
Missense Mutations in the KAT Domain of CREBBP Gene in Patients with Follicular Lymphoma: Implications for Differential Diagnosis and Prognosis
 , , , , , , , ,
, , , , , , , ,
Abstract
1. Introduction
2. Results
2.1. Clinical Data
2.2. FISH
2.3. Structural Changes in the CREBBP Gene
2.4. Comparison of the Main Characteristics of Patients in the FL 1–3B (14;18)-Positive Group With and Without CREBBP KAT Missense Mutations (KATmiss)
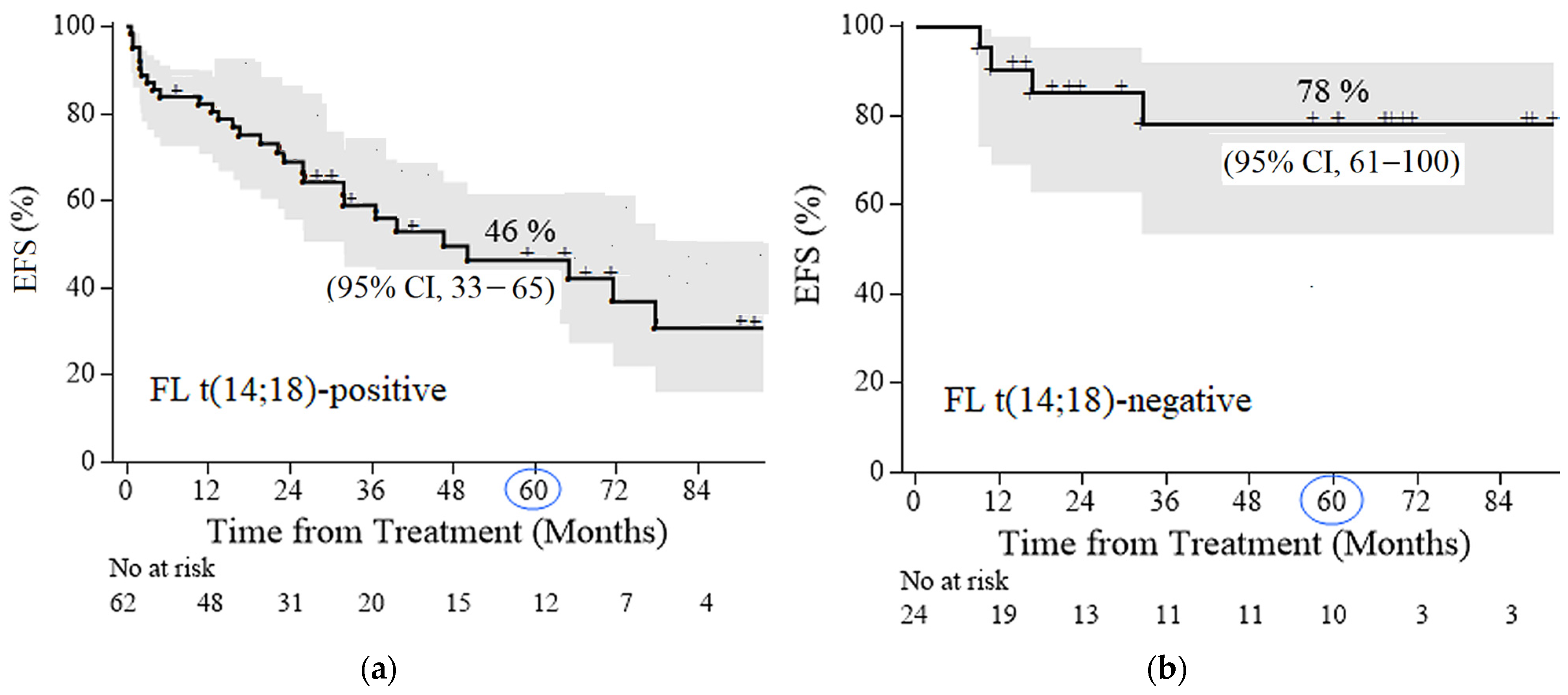
2.5. Survival Analysis
2.6. Causes of Death
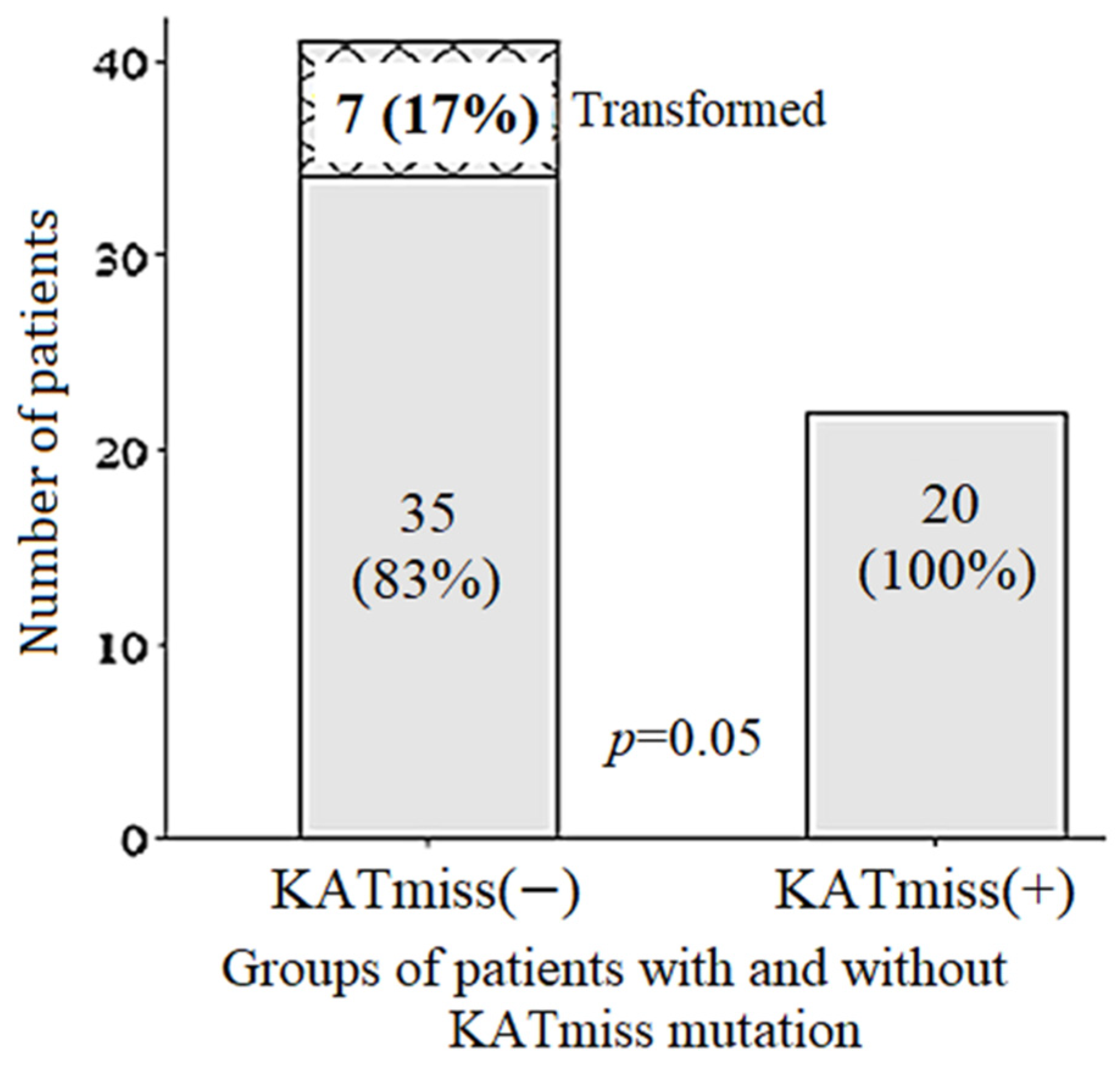
2.7. Frequency and Time to Transformation, Frequency of Progression
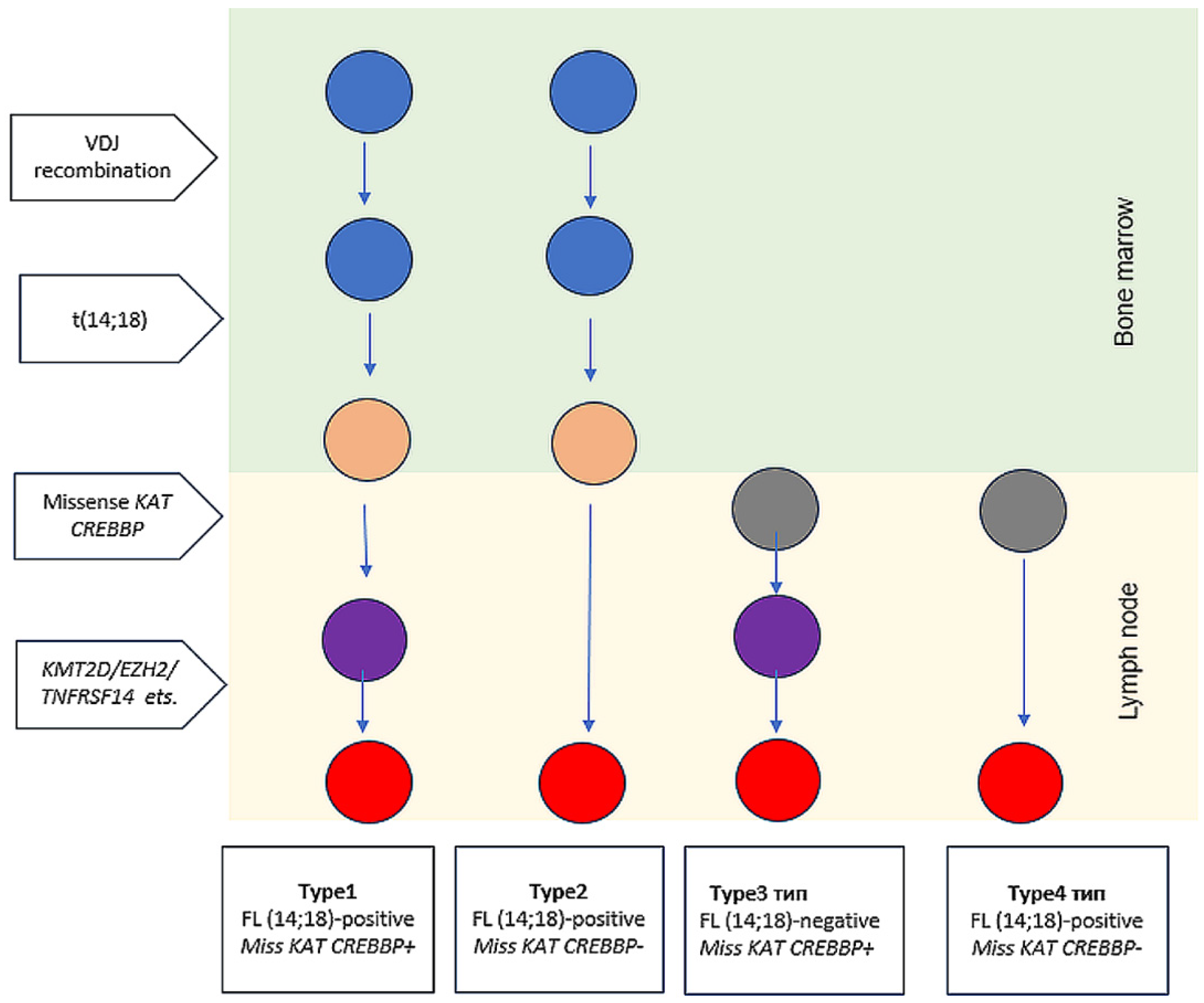
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. DNA Isolation, Clonality Assessment, and BCL2::IGH Breakpoints Evaluation
4.3. Fluorescence In Situ Hybridization (FISH)
4.4. NGS
4.5. LOH
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FL | Follicular lymphoma |
| POD24 | Progression of disease within 24 months of treatment initiation |
| POD12 | Progression of disease within 12 months of treatment initiation |
| DLBCL | Diffuse large B-cell lymphoma |
| KAT | Lysine acetyltransferase domain |
| CCT | Conventional chemotherapy programs |
| HDCT/auto-HSCT | High-dose chemotherapy with autologous hematopoietic stem cell transplantation |
| VAF | Variant allele frequency |
| LOH | Loss of heterozygosity |
| FFPE | Formalin-fixed paraffin-embedded tumor samples |
| OS | Overall survival |
| EFS | Event-free survival |
| SD | Standard deviations |
| SUV | Standard uptake value |
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; WHO Press: Geneva, Switzerland, 2008. [Google Scholar]
- Brady, J.L.; Binkley, M.S.; Hajj, C.; Chelius, M.; Chau, K.; Balogh, A.; Levis, M.; Filippi, A.R.; Jones, M.; Mac Manus, M.; et al. Definitive radiotherapy for localized follicular lymphoma staged by 18F-FDG PET-CT: A collaborative study by ILROG. Blood 2019, 133, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Casulo, C.; Dixon, J.G.; Le-Rademacher, J.; Hoster, E.; Hochster, H.S.; Hiddemann, W.; Marcus, R.; Kimby, E.; Herold, M.; Sebban, C.; et al. Validation of POD24 as a robust early clinical end point of poor survival in FL from 5225 patients on 13 clinical trials. Blood 2022, 139, 1684–1693. [Google Scholar] [CrossRef] [PubMed]
- Casulo, C.; Byrtek, M.; Dawson, K.L.; Zhou, X.; Farber, C.M.; Flowers, C.R.; Hainsworth, J.D.; Maurer, M.J.; Cerhan, J.R.; Link, B.K.; et al. Early Relapse of Follicular Lymphoma After Rituximab Plus Cyclophosphamide, Doxorubicin, Vincristine, and Prednisone Defines Patients at High Risk for Death: An Analysis From the National LymphoCare Study. J. Clin. Oncol. 2015, 33, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
- Casulo, C.; Herold, M.; Hiddemann, W.; Iyengar, S.; Marcus, R.E.; Seymour, J.F.; Launonen, A.; Knapp, A.; Nielsen, T.G.; Mir, F. Risk Factors for and Outcomes of Follicular Lymphoma Histological Transformation at First Progression in the GALLIUM Study. Clin. Lymphoma Myeloma Leuk. 2023, 23, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Magnoli, F.; Tibiletti, M.G.; Uccella, S. Unraveling Tumor Heterogeneity in an Apparently Monolithic Disease: BCL2 and Other Players in the Genetic Landscape of Nodal Follicular Lymphoma. Front. Med. 2019, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Salaverria, I.; Weigert, O.; Quintanilla-Martinez, L. The clinical and molecular taxonomy of t(14;18)-negative follicular lymphomas. Blood Adv. 2023, 7, 5258–5271. [Google Scholar] [CrossRef] [PubMed]
- Laurent, C.; Cook, J.R. Diagnosis and Classification of Follicular Lymphoma and Related Entities. Adv. Anat. Pathol. 2025, 32, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Dreval, K.; Hilton, L.K.; Cruz, M.; Shaalan, H.; Ben-Neriah, S.; Boyle, M.; Collinge, B.; Coyle, K.M.; Duns, G.; Farinha, P.; et al. Genetic subdivisions of follicular lymphoma defined by distinct coding and noncoding mutation patterns. Blood 2023, 142, 561–573. [Google Scholar] [CrossRef]
- Green, M.R.; Gentles, A.J.; Nair, R.V.; Irish, J.M.; Kihira, S.; Liu, C.L.; Kela, I.; Hopmans, E.S.; Myklebust, J.H.; Ji, H.; et al. Hierarchy in somatic mutations arising during genomic evolution and progression of follicular lymphoma. Blood 2013, 121, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Kihira, S.; Liu, C.L.; Nair, R.V.; Salari, R.; Gentles, A.J.; Irish, J.; Stehr, H.; Vicente-Dueñas, C.; Romero-Camarero, I.; et al. Mutations in early follicular lymphoma progenitors are associated with suppressed antigen presentation. Proc. Natl. Acad. Sci. USA 2015, 112, E1116–E1125. [Google Scholar] [CrossRef] [PubMed]
- Nann, D.; Ramis-Zaldivar, J.E.; Müller, I.; Gonzalez-Farre, B.; Schmidt, J.; Egan, C.; Salmeron-Villalobos, J.; Clot, G.; Mattern, S.; Otto, F.; et al. Follicular lymphoma t(14;18)-negative is genetically a heterogeneous disease. Blood Adv. 2020, 4, 5652–5665. [Google Scholar] [CrossRef] [PubMed]
- Smolyaninova, A.K.; Belyayeva, A.V.; Sidorova, Y.V.; Gabeeva, N.G.; Tatarnikova, S.A.; Badmazhapova, D.S.; Koroleva, D.A.; Gemdzhian, E.G.; Kovrigina, A.M.; Sudarikov, A.B.; et al. High-dose chemotherapy with transplantation of autologous hematopoietic stem cells in the first line of follicular lymphoma therapy. Russ. J. Hematol. Transfusiol. 2023, 68, 344–362. [Google Scholar] [CrossRef]
- Gabeeva, N.G.; Koroleva, D.A.; Smolyaninova, A.K.; Belyaeva, A.V.; Tatarnikova, C.A.; Gemdzhian, E.G.; Tsygankova, S.V.; Bulygina, E.S.; Rastorguev, S.M.; Nedoluzhko, A.V.; et al. Chemotherapy According to the R-mNHL-BFM-90 Protocol in Combination with Lenalidomide as the First Line Therapy in Patients with Mum1-Positive Diffusive Large B-Cell Lymphoma and Follicular Lymphoma Grade 3B. Russ. J. Hematol. Transfusiol. 2019, 64, 150–164. [Google Scholar] [CrossRef]
- Schroers-Martin, J.G.; Soo, J.; Brisou, G.; Scherer, F.; Kurtz, D.M.; Sworder, B.J.; Khodadoust, M.S.; Jin, M.C.; Bru, A.; Liu, C.L.; et al. Tracing Founder Mutations in Circulating and Tissue-Resident Follicular Lymphoma Precursors. Cancer Discov. 2023, 13, 1310–1323. [Google Scholar] [CrossRef] [PubMed]
- Mar, N.; Digiuseppe, J.A.; Dailey, M.E. Rubinstein–Taybi syndrome—A window into follicular lymphoma biology. Leuk. Lymphoma 2016, 57, 2908–2910. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Rodova, M.; Miska, E.A.; Calvet, J.P.; Kouzarides, T. Acetylation of β-Catenin by CREB-binding Protein (CBP). J. Biol. Chem. 2002, 277, 25562–25567. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, W.; Ravanmehr, V.; Cui, G.; Bowman, K.; Chen, R.; Henderson, J.M.; Lockman, S.; Rojas, E.; Wilson, A.; et al. Blunted CD40-responsive enhancer activation in CREBBP-mutant lymphomas can be restored by enforced CD4 T-cell engagement. Blood J. 2025, 146, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Holloman, M.; Kim, A.; Chang, Y.; Holmes, A.; Cai, B.; Meyer, S.N.; Basso, K.; Bhagat, G.; Dalla-Favera, R.; et al. Differential Role of Crebbp Missense and Truncating Mutations in the Malignant Transformation of Germinal Center B Cells. Blood. 2024, 144 (Suppl. S1), 47. [Google Scholar] [CrossRef]
- García-Ramírez, I.; Tadros, S.; González-Herrero, I.; Martín-Lorenzo, A.; Rodríguez-Hernández, G.; Moore, D.; Ruiz-Roca, L.; Blanco, O.; Alonso-López, D.; Rivas, J.D.L.; et al. Crebbp loss cooperates with Bcl2 overexpression to promote lymphoma in mice. Blood 2017, 129, 2645–2656. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Zhang, W.; Ravanmehr, V.; Mechaly, A.; Regad, L.; Haouz, A.; Chen, R.; Henderson, J.M.; Rojas, E.; Wilson, A.; et al. CREBBP lysine acetyltransferase domain mutations create zombie enzymes that alter chromatin loading dynamics and prevent EP300 redundancy. bioRxiv 2023. [Google Scholar] [CrossRef]
- Schroers-Martin, J.G.; Soo, J.; Brisou, G.; Scherer, F.; Kurtz, D.M.; Sworder, B.; Khodadoust, M.S.; Jin, M.C.; Bru, A.; Liu, C.L.; et al. Recurrent Crebbp Mutations in Follicular Lymphoma Appear Localized to the Committed B-Cell Lineage. Blood 2020, 136 (Suppl. S1), 30–31. [Google Scholar] [CrossRef]
- Adolph, L.C.; Fichaux, Q.; Strobl, C.D.; Antoniolli, M.; Passerini, V.; Keay, W.D.; Jurinovic, V.; Lechner, A.; Hodson, D.J.; von Bergwelt, M.; et al. CHOP but Not Bendamustine Reverses EZH2 Y641 Mutation Induced MHC-I/II Loss in Human Lymphoma Models. Blood 2021, 138 (Suppl. S1), 2391. [Google Scholar] [CrossRef]
- Sun, C.; Li, W.; Yu, J.; Zhang, T.; Gong, W.; Liu, H.; Gao, F.; Song, Z.; Li, L.; Qiu, L.; et al. Molecular landscape of distinct follicular lymphoma histologic grades: Insights from genomic and transcriptome analyses. Leukemia 2025, 39, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, J.V.; Biderman, B.V.; Nikulina, E.E.; Sudarikov, A.B. A simple and efficient method for DNA extraction from skin and paraffin-embedded tissues applicable to T-cell clonality assays. Exp. Dermatol. 2012, 21, 57–60. [Google Scholar] [CrossRef] [PubMed]
- van Dongen, J.J.M.; Langerak, A.W.; Brüggemann, M.; Evans, P.A.S.; Hummel, M.; Lavender, F.L.; Delabesse, E.; Davi, F.; Schuuring, E.; García-Sanz, R.; et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: Report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia 2003, 17, 2257–2317. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Markovets, A.; Ahdesmaki, M.; Chapman, B.; Hofmann, O.; McEwen, R.; Johnson, J.; Dougherty, B.; Barrett, J.C.; Dry, J.R. VarDict: A novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016, 44, e108. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Sherry, S.T.; Ward, M.; Sirotkin, K. dbSNP—database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999, 9, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [PubMed]
- Petrackova, A.; Vasinek, M.; Sedlarikova, L.; Dyskova, T.; Schneiderova, P.; Novosad, T.; Papajik, T.; Kriegova, E. Standardization of Sequencing Coverage Depth in NGS: Recommendation for Detection of Clonal and Subclonal Mutations in Cancer Diagnostics. Front. Oncol. 2019, 9, 851. [Google Scholar] [CrossRef] [PubMed]
- Horak, P.; Griffith, M.; Danos, A.M.; Pitel, B.A.; Madhavan, S.; Liu, X.; Chow, C.; Williams, H.; Carmody, L.; Barrow-Laing, L.; et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genet. Med. 2022, 24, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Abdurashidova, R.R.; Risinskaya, N.V.; Mangasarova, Y.K.; Surin, V.L.; Shupletsova, I.A.; Chabaeva, Y.A.; Magomedova, A.U.; Abramova, T.V.; Nikulina, E.E.; Yusupov, R.Y.; et al. Analysis of microsatellite instability in primary mediastinal large B-cell lymphoma: Focus on PD-L1/PD-L2 and CIITA. Russ. J. Hematol. Transfusiol. 2024, 69, 297–318. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | FL 1–3B (14;18)-Positive (n = 62) | FL 3A–3B (14;18)-Negative (n = 24) | p-Value |
|---|---|---|---|
| Male/Female | 31/31 | 14/10 | 0.63 |
| Age, median (range). years | 42 (24–77) | 47 (34–71) | 0.21 |
| IV stage | 2/62 (84%) | 6/23 (26%) | 0.001 |
| Local involvement of the lymph. nodes of the head and neck (tonsils +/−) | 0/62 (0%) | 12/24 (50%) | 0.006 |
| ECOG: | 0.94 | ||
| 0–1 | 38/62 (61%) | 9/23 (39%) | |
| 2 | 16/62 (25%) | 7/23 (30%) | |
| 3 | 7/62 (11%) | 3/23 (13%) | |
| 4 | 2/62 (3%) | 4/23 (18%) | |
| Bulky (>6 cm) | 38/61 (62%) | 11/24 (45%) | 0.22 |
| LDG, higher/total | 32/58 (55%) | 11/24 (52%) | 0.47 |
| Hemoglobin ˂120 g/L | 39/62 (63%) | 8/24 (33%) | 0.03 |
| Bone marrow involvement | 51/62 (82%) | 2/24 (9%) | 0.001 |
| FLIPI: | 0.01 | ||
| low | 6/59 (10%) | 12/23 (52%) | |
| intermediate | 18/59 (31%) | 3/23 (13%) | |
| high risk | 35/59 (59%) | 8/23 (35%) | |
| SUVmax, median (range) [interquartile range] | 10.8 (3.9–49) [8.8–13.6] | 18.0 (6.8–49) [10.5–23.9] | 0.01 |
| SUVmax, mean (SD) | 11.7 (5.0) | 18.3 (5.0) | 0.01 |
| SUVmax > 14 | 12/47 (25%) | 11/18 (61%) | 0.01 |
| SUVmax > 20 | 7/47 (15%) | 8/18 (44%) | 0.02 |
| Characteristics | FL 1–3B (14;18)-Positive With KATmiss (n = 20) | FL 1–3B (14;18)-Positive Without Katmiss (n = 42) | p-Value |
|---|---|---|---|
| Male/Female | 9/11 | 27/15 | 0.18 |
| Age, median (range). years | 47 (32–77) | 45 (24–71) | 0.31 |
| IV stage | 17/20 (85%) | 33/42 (79%) | 0.80 |
| ECOG: | 0.98 | ||
| 0–1 | 14/19 (74%) | 23/42 (55%) | |
| 2 | 4/19 (21%) | 11/42 (26%) | |
| 3 | 0/19 (0%) | 7/42 (17%) | |
| 4 | 1/19 (5%) | 1/42 (2%) | |
| Bulky (>6 cm) | 13/20 (65%) | 27/40 (68%) | 0.85 |
| LDG. higher/total | 13/18 (72%) | 21/37 (57%) | 0.42 |
| Hemoglobin ˂120 g/L | 8/20 (40%) | 11/38 (29%) | 0.58 |
| Grade: | 0.96 | ||
| 1–2 | 15/20 (75%) | 29/42 (69%) | |
| 3A | 2/20 (10%) | 6/42 (14%) | |
| mix (1–2 + 3A, 1–2 + 3A) | 3/20 (15%) | 7/42 (17%) | |
| Bone marrow involvement | 17/20 (85%) | 35/41 (85%) | 1.00 |
| FLIPI: | 0.99 | ||
| low | 3/18 (17%) | 4/40 (10%) | |
| intermediate | 5/18 (28%) | 13/40 (33%) | |
| high-risk | 10/18 (56%) | 23/40 (57%) | |
| SUVmax, median (range), [interquartile range] | 8.6 (4.4–20.1), [5.14–13.18] | 11.7 (3.9–22.3), [10.1–18.0] | 0.05 |
| SUVmax, mean (SD) | 9.7 (4.6) | 12.6 (5.0) | 0.05 |
| SUVmax > 14 | 2/14 (25%) | 8/31 (29%) | 0.71 |
| SUVmax > 20 | 1/14 (0%) | 4/31 (16%) | 1.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smolianinova, A.; Bolshakov, I.; Sidorova, Y.; Kovrigina, A.; Obukhova, T.; Gabeeva, N.; Gemdzhian, E.; Nikulina, E.; Biderman, B.; Severina, N.; et al. Missense Mutations in the KAT Domain of CREBBP Gene in Patients with Follicular Lymphoma: Implications for Differential Diagnosis and Prognosis. Int. J. Mol. Sci. 2025, 26, 6913. https://doi.org/10.3390/ijms26146913
Smolianinova A, Bolshakov I, Sidorova Y, Kovrigina A, Obukhova T, Gabeeva N, Gemdzhian E, Nikulina E, Biderman B, Severina N, et al. Missense Mutations in the KAT Domain of CREBBP Gene in Patients with Follicular Lymphoma: Implications for Differential Diagnosis and Prognosis. International Journal of Molecular Sciences. 2025; 26(14):6913. https://doi.org/10.3390/ijms26146913
Chicago/Turabian StyleSmolianinova, Anna, Ivan Bolshakov, Yulia Sidorova, Alla Kovrigina, Tatiana Obukhova, Nelli Gabeeva, Eduard Gemdzhian, Elena Nikulina, Bella Biderman, Nataliya Severina, and et al. 2025. "Missense Mutations in the KAT Domain of CREBBP Gene in Patients with Follicular Lymphoma: Implications for Differential Diagnosis and Prognosis" International Journal of Molecular Sciences 26, no. 14: 6913. https://doi.org/10.3390/ijms26146913
APA StyleSmolianinova, A., Bolshakov, I., Sidorova, Y., Kovrigina, A., Obukhova, T., Gabeeva, N., Gemdzhian, E., Nikulina, E., Biderman, B., Severina, N., Risinskaya, N., Sudarikov, A., Zvonkov, E., & Parovichnikova, E. (2025). Missense Mutations in the KAT Domain of CREBBP Gene in Patients with Follicular Lymphoma: Implications for Differential Diagnosis and Prognosis. International Journal of Molecular Sciences, 26(14), 6913. https://doi.org/10.3390/ijms26146913