
Single-Cell Dissection of the Serrated Pathway: Cellular Heterogeneity and Genetic Causality in Colorectal Cancer


Abstract
1. Introduction
2. Results
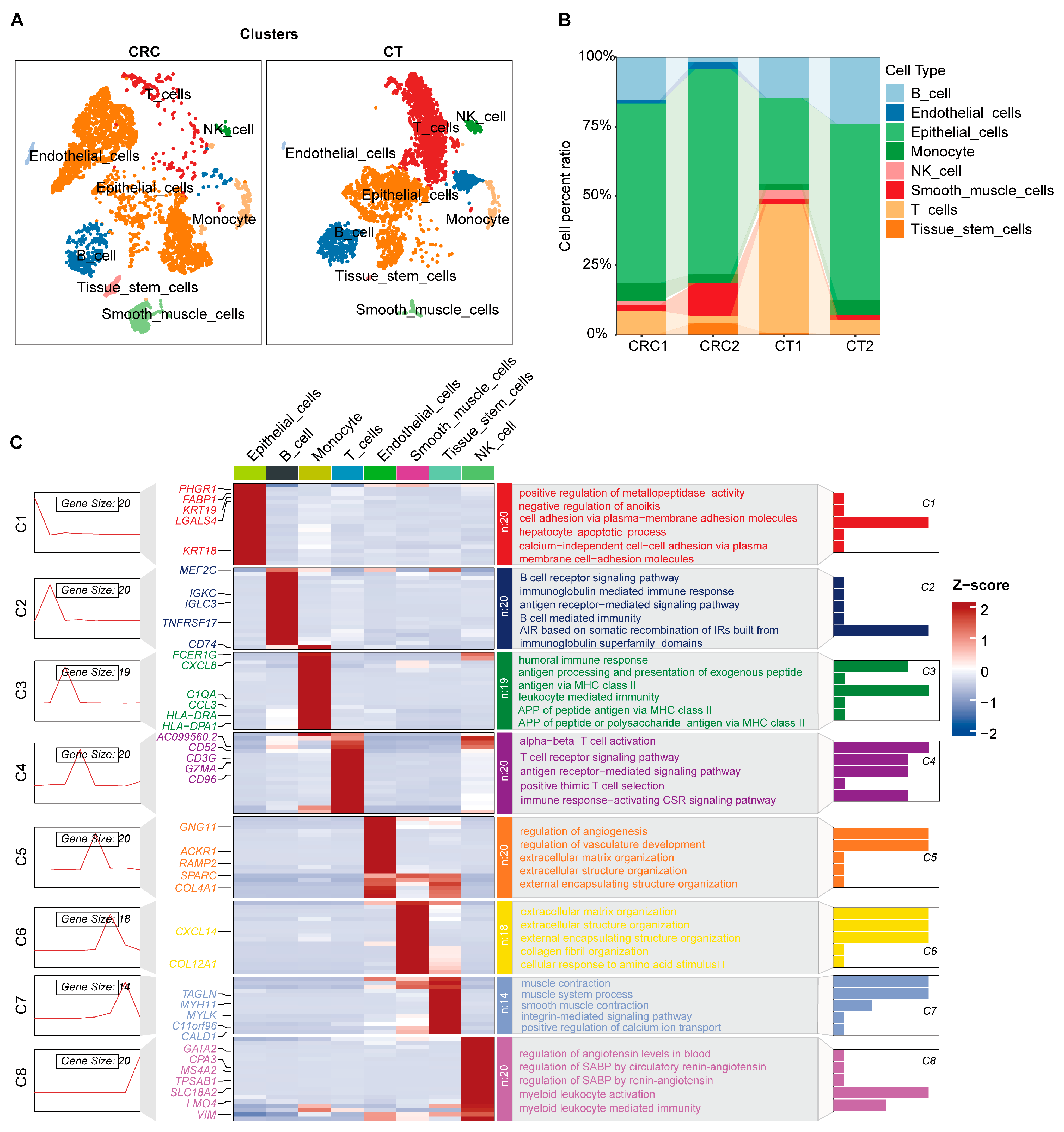
2.1. Single-Cell Atlas and Cellular Functional Exploration in CRC
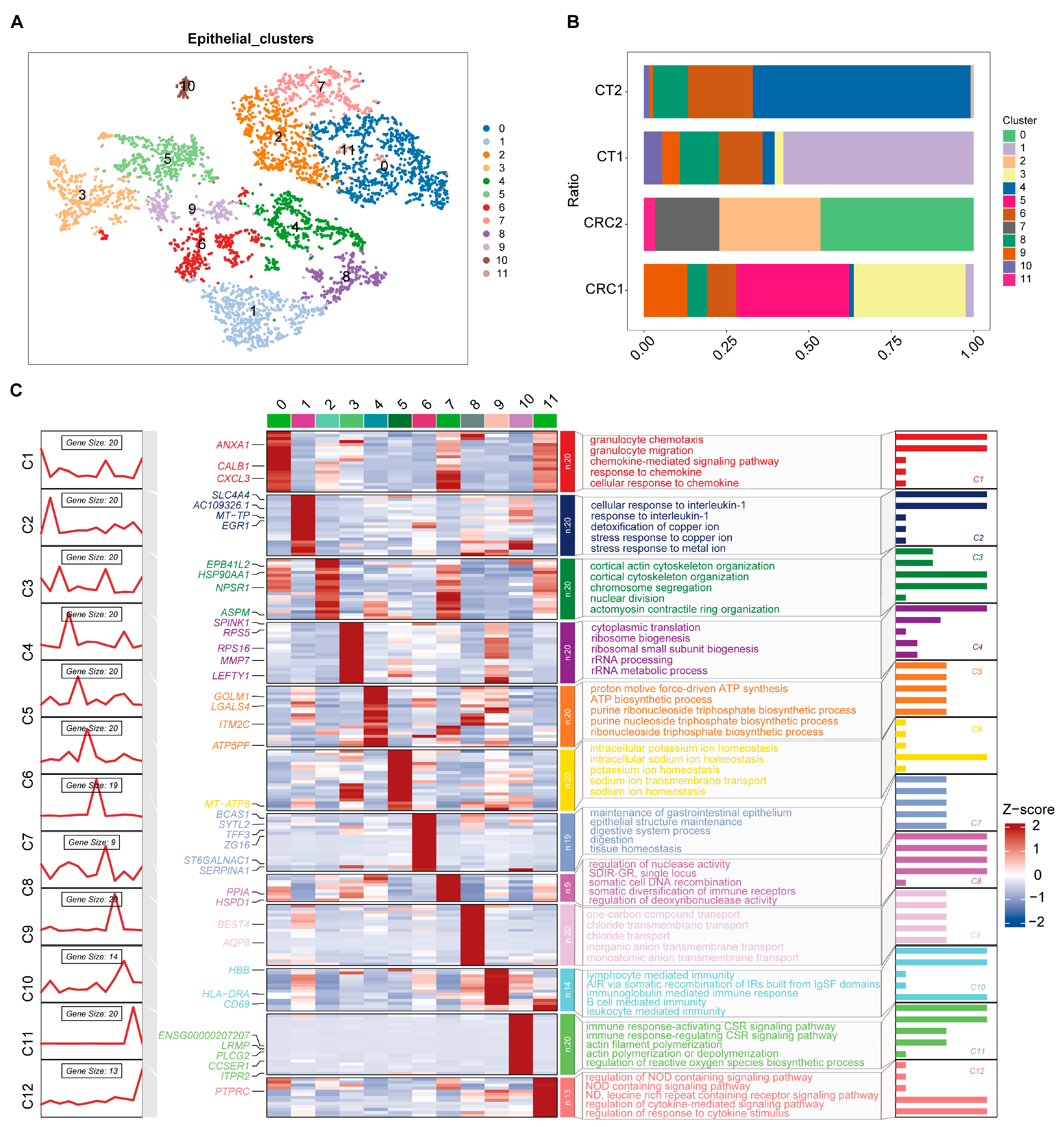
2.2. Characteristics of CRC Epithelial Cell Subgroups
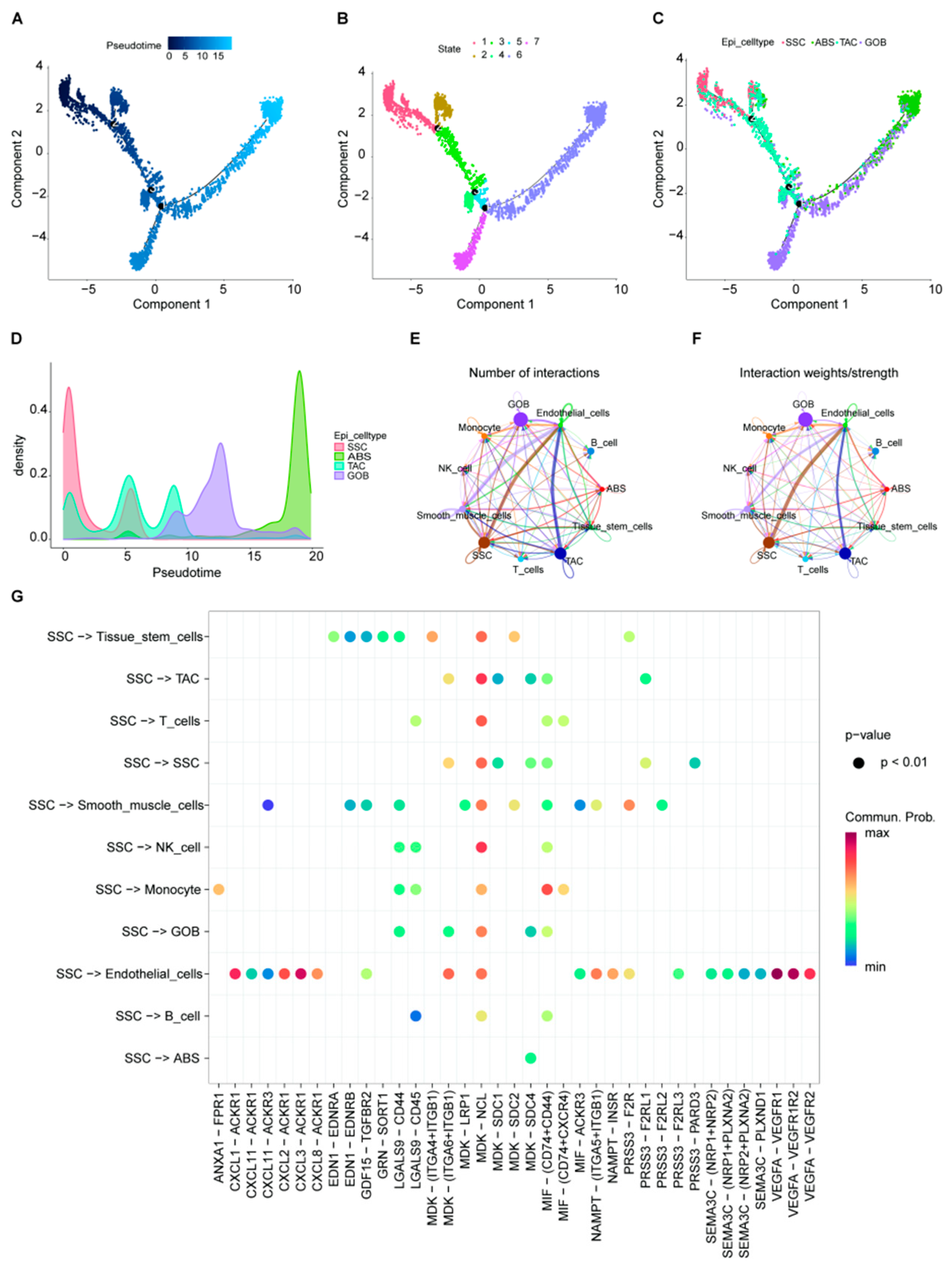
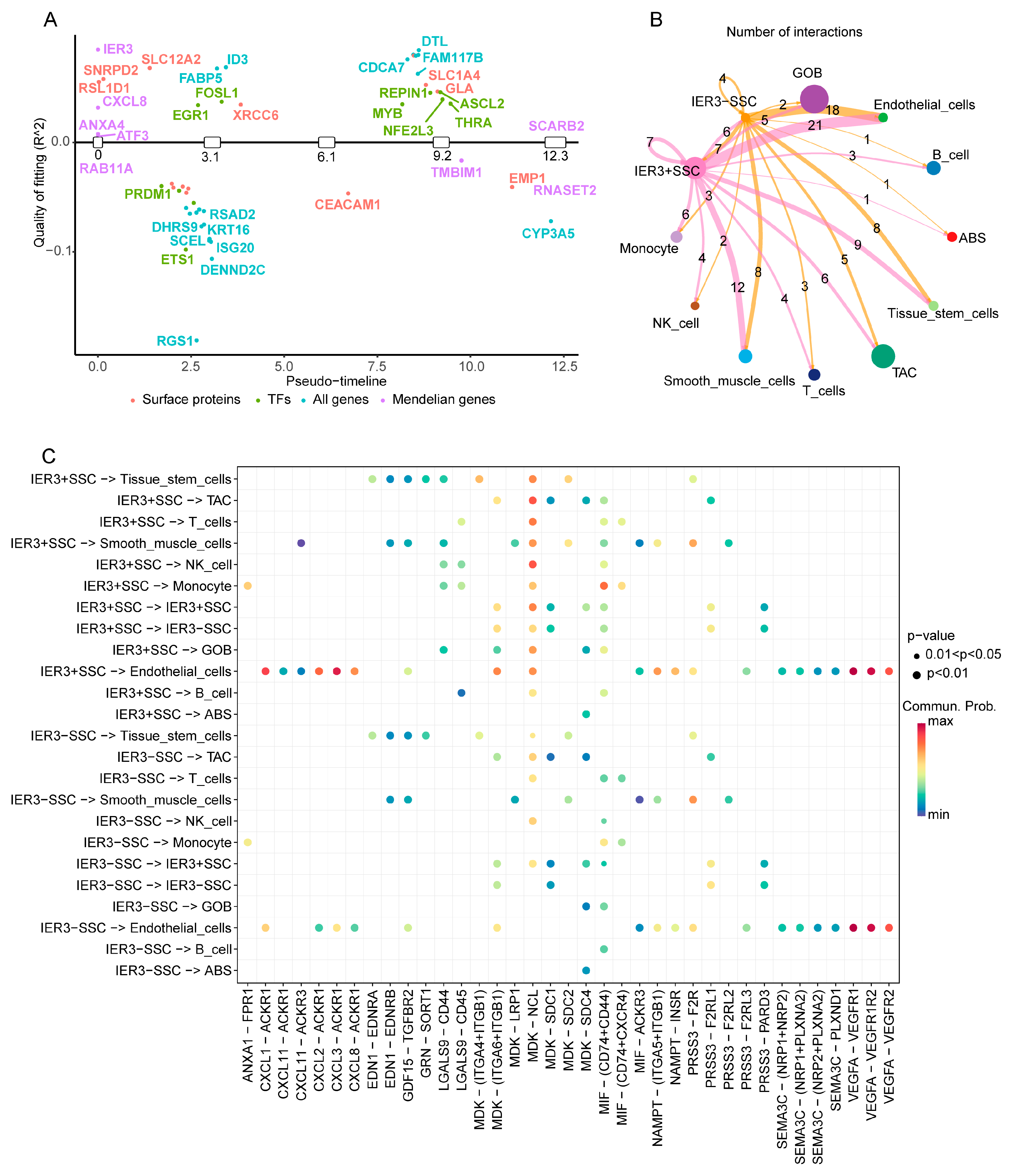
2.3. Epithelial Cell Trajectory Evolution and Interactions with Other Cell Types
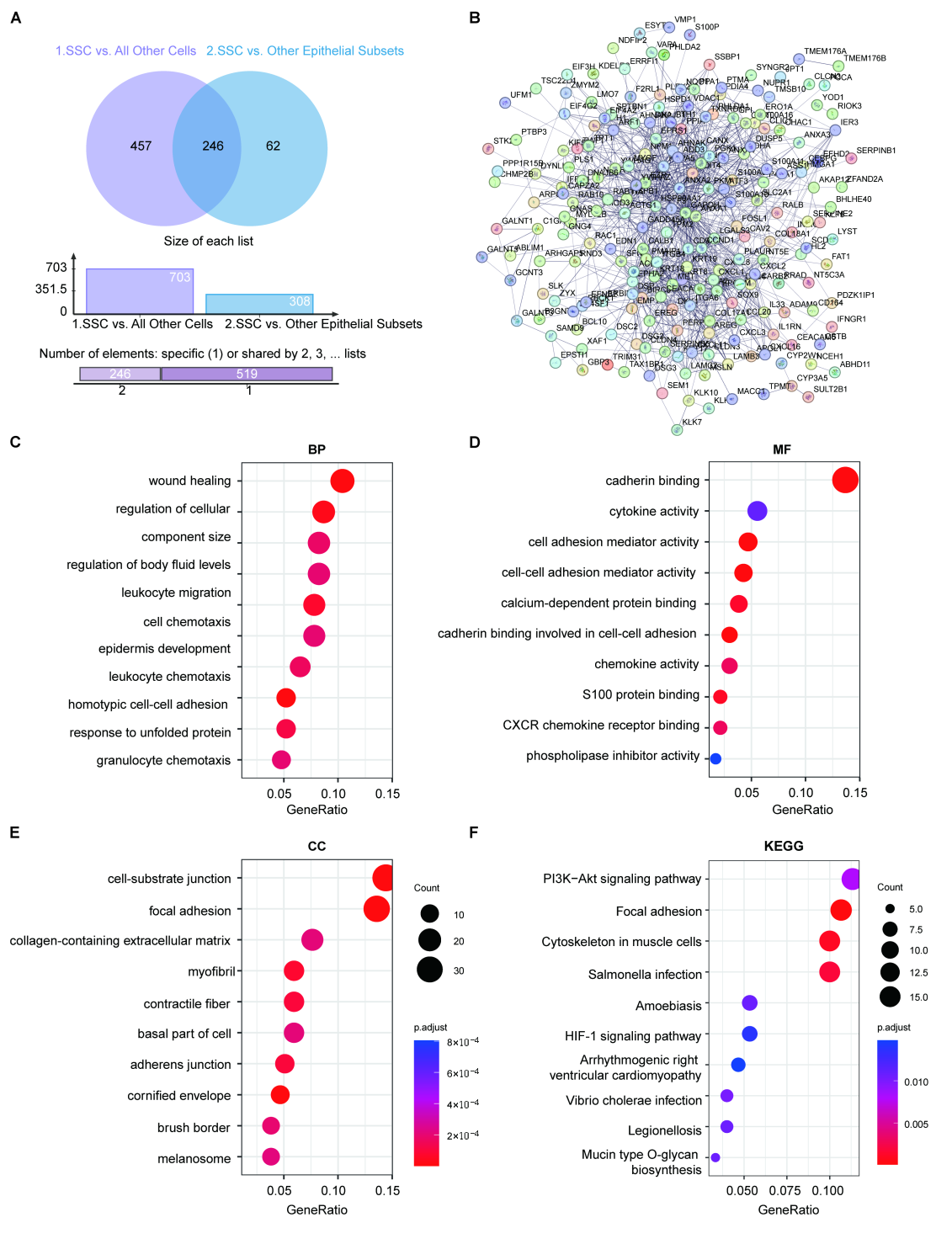
2.4. Gene Characterization in SSCs
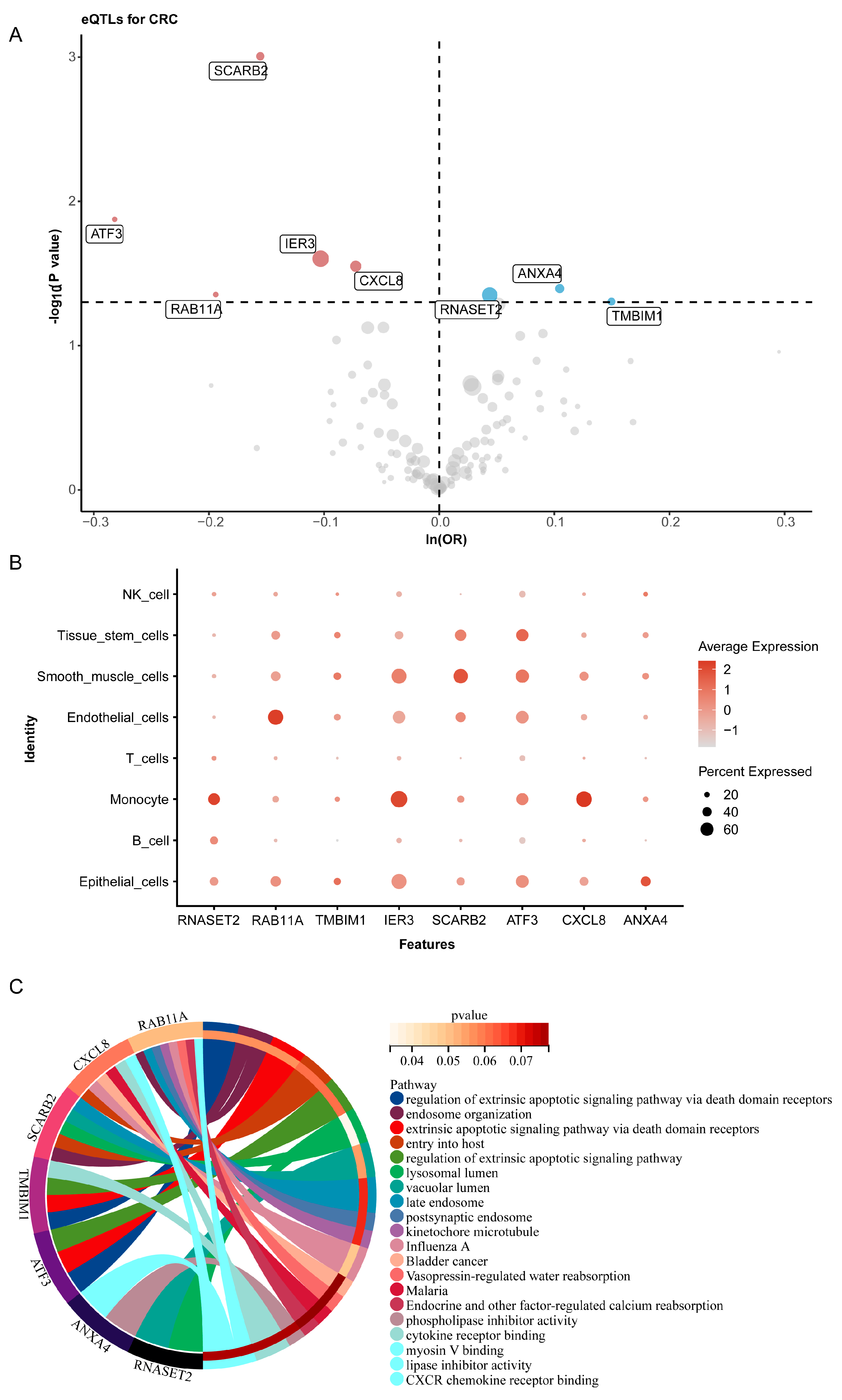
2.5. Shared Differential Gene eQTLs and Colorectal Cancer Risk
2.6. Switch Genes and Their Impact on Colorectal Cancer Epithelial Cells
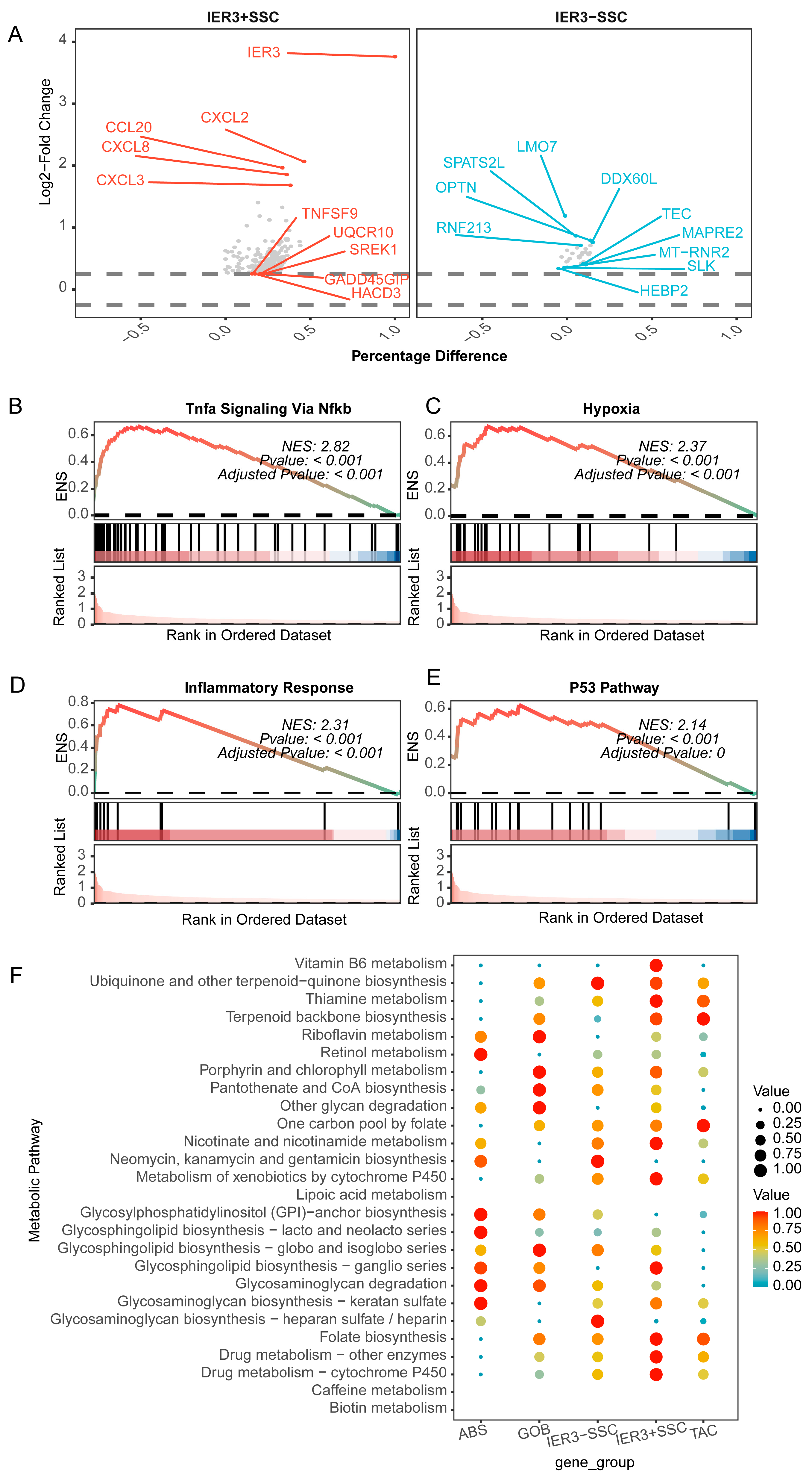
2.7. Impact of IER3 on CRC Epithelial Cells
3. Discussion
4. Materials and Methods
4.1. Single-Cell RNA Sequencing Data Retrieval and Processing for CRC
4.2. Identification and Functional Enrichment Analysis of CRC-Related Serrated Characteristic Cells
4.3. Expression and Functional Analysis of Core Genes at the Single-Cell Level in CRC
4.4. MR Analysis of SSCs Marker Genes and CRC Expression Quantitative Trait Loci (eQTLs)
4.5. GeneSwitches Analysis of Core Genes
4.6. Impact of Key Switch Genes on CRC Cell Interactions and Biological Processes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Zheng, R.; Zeng, H.; Wang, S.; Sun, K.; Chen, R.; Li, L.; Wei, W.; He, J. Cancer incidence and mortality in China, 2022. J. Natl. Cancer Cent. 2024, 4, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Crockett, S.D.; Nagtegaal, I.D. Terminology, Molecular Features, Epidemiology, and Management of Serrated Colorectal Neoplasia. Gastroenterology 2019, 157, 949–966.e4. [Google Scholar] [CrossRef] [PubMed]
- Ijspeert, J.E.G.; Vermeulen, L.; Meijer, G.A.; Dekker, E. Serrated neoplasia—Role in colorectal carcinogenesis and clinical implications. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Doherty, A.R.; Raju, M.; Liu, L.; Lei, N.Y.; Amsden, L.B.; Lee, J.K.; Levin, T.R.; Corley, D.A.; Herrinton, L.J. Risk stratification for colorectal cancer in individuals with subtypes of serrated polyps. Gut 2021, 71, 324301. [Google Scholar] [CrossRef] [PubMed]
- East, J.E.; Vieth, M.; Rex, D.K. Serrated lesions in colorectal cancer screening: Detection, resection, pathology and surveillance. Gut 2015, 64, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, T.; Okamoto, K.; Fujimoto, S.; Bando, M.; Wada, H.; Miyamoto, H.; Sato, Y.; Muguruma, N.; Horimoto, K.; Takayama, T. Lansoprazole inhibits the development of sessile serrated lesions by inducing G1 arrest via Skp2/p27 signaling pathway. J. Gastroenterol. 2024, 59, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Park, J.H.; Park, C.H. Serrated Polyps and the Risk of Metachronous Colorectal Advanced Neoplasia: A Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2022, 20, 31–43.e1. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.; Yamada, A.; Ikegami, S.; Haider, H.; Komaki, Y.; Komaki, F.; Micic, D.; Sakuraba, A. Risk of Colorectal Cancer in Serrated Polyposis Syndrome: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2022, 20, 622–630.e7. [Google Scholar] [CrossRef] [PubMed]
- Aiderus, A.; Barker, N.; Tergaonkar, V. Serrated colorectal cancer: Preclinical models and molecular pathways. Trends Cancer 2024, 10, 76–91. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, M.; Mohamed, M.F.H.; Abdalla, A.O.; Jaber, F.; Baliss, M.; Ahmed, K.; Eckmann, J.; Bilal, M.; Shaukat, A. Adenomas and Sessile Serrated Lesions in 45- to 49-Year-Old Individuals Undergoing Colonoscopy: A Systematic Review and Meta-Analysis. Am. J. Gastroenterol. 2024, 119, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Kambara, T.; Simms, L.A.; Whitehall, V.L.J.; Spring, K.J.; Wynter, C.V.A.; Walsh, M.D.; Barker, M.A.; Arnold, S.; McGivern, A.; Matsubara, N.; et al. BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut 2004, 53, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Fennell, L.; Kane, A.; Liu, C.; McKeone, D.; Hartel, G.; Su, C.; Bond, C.; Bettington, M.; Leggett, B.; Whitehall, V. Braf mutation induces rapid neoplastic transformation in the aged and aberrantly methylated intestinal epithelium. Gut 2022, 71, 1127–1140. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.D.; Xu, G.S.; Hu, X.L.; Li, W.Q.; Yao, N.; Han, F.Z.; Zhang, Y.; Qu, J. The histologic features, molecular features, detection and management of serrated polyps: A review. Front. Oncol. 2024, 14, 1356250. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, F.; Sato, Y.; Okamoto, K.; Fujino, Y.; Mitsui, Y.; Kagemoto, K.; Kawaguchi, T.; Miyamoto, H.; Muguruma, N.; Sonoda, T.; et al. Colorectal carcinoma occurring via the adenoma-carcinoma pathway in patients with serrated polyposis syndrome. J. Gastroenterol. 2022, 57, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Utsumi, T.; Yamada, Y.; Diaz-Meco, M.T.; Moscat, J.; Nakanishi, Y. Sessile serrated lesions with dysplasia: Is it possible to nip them in the bud? J. Gastroenterol. 2023, 58, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Gagrat, Z.D.; Krockenberger, M.; Bhattacharya, A.; Gagrat, B.Z.; Leduc, C.M.; Matter, M.B.; Fourrier, K.D.; Mahoney, D.W.; Edwards, V.D.; Lidgard, G.P.; et al. Next-generation Multi-target Stool DNA Panel Accurately Detects Colorectal Cancer and Advanced Precancerous Lesions. Cancer Prev. Res. (Phila.) 2024, 17, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Aniwan, S.; Chiu, H.M.; Laohavichitra, K.; Chirapongsathorn, S.; Yamamura, T.; Kuo, C.Y.; Yoshida, N.; Ang, T.L.; Takezawa, T.; et al. Linked-Color Imaging Detects More Colorectal Adenoma and Serrated Lesions: An International Randomized Controlled Trial. Clin. Gastroenterol. Hepatol. 2023, 21, 1493–1502.e4. [Google Scholar] [CrossRef] [PubMed]
- Kanth, P.; Yu, Z.; Keener, M.B.; Koptiuch, C.; Kohlmann, W.K.; Neklason, D.W.; Westover, M.; Curtin, K. Cancer Risk in Patients with and Relatives of Serrated Polyposis Syndrome and Sporadic Sessile Serrated Lesions. Am. J. Gastroenterol. 2022, 117, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Rouphael, C.; El Halabi, J.; Bena, J.; McMichael, J.; Burke, C.A. Impact of Clinical and Endoscopic Features on the Development of Metachronous Colorectal Advanced Serrated Lesions. Clin. Gastroenterol. Hepatol. 2024, 22, 1117–1126.e6. [Google Scholar] [CrossRef] [PubMed]
- Djinbachian, R.; von Renteln, D. Virtual scale endoscope-assisted wide-field resection of large sessile serrated lesions. Endoscopy 2023, 55, E314–E315. [Google Scholar] [CrossRef] [PubMed]
- Sumida, T.S.; Hafler, D.A. Population genetics meets single-cell sequencing. Science 2022, 376, 134–135. [Google Scholar] [CrossRef] [PubMed]
- Hagemann-Jensen, M.; Ziegenhain, C.; Sandberg, R. Scalable single-cell RNA sequencing from full transcripts with Smart-seq3xpress. Nat. Biotechnol. 2022, 40, 1452–1457. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Tang, R.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Applications of single-cell sequencing in cancer research: Progress and perspectives. J. Hematol. Oncol. 2021, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Brink, S.C.v.D.; Sage, F.; Vértesy, Á.; Spanjaard, B.; Peterson-Maduro, J.; Baron, C.S.; Robin, C.; van Oudenaarden, A. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods 2017, 14, 935–936. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Ebrahim, S. ‘Mendelian randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Emdin, C.A.; Khera, A.V.; Kathiresan, S. Mendelian Randomization. JAMA 2017, 318, 1925–1926. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Harbord, R.M.; Sterne, J.A.; Timpson, N.; Smith, G.D. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 2008, 27, 1133–1163. [Google Scholar] [CrossRef] [PubMed]
- Joshi, B.P.; Dai, Z.; Gao, Z.; Lee, J.H.; Ghimire, N.; Chen, J.; Prabhu, A.; Wamsteker, E.J.; Kwon, R.S.; Elta, G.H.; et al. Detection of Sessile Serrated Adenomas in the Proximal Colon Using Wide-Field Fluorescence Endoscopy. Gastroenterology 2017, 152, 1002–1013.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Lauwers, G.Y.; Choi, W.-T. Increased Risk of Non-conventional and Invisible Dysplasias in Patients with Primary Sclerosing Cholangitis and Inflammatory Bowel Disease. J. Crohn’s Colitis 2022, 16, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Gómez, M.; Costas-Ríos, L.; Garcia-Prieto, C.A.; Álvarez-Rodríguez, L.; Bujanda, L.; Barrero, M.; Castells, A.; Balaguer, F.; Jover, R.; Esteller, M.; et al. Serum DNA methylome of the colorectal cancer serrated pathway enables non-invasive detection. Mol. Oncol. 2023, 18, 2696–2713. [Google Scholar] [CrossRef] [PubMed]
- Bahceci, D.; Alpert, L.; Storozuk, T.; Liao, X.; Yozu, M.; Westerhoff, M.; Kővári, B.P.; Lauwers, G.Y.; Choi, W.T. Dysplasia Detected in Patients with Serrated Epithelial Change Is Frequently Associated with an Invisible or Flat Endoscopic Appearance, Nonconventional Dysplastic Features, and Advanced Neoplasia. Am. J. Surg. Pathol. 2024, 48, 1326–1334. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.N.; Grasso, D.; Lopez-Millan, M.B.; Hamidi, T.; Loncle, C.; Tomasini, R.; Lomberk, G.; Porteu, F.; Urrutia, R.; Iovanna, J.L. IER3 supports KRASG12D-dependent pancreatic cancer development by sustaining ERK1/2 phosphorylation. J. Clin. Investig. 2014, 124, 4709–4722. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Lee, K.; Kim, Y.-H.; Oh, H.K.; Maeng, Y.-I.; Kim, T.-H.; Suh, D.-S.; Bae, J. Scaffold protein FHL2 facilitates MDM2-mediated degradation of IER3 to regulate proliferation of cervical cancer cells. Oncogene 2016, 35, 5106–5118. [Google Scholar] [CrossRef] [PubMed]
- Mahale, S.; Setia, M.; Prajapati, B.; Subhash, S.; Yadav, M.P.; Kosalai, S.T.; Deshpande, A.; Kuchlyan, J.; Di Marco, M.; Westerlund, F.; et al. HnRNPK maintains single strand RNA through controlling double-strand RNA in mammalian cells. Nat. Commun. 2022, 13, 4865. [Google Scholar] [CrossRef] [PubMed]
- Ria, R.; Todoerti, K.; Berardi, S.; Coluccia, A.M.; De Luisi, A.; Mattioli, M.; Ronchetti, D.; Morabito, F.; Guarini, A.; Petrucci, M.T.; et al. Gene expression profiling of bone marrow endothelial cells in patients with multiple myeloma. Clin. Cancer Res. 2009, 15, 5369–5378. [Google Scholar] [CrossRef] [PubMed]
- Vyas, K.; Patel, M.M. Insights on drug and gene delivery systems in liver fibrosis. Asian J. Pharm. Sci. 2023, 18, 100779. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Lu, Y.; Wang, K.; Bai, L.; Shi, G.; Huang, Y.; Li, Y. Transplantation of bone marrow mesenchymal stem cells improves cognitive deficits and alleviates neuropathology in animal models of Alzheimer’s disease: A meta-analytic review on potential mechanisms. Transl. Neurodegener. 2020, 9, 20. [Google Scholar] [CrossRef] [PubMed]
- Crockett, S.D. Sessile Serrated Polyps and Colorectal Cancer. JAMA 2017, 317, 975–976. [Google Scholar] [CrossRef] [PubMed]
- van Toledo, D.E.F.W.M.; Ijspeert, J.E.G.; Bossuyt, P.M.M.; Bleijenberg, A.G.C.; van Leerdam, M.E.; van der Vlugt, M.; Lansdorp-Vogelaar, I.; Spaander, M.C.W.; Dekker, E. Serrated polyp detection and risk of interval post-colonoscopy colorectal cancer: A population-based study. Lancet Gastroenterol. Hepatol. 2022, 7, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Egoavil, C.; Juárez, M.; Guarinos, C.; Rodríguez-Soler, M.; Hernández-Illán, E.; Alenda, C.; Payá, A.; Castillejo, A.; Serradesanferm, A.; Bujanda, L.; et al. Increased Risk of Colorectal Cancer in Patients with Multiple Serrated Polyps and Their First-Degree Relatives. Gastroenterology 2017, 153, 106–112.e2. [Google Scholar] [CrossRef] [PubMed]
- Muta, Y.; Linares, J.F.; Martinez-OrdoñeZ, A.; Duran, A.; Cid-Diaz, T.; Kinoshita, H.; Zhang, X.; Han, Q.; Nakanishi, Y.; Nakanishi, N.; et al. Enhanced SREBP2-driven cholesterol biosynthesis by PKCλ/ι deficiency in intestinal epithelial cells promotes aggressive serrated tumorigenesis. Nat. Commun. 2023, 14, 8075. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Miyanishi, K.; Hayashi, T.; Sato, Y.; Niitsu, Y. Colorectal cancer: Genetics of development and metastasis. J. Gastroenterol. 2006, 41, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Heino, S.; Fang, S.; Lähde, M.; Högström, J.; Nassiri, S.; Campbell, A.; Flanagan, D.; Raven, A.; Hodder, M.; Nasreddin, N.; et al. Lef1 restricts ectopic crypt formation and tumor cell growth in intestinal adenomas. Sci. Adv. 2021, 7, eabj0512. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Okamoto, K.; Tanahashi, T.; Fujimoto, S.; Nakagawa, T.; Bando, M.; Ma, B.; Kawaguchi, T.; Fujino, Y.; Mitsui, Y.; et al. S100P Expression via DNA Hypomethylation Promotes Cell Growth in the Sessile Serrated Adenoma/Polyp-Cancer Sequence. Digestion 2021, 102, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, D.M.; Chen, B.; Iticovici, M.; Thaker, A.I.; Dai, N.; VanDussen, K.L.; Shaikh, N.; Lim, C.K.; Guillemin, G.J.; Tarr, P.I.; et al. Epithelial Indoleamine 2,3-Dioxygenase 1 Modulates Aryl Hydrocarbon Receptor and Notch Signaling to Increase Differentiation of Secretory Cells and Alter Mucus-Associated Microbiota. Gastroenterology 2019, 157, 1093–1108.e11. [Google Scholar] [CrossRef] [PubMed]
- Bohere, J.; Eldridge-Thomas, B.L.; Kolahgar, G. Vinculin recruitment to α-catenin halts the differentiation and maturation of enterocyte progenitors to maintain homeostasis of the Drosophila intestine. Elife 2022, 11, 72836. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.C.; Rex, D.K.; Mackenzie, T.A.; Hisey, W.; Robinson, C.M.; Butterly, L.F. Higher Serrated Polyp Detection Rates Are Associated with Lower Risk of Postcolonoscopy Colorectal Cancer: Data from the New Hampshire Colonoscopy Registry. Am. J. Gastroenterol. 2023, 118, 1927–1930. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.-J.; Lu, X.-F.; Chen, H.; Wang, X.-Y.; Cheng, W.; Zhang, Q.-W.; Chen, J.-N.; Wang, X.-Y.; Jin, J.-Z.; Yan, F.-R.; et al. Single-cell Transcriptomics Reveals Early Molecular and Immune Alterations Underlying the Serrated Neoplasia Pathway Toward Colorectal Cancer. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 393–424. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.W.; Bledsoe, J.R.; Morales-Oyarvide, V.; Huynh, T.G.; Mino-Kenudson, M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod. Pathol. 2016, 29, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Rau, T.T.; Atreya, R.; Aust, D.; Baretton, G.; Eck, M.; Erlenbach-Wünsch, K.; Hartmann, A.; Lugli, A.; Stöhr, R.; Vieth, M.; et al. Inflammatory response in serrated precursor lesions of the colon classified according to WHO entities, clinical parameters and phenotype-genotype correlation. J. Pathol. Clin. Res. 2016, 2, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Arlt, A.; Schäfer, H. Role of the immediate early response 3 (IER3) gene in cellular stress response, inflammation and tumorigenesis. Eur. J. Cell Biol. 2011, 90, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ma, J.; Zhou, D.; Zhou, J.; Hu, B.; Ma, X.; Tang, J.; Bai, Y.; Chen, H.; Jing, Y. Single-Nucleus Transcriptome Profiling of Locally Advanced Cervical Squamous Cell Cancer Identifies Neural-Like Progenitor Program Associated with the Efficacy of Radiotherapy. Adv. Sci. 2023, 10, e2300348. [Google Scholar] [CrossRef] [PubMed]
- Jordan, K.R.; Sikora, M.J.; Slansky, J.E.; Minic, A.; Richer, J.K.; Moroney, M.R.; Hu, J.; Wolsky, R.J.; Watson, Z.L.; Yamamoto, T.M.; et al. The Capacity of the Ovarian Cancer Tumor Microenvironment to Integrate Inflammation Signaling Conveys a Shorter Disease-free Interval. Clin. Cancer Res. 2020, 26, 6362–6373. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, C.; Ohuchida, K.; Katayama, N.; Yamada, Y.; Nakamura, S.; Okuda, S.; Otsubo, Y.; Iwamoto, C.; Torata, N.; Horioka, K.; et al. Tumor-infiltrating monocytic myeloid-derived suppressor cells contribute to the development of an immunosuppressive tumor microenvironment in gastric cancer. Gastric Cancer 2024, 27, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, Z.; Chen, S.; Tan, L.; He, L.; Yuan, D.; Zheng, L.; Zhong, J.H.; Li, A.; Zhang, H.; et al. Immediate early response 3 gene promotes aggressive progression and autophagy of AML by negatively regulating AKT/mTOR. Transl. Oncol. 2023, 35, 101711. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Song, L.; Lin, Y.; Nowialis, P.; Gao, Q.; Li, T.; Li, B.; Mao, X.; Song, Q.; Xing, C.; et al. ROS-mediated SRMS activation confers platinum resistance in ovarian cancer. Oncogene 2023, 42, 1672–1684. [Google Scholar] [CrossRef] [PubMed]
- Clough, E.; Barrett, T. The Gene Expression Omnibus Database. Methods Mol. Biol. 2016, 1418, 93–110. [Google Scholar] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, D.; Li, Y.; Qi, L.; Si, W.; Bo, Y.; Chen, X.; Ye, Z.; Fan, H.; Liu, B.; et al. Zhang, Spatiotemporal single-cell analysis decodes cellular dynamics underlying different responses to immunotherapy in colorectal cancer. Cancer Cell 2024, 42, 1268–1285.e7. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wang, Y.; Yang, W.; Wang, C.; Guo, T.; Yang, J.; Shao, Z.; Cai, G.; Cai, S.; Zhang, L.; et al. Molecular Profiling Provides Clinical Insights into Targeted and Immunotherapies as Well as Colorectal Cancer Prognosis. Gastroenterology 2023, 165, 414–428.e7. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterprofiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S.; Hattori, M.; Aoki-Kinoshita, K.F.; Itoh, M.; Kawashima, S.; Katayama, T.; Araki, M.; Hirakawa, M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006, 34, D354–D357. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Cacchiarelli, D.; Grimsby, J.; Pokharel, P.; Li, S.; Morse, M.; Lennon, N.J.; Livak, K.J.; Mikkelsen, T.S.; Rinn, J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014, 32, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Hill, A.; Packer, J.; Lin, D.; Ma, Y.-A.; Trapnell, C. Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 2017, 14, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018, 7, e34408. [Google Scholar] [CrossRef] [PubMed]
- Võsa, U.; Claringbould, A.; Westra, H.-J.; Bonder, M.J.; Deelen, P.; Zeng, B.; Kirsten, H.; Saha, A.; Kreuzhuber, R.; Yazar, S.; et al. Large-scale cis- and trans-eQTL analyses identify thousands of genetic loci and polygenic scores that regulate blood gene expression. Nat. Genet. 2021, 53, 1300–1310. [Google Scholar] [CrossRef] [PubMed]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Smith, G.D.; Davies, N.M.; Dudbridge, F.; Gill, D.; Glymour, M.M.; Hartwig, F.P.; Kutalik, Z.; Holmes, M.V.; Minelli, C.; et al. Guidelines for performing Mendelian randomization investigations: Update for summer 2023. Wellcome Open Res. 2019, 4, 186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Lian, P.; Liu, H.; Wang, Y.; Zhou, M.; Feng, Z. Causal Associations between Gut Microbiota and Different Types of Dyslipidemia: A Two-Sample Mendelian Randomization Study. Nutrients 2023, 15, 4445. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Gao, X.; Pan, X.-F.; Zhou, T.; Zhu, C.; Li, F.; Fan, J.-G.; Targher, G.; Zhao, J. The hepato-ovarian axis: Genetic evidence for a causal association between non-alcoholic fatty liver disease and polycystic ovary syndrome. BMC Med. 2023, 21, 62. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Wang, Y.; Fu, Z.; Liao, J.; Liu, H.; Zhou, M. Exploring the Causal Effects of Mineral Metabolism Disorders on Telomere and Mitochondrial DNA: A Bidirectional Two-Sample Mendelian Randomization Analysis. Nutrients 2024, 16, 1417. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Butterworth, A.; Thompson, S.G. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet. Epidemiol. 2013, 37, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Deng, Y.; Liu, H.; Fu, Z.; Wang, Y.; Zhou, M.; Feng, Z. Causal Relationship between Meat Intake and Biological Aging: Evidence from Mendelian Randomization Analysis. Nutrients 2024, 16, 2433. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Shi, M.; Zhong, Z.; Hu, H.; Sang, H.; Zhou, M.; Feng, Z. Causal Relationship between Aging and Anorexia Nervosa: A White-Matter-Microstructure-Mediated Mendelian Randomization Analysis. Biomedicines 2024, 12, 1874. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Bowden, J.; Fall, T.; Ingelsson, E.; Thompson, S.G. Sensitivity Analyses for Robust Causal Inference from Mendelian Randomization Analyses with Multiple Genetic Variants. Epidemiology 2017, 28, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Cao, E.Y.; Ouyang, J.F.; Rackham, O.J.L. GeneSwitches: Ordering gene expression and functional events in single-cell experiments. Bioinformatics 2020, 36, 3273–3275. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | Time | CI | p-Values | FDR | R2 | Estimates | Direction |
|---|---|---|---|---|---|---|---|
| IER3 | 0.00 | 1.75 | 3.89 × 10−10 | 4.85 × 10−8 | 8.48 × 10−2 | 0.32 | up |
| CXCL8 | 0.00 | 1.93 | 3.31 × 10−9 | 2.66 × 10−7 | 3.18 × 10−2 | 0.17 | up |
| ANXA4 | 0.00 | 5.92 | 2.62 × 10−3 | 2.00 × 10−2 | 7.71 × 10−3 | 0.08 | up |
| ATF3 | 0.00 | 19.81 | 3.65 × 10−2 | 1.43 × 10−1 | 5.57 × 10−3 | 0.07 | up |
| RAB11A | 0.00 | 26.42 | 9.22 × 10−2 | 2.66 × 10−1 | 2.75 × 10−3 | 0.05 | up |
| SCARB2 | 12.28 | 946.42 | 7.95 × 10−1 | 9.02 × 10−1 | 5.34 × 10−5 | −0.01 | down |
| RNASET2 | 12.28 | 59.52 | 2.60 × 10−1 | 4.96 × 10−1 | 1.01 × 10−3 | −0.03 | down |
| TMBIM1 | 9.76 | 4.90 | 3.00 × 10−6 | 8.16 × 10−5 | 1.65 × 10−2 | −0.12 | down |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cen, M.; Wen, Y.; Feng, Z.; Shu, Y.; Hu, C. Single-Cell Dissection of the Serrated Pathway: Cellular Heterogeneity and Genetic Causality in Colorectal Cancer. Int. J. Mol. Sci. 2025, 26, 7187. https://doi.org/10.3390/ijms26157187
Cen M, Wen Y, Feng Z, Shu Y, Hu C. Single-Cell Dissection of the Serrated Pathway: Cellular Heterogeneity and Genetic Causality in Colorectal Cancer. International Journal of Molecular Sciences. 2025; 26(15):7187. https://doi.org/10.3390/ijms26157187
Chicago/Turabian StyleCen, Ming, Yunhan Wen, Zhijun Feng, Yahai Shu, and Chuanxia Hu. 2025. "Single-Cell Dissection of the Serrated Pathway: Cellular Heterogeneity and Genetic Causality in Colorectal Cancer" International Journal of Molecular Sciences 26, no. 15: 7187. https://doi.org/10.3390/ijms26157187
APA StyleCen, M., Wen, Y., Feng, Z., Shu, Y., & Hu, C. (2025). Single-Cell Dissection of the Serrated Pathway: Cellular Heterogeneity and Genetic Causality in Colorectal Cancer. International Journal of Molecular Sciences, 26(15), 7187. https://doi.org/10.3390/ijms26157187