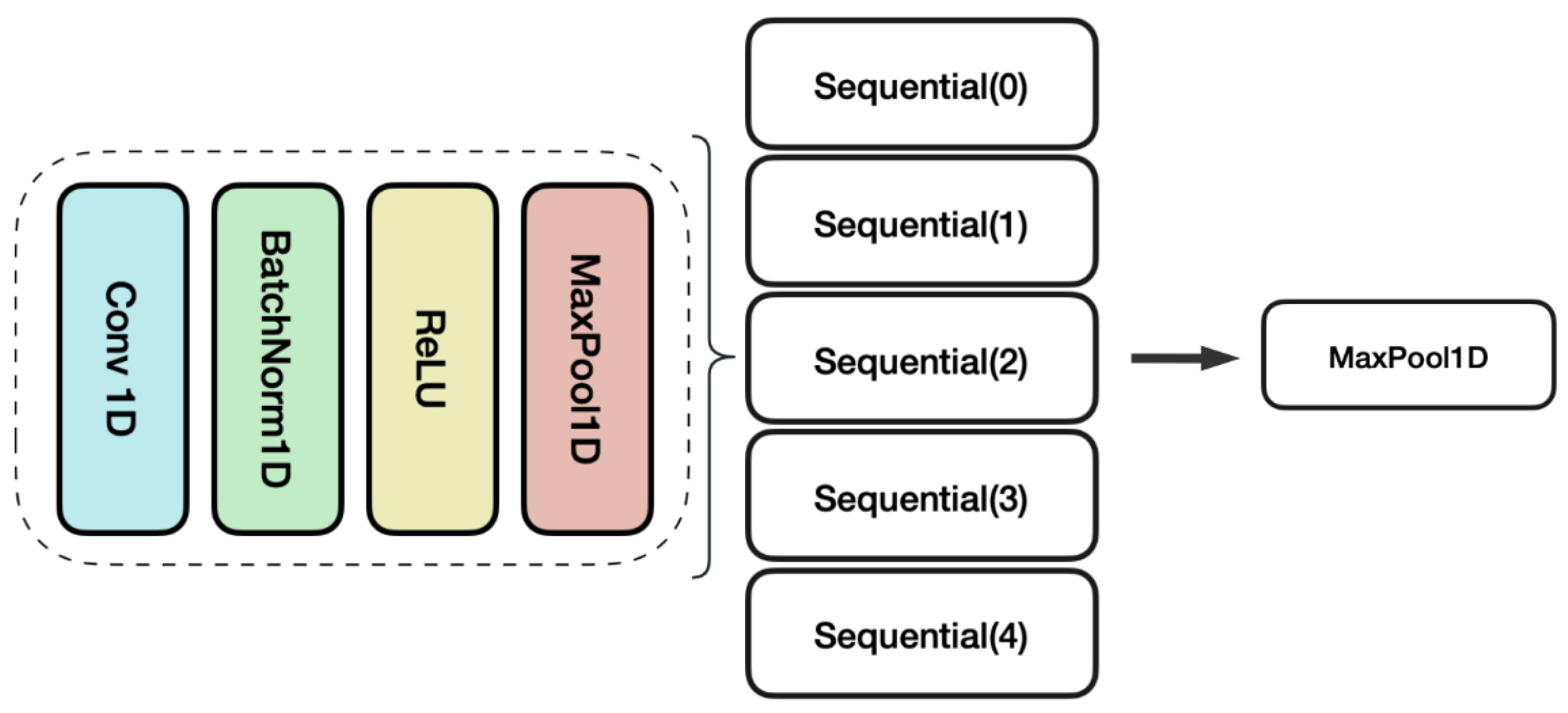
3.2. GAN Model Architecture
3.3. AMP Activity Predictor
3.4. AMP Multifunction Predictor
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AMPs | Antimicrobial peptides |
| FBGAN | Feedback Generative Adversarial Network |
| GAN | Generative Adversarial Network |
| VAE | Variational Autoencoder |
| MIC | Minimum Inhibitory Concentrations |
| APD3 | Antimicrobial Peptide Database |
| DRAMP | Data Repository of Antimicrobial Peptides |
| DNN | Deep Neural Network |
| CNN | Convolutional Neural Network |
| MSE | Mean Squared Error |
| GRAMPA | Giant Repository of AMP Activities |
| CAMPR4 | Collection of Anti-Microbial Peptides |
| SEN | Sensitivity |
| SPE | Specificity |
| ACC | Accuracy |
| PRE | Precision |
| MCC | Matthews Correlation Coefficient |
References
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic resistance is ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A.R.B. From antimicrobial to anticancer peptides: A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The value of antimicrobial peptides in the age of resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919. [Google Scholar]
- Wan, F.; Wong, F.; Collins, J.J.; Fuente-Nunez, C.D. Machine learning for antimicrobial peptide identification and design. Nat. Rev. Bioeng. 2024, 2, 392–407. [Google Scholar] [CrossRef]
- Das, P.; Sercu, T.; Wadhawan, K.; Padhi, I.; Gehrmann, S.; Cipcigan, F.; Chenthamarakshan, V.; Strobelt, H.; Santos, C.D.; Chen, P.; et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat. Biomed. Eng. 2021, 5, 613–623. [Google Scholar] [CrossRef]
- Tucs, A.; Tran, D.P.; Yumoto, A.; Ito, Y.; Uzawa, T.; Tsuda, K. Generating ampicillin-level antimicrobial peptides with activity-aware generative adversarial networks. ACS Omega 2020, 5, 22847–22851. [Google Scholar] [CrossRef]
- Oort, C.M.V.; Ferrell, J.B.; Remington, J.M.; Wshah, S.; Li, J. AMPGAN v2: Machine learning-guided design of antimicrobial peptides. J. Chem. Inf. Model. 2021, 61, 2198–2207. [Google Scholar] [CrossRef]
- Surana, S.; Arora, P.; Singh, D.; Sahasrabuddhe, D.; Valadi, J. PandoraGAN: Generating antiviral peptides using generative adversarial network. SN Comput. Sci. 2023, 4, 607. [Google Scholar] [CrossRef]
- Dean, S.N.; Alvarez, J.A.E.; Zabetakis, D.; Walper, S.A.; Malanoski, A.P. PepVAE: Variational autoencoder framework for antimicrobial peptide generation and activity prediction. Front. Microbiol. 2021, 12, 725727. [Google Scholar] [CrossRef] [PubMed]
- Lazzaro, B.P.; Zasloff, M.; Rolff, J. Antimicrobial peptides: Application informed by evolution. Science 2020, 368, eaau5480. [Google Scholar] [CrossRef]
- Gupta, A.; Zou, J. Feedback GAN (FBGAN) for DNA: A novel feedback-loop architecture for optimizing protein functions. arXiv 2018, arXiv:1804.01694. [Google Scholar]
- Witten, J.; Witten, Z. Deep learning regression model for antimicrobial peptide design. BioRxiv 2019. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pu, Y.; Tang, J.; Zou, Q.; Guo, F. DeepAVP: A dual-channel deep neural network for identifying variable-length antiviral peptides. IEEE J. Biomed. Health Inform. 2020, 24, 3012–3019. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Shrivastava, S.; Singh, S.K.; Kumar, A.; Saxena, S.; Singh, R.K. Deep-AFPPred: Identifying novel antifungal peptides using pre-trained embeddings from Seq2Vec with 1D-CNN-BiLSTM. Briefings Bioinform. 2022, 23, bbab422. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Shrivastava, S.; Singh, S.K.; Kumar, A.; Saxena, S. StaBle-ABPpred: A stacked ensemble predictor based on biLSTM and attention mechanism for accelerated discovery of antibacterial peptides. Briefings Bioinform. 2022, 23, bbab439. [Google Scholar] [CrossRef]
- Sharma, R.; Shrivastava, S.; Singh, S.K.; Kumar, A.; Saxena, S.; Singh, R.K. Deep-ABPpred: Identifying antibacterial peptides in protein sequences using bidirectional LSTM with word2vec. Briefings Bioinform. 2021, 22, bbab065. [Google Scholar] [CrossRef]
- Xu, J.; Li, F.; Li, C.; Guo, X.; Landersdorfer, C.; Shen, H.H.; Peleg, A.Y.; Li, J.; Imoto, S.; Yao, J.; et al. IAMP-CN: A deep-learning approach for identifying antimicrobial peptides and their functional activities. Briefings Bioinform. 2023, 24, bbad240. [Google Scholar] [CrossRef]
- Wang, X.-F.; Tang, J.-Y.; Liang, H.; Sun, J.; Dorje, S.; Peng, B.; Ji, X.-W.; Li, Z.; Zhang, X.-E.; Wang, D.-B. ProT-Diff: A Modularized and Efficient Approach to De Novo Generation of Antimicrobial Peptide Sequences through Integration of Protein Language Model and Diffusion Model. bioRxiv 2024. [Google Scholar] [CrossRef]
- Tucs, A.; Berenger, F.; Yumoto, A.; Tamura, R.; Uzawa, T.; Tsuda, K. Quantum Annealing Designs Nonhemolytic Antimicrobial Peptides in a Discrete Latent Space. ACS Med. Chem. Lett. 2023, 14, 577–582. [Google Scholar] [CrossRef]
- Cai, J.; Yan, J.; Un, C.; Wang, Y.; Campbell-Valois, F.-X.; Siu, S.W.I. BERT-AmPEP60: A BERT-Based Transfer Learning Approach to Predict the Minimum Inhibitory Concentrations of Antimicrobial Peptides for Escherichia coli and Staphylococcus aureus. J. Chem. Inf. Model. 2025, 65, 3186–3202. [Google Scholar] [CrossRef]
- Lee, H.T.; Lee, C.C.; Yang, J.R.; Lai, J.Z.; Chang, K.Y. A large-scale structural classification of antimicrobial peptides. Biomed. Res. Int. 2015, 2015, 475062. [Google Scholar] [CrossRef]
- Gawde, U.; Chakraborty, S.; Waghu, F.H.; Barai, R.S.; Khanderkar, A.; Indraguru, R.; Shirsat, T.; Idicula-Thomas, S. CAMPR4: A database of natural and synthetic antimicrobial peptides. Nucleic Acids Res. 2023, 51, D377–D383. [Google Scholar] [CrossRef]
- Thomas, S.; Karnik, S.; Barai, R.S.; Jayaraman, V.K.; Idicula-Thomas, S. CAMP: A useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2010, 38, D774–D780. [Google Scholar] [CrossRef]
- Xiao, X.; Wang, P.; Lin, W.Z.; Jia, J.H.; Chou, K.C. IAMP-2L: A two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013, 436, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Shao, Y.T.; Cheng, X.; Stamatovic, B. IAMP-CA2L: A new CNN-BiLSTM-SVM classifier based on cellular automata image for identifying antimicrobial peptides and their functional types. Briefings Bioinform. 2021, 22, bbab209. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.R.; Kuo, T.R.; Wu, L.C.; Lee, T.Y.; Horng, J.T. Characterization and identification of antimicrobial peptides with different functional activities. Briefings Bioinform. 2020, 21, 1098–1114. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Kang, X.; Dong, F.; Liu, Y.; Zhu, N.; Hu, Y.; Xu, H.; Lao, X.; Zheng, H. DRAMP 3.0: An enhanced comprehensive data repository of antimicrobial peptides. Nucleic Acids Res. 2022, 50, D488–D496. [Google Scholar] [CrossRef] [PubMed]
- Jhong, J.-H.; Chi, Y.-H.; Li, W.-C.; Lin, T.-H.; Huang, K.-Y.; Lee, T.-Y. dbAMP: An integrated resource for exploring antimicrobial peptides with functional activities and physicochemical properties on transcriptome and proteome data. Nucleic Acids Res. 2019, 47, D285–D297. [Google Scholar] [CrossRef]
- Gogoladze, G.; Grigolava, M.; Vishnepolsky, B.; Chubinidze, M.; Duroux, P.; Lefranc, M.-P.; Pirtskhalava, M. dbaasp: Database of antimicrobial activity and structure of peptides. FEMS Microbiol. Lett. 2014, 357, 63–68. [Google Scholar] [CrossRef]
- Zhao, X.; Wu, H.; Lu, H.; Li, G.; Huang, Q. LAMP: A Database Linking Antimicrobial Peptides. PLoS ONE 2013, 8, e66557. [Google Scholar] [CrossRef]
- Mehta, D.; Anand, P.; Kumar, V.; Joshi, A.; Mathur, D.; Singh, S.; Tuknait, A.; Chaudhary, K.; Gautam, S.K.; Gautam, A.; et al. ParaPep: A web resource for experimentally validated antiparasitic peptide sequences and their structures. Database 2014, 2014, bau051. [Google Scholar] [CrossRef]
- Hammami, R.; Hamida, J.B.; Vergoten, G.; Fliss, I. PhytAMP: A database dedicated to antimicrobial plant peptides. Nucleic Acids Res. 2008, 37, D963–D968. [Google Scholar] [CrossRef]
- Qureshi, A.; Thakur, N.; Tandon, H.; Kumar, M. AVPdb: A database of experimentally validated antiviral peptides targeting medically important viruses. Nucleic Acids Res. 2013, 42, D1147–D1153. [Google Scholar] [CrossRef]
- Tyagi, A.; Tuknait, A.; Anand, P.; Gupta, S.; Sharma, M.; Mathur, D.; Joshi, A.; Singh, S.; Gautam, A.; Raghava, G.P.S. CancerPPD: A database of anticancer peptides and proteins. Nucleic Acids Res. 2015, 43, D837–D843. [Google Scholar] [CrossRef] [PubMed]
- Théolier, J.; Fliss, I.; Jean, J.; Hammami, R. MilkAMP: A comprehensive database of antimicrobial peptides of dairy origin. Dairy Sci. Technol. 2014, 94, 181–193. [Google Scholar] [CrossRef]
- Wang, R.; Wang, T.; Zhuo, L.; Wei, J.; Fu, X.; Zou, Q.; Yao, X. Diff-AMP: Tailored designed antimicrobial peptide framework with all-in-one generation, identification, prediction and optimization. Briefings Bioinform. 2024, 25, bbae078. [Google Scholar] [CrossRef]
- Meher, P.K.; Sahu, T.K.; Saini, V.; Rao, A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physicochemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017, 7, 42362. [Google Scholar] [CrossRef] [PubMed]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef]
- Goodfellow, I.; Pouget-Abadie, J.; Mirza, M.; Xu, B.; Warde-Farley, D.; Ozair, S.; Courville, A.; Bengio, Y. Generative adversarial nets. In Advances in Neural Information Processing Systems; Curran Associates, Inc.: Red Hook, NY, USA, 2014; Volume 27. [Google Scholar]
- Arjovsky, M.; Chintala, S.; Bottou, L. Wasserstein generative adversarial networks. In International Conference on Machine Learning; PMLR: Sydney, Australia, 2017; pp. 214–223. [Google Scholar]
- Kawashima, S.; Ogata, H.; Kanehisa, M. AAIndex: Amino acid index database. Nucleic Acids Res. 1999, 27, 368–369. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Henikoff, J. Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA 1992, 89, 10915–10919. [Google Scholar] [CrossRef]
- Chou, K.C. Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology. Curr. Proteom. 2009, 6, 262–274. [Google Scholar] [CrossRef]
- Gull, S.; Shamim, N.; Minhas, F. AMAP: Hierarchical multi-label prediction of biologically active and antimicrobial peptides. Comput. Biol. Med. 2019, 107, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Qiaozhen, M.; Tang, J.; Guo, F. Multi-AMP: Detecting the antimicrobial peptides and their activities using multi-task learning. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021; pp. 710–713. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Core Functional Categories | Original Labels Included |
|---|---|
| Antibacterial | Antibacterial, anti-Gram-positive, Anti-Gram-negative, anti-TB, antibiofilm |
| Antifungal | Antifungal, anticandida |
| Anticancer | Anticancer, anti-mammalian-cell |
| Antiviral | Antiviral, anti-HIV |
| Antiparasitic | Antiparasitic, antimalarial, antiplasmodial, antiprotozoal |
| Functions | Classification Results | Training Dataset | ||
|---|---|---|---|---|
| Positive | Negative | Positive | Negative | |
| AMPs | 35,448 | 103,941 | 14,731 | 19,793 |
| Antibacterial | 17,288 | 18,160 | 8865 | 7865 |
| Antifungal | 13,431 | 22,017 | 3926 | 12,993 |
| Antiviral | 16,748 | 18,700 | 3525 | 13,384 |
| Anticancer | 21,664 | 13,784 | 3262 | 13,630 |
| Antiparasitic | 14,348 | 21,100 | 313 | 16,287 |
| Identification Task | |||||
|---|---|---|---|---|---|
| Model | SEN ↑ | SPE ↑ | ACC ↑ | PRE ↑ | MCC ↑ |
| CAMP-SVM [25] | 0.826 | 0.870 | 0.848 | 0.864 | 0.696 |
| CAMP-RF [25] | 0.876 | 0.926 | 0.901 | 0.922 | 0.803 |
| CAMP-ANN [25] | 0.852 | 0.854 | 0.853 | 0.853 | 0.705 |
| CAMP-DA [25] | 0.876 | 0.902 | 0.889 | 0.899 | 0.778 |
| diff-AMP [38] | 0.830 | 0.914 | 0.869 | 0.915 | 0.741 |
| iAMPpred [39] | 0.860 | 0.887 | 0.873 | 0.885 | 0.747 |
| AMPscannerv2 [40] | 0.924 | 0.928 | 0.926 | 0.928 | 0.852 |
| ours | 0.962 | 0.862 | 0.912 | 0.874 | 0.828 |
| Performance of Function-Specific AMP Classifiers | |||||
|---|---|---|---|---|---|
| Functions | Accuracy ↑ | Precision ↑ | F1-Score ↑ | MCC ↑ | AUC↑ |
| Antibacterial | 0.8722 | 0.8706 | 0.8750 | 0.7445 | 0.9445 |
| Antifungal | 0.8763 | 0.8745 | 0.8731 | 0.7429 | 0.9423 |
| Antiviral | 0.9403 | 0.9446 | 0.9408 | 0.8806 | 0.9758 |
| Anticancer | 0.8934 | 0.9014 | 0.8926 | 0.7865 | 0.9561 |
| Antiparasitic | 0.9256 | 0.9221 | 0.9261 | 0.8518 | 0.9683 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ying, F.; Go, W.; Li, Z.; Ouyang, C.; Phaphuangwittayakul, A.; Dhuny, R. Computational Design of Potentially Multifunctional Antimicrobial Peptide Candidates via a Hybrid Generative Model. Int. J. Mol. Sci. 2025, 26, 7387. https://doi.org/10.3390/ijms26157387
Ying F, Go W, Li Z, Ouyang C, Phaphuangwittayakul A, Dhuny R. Computational Design of Potentially Multifunctional Antimicrobial Peptide Candidates via a Hybrid Generative Model. International Journal of Molecular Sciences. 2025; 26(15):7387. https://doi.org/10.3390/ijms26157387
Chicago/Turabian StyleYing, Fangli, Wilten Go, Zilong Li, Chaoqian Ouyang, Aniwat Phaphuangwittayakul, and Riyad Dhuny. 2025. "Computational Design of Potentially Multifunctional Antimicrobial Peptide Candidates via a Hybrid Generative Model" International Journal of Molecular Sciences 26, no. 15: 7387. https://doi.org/10.3390/ijms26157387
APA StyleYing, F., Go, W., Li, Z., Ouyang, C., Phaphuangwittayakul, A., & Dhuny, R. (2025). Computational Design of Potentially Multifunctional Antimicrobial Peptide Candidates via a Hybrid Generative Model. International Journal of Molecular Sciences, 26(15), 7387. https://doi.org/10.3390/ijms26157387