

Long-Chain Fatty Acids Alter Estrogen Receptor Expression in Breast Cancer Cells

Abstract
1. Introduction
2. Results
2.1. Effect of LCFAs on Cell Viability in BCA Cells
2.2. Effect of LCFAs on Mitochondrial Function in BCA Cells
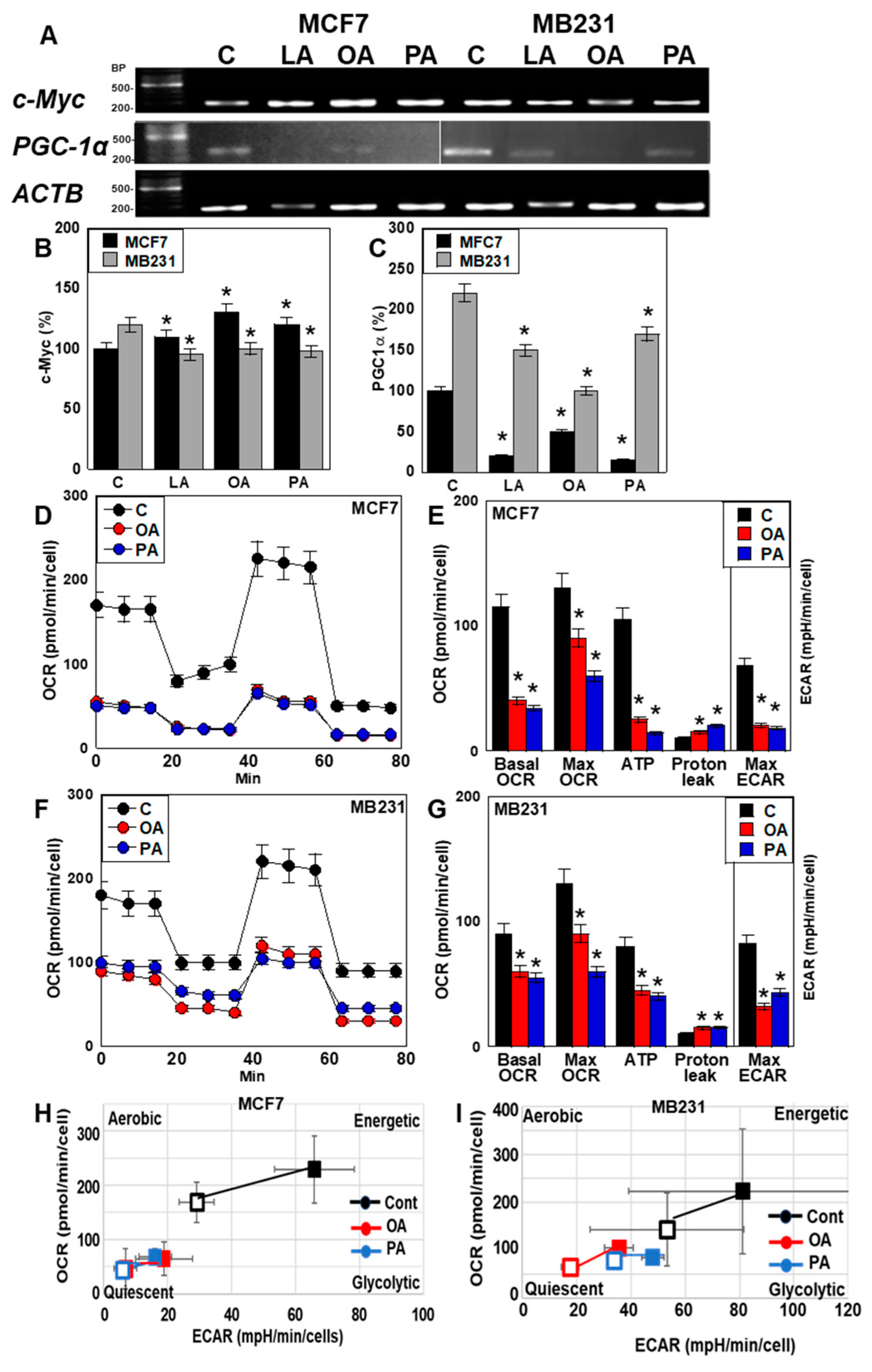
2.3. Effect of LCFAs on Stemness and Differentiation of BCA Cells
2.4. Effect of LCFAs on Energy Metabolism of BCA Cells
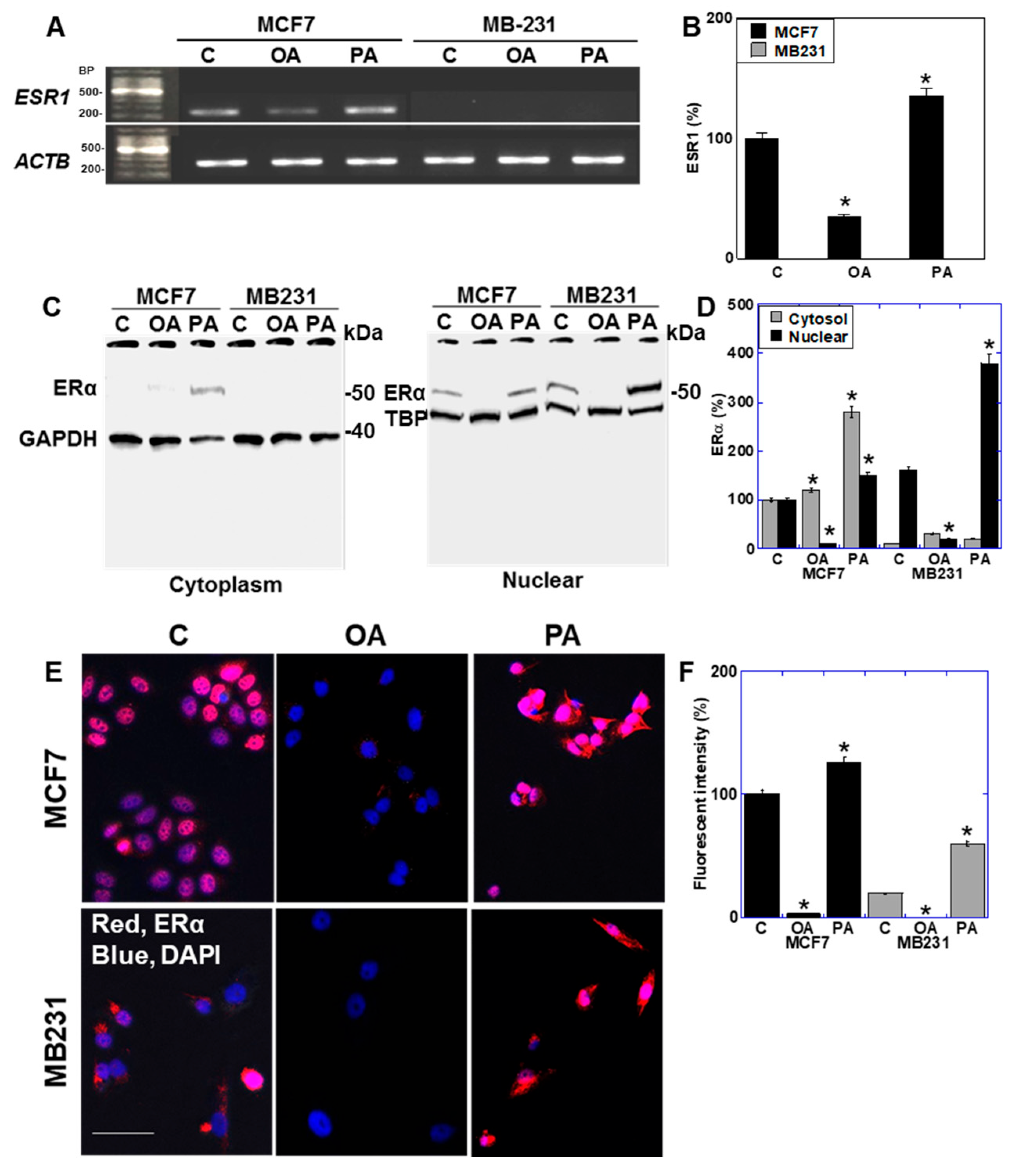
2.5. Effect of LCFAs on Expression of Estrogen Receptor in BCA Cells
2.6. Function of ER Expressed in BCA Cells
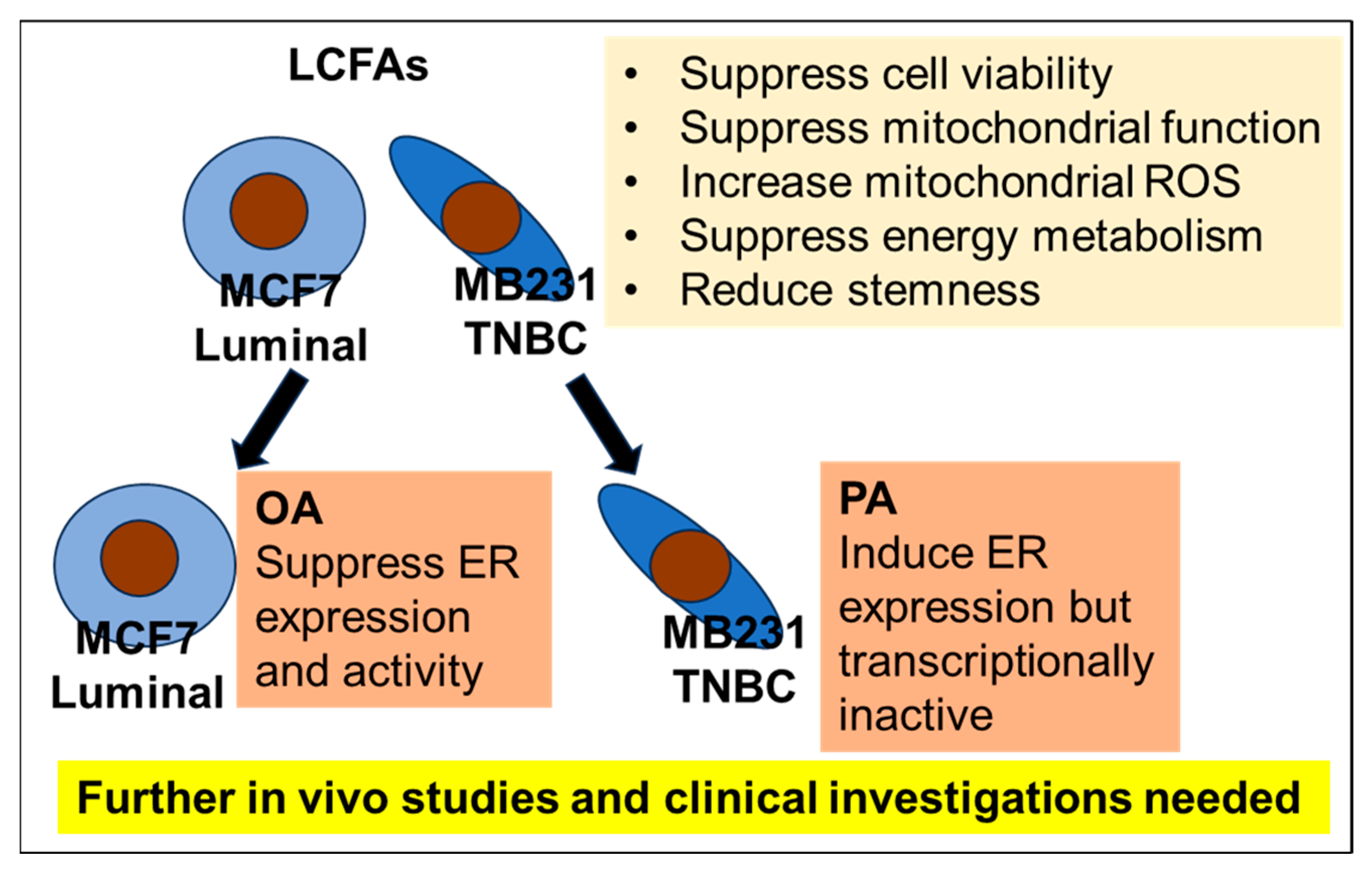
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. MTS [3-(4,5-Dimethylthiazol-2-yl)-5-(3-Carboxymethoxyphenyl)-2-(4-Sulfophenyl)-2H-Tetrazolium] Assay
4.3. Mitochondrial Imaging
4.4. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
4.5. Sphere Assay
4.6. Protein Extraction
4.7. Immunoblot Analysis
4.8. Immunocytochemistry
4.9. Mitochondrial Stress Test (Seahorse Assay)
4.10. Glycolytic Stress Test
4.11. Detection of microRNA
4.12. Inhibition of microRNA
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviation
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Reis-Filho, J.S.; Pusztai, L. Gene expression profiling in breast cancer: Classification, prognostication, and prediction. Lancet 2011, 378, 1812–1823. [Google Scholar] [CrossRef]
- Prat, A.; Cheang, M.C.; Martín, M.; Parker, J.S.; Carrasco, E.; Caballero, R.; Tyldesley, S.; Gelmon, K.; Bernard, P.S.; Nielsen, T.O.; et al. Prognostic significance of progesterone receptor-positive tumor cells within immunohistochemically defined luminal A breast cancer. J. Clin. Oncol. 2013, 31, 203–209. [Google Scholar] [CrossRef]
- Carey, L.A.; Berry, D.A.; Cirrincione, C.T.; Barry, W.T.; Pitcher, B.N.; Harris, L.N.; Ollila, D.W.; Krop, I.E.; Henry, N.L.; Weckstein, D.J.; et al. Molecular Heterogeneity and Response to Neoadjuvant Human Epidermal Growth Factor Receptor 2 Targeting in CALGB 40601, a Randomized Phase III Trial of Paclitaxel Plus Trastuzumab With or Without Lapatinib. J. Clin. Oncol. 2016, 34, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Baselga, J.; Kim, S.B.; Ro, J.; Semiglazov, V.; Campone, M.; Ciruelos, E.; Ferrero, J.M.; Schneeweiss, A.; Heeson, S.; et al. Pertuzumab, trastuzumab, and docetaxel in HER2-positive metastatic breast cancer. N. Engl. J. Med. 2015, 372, 724–734. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Harkins, B.; Geyer, C.E., Jr. Overcoming treatment challenges in advanced breast cancer. Semin. Oncol. Nurs. 2007, 23 (Suppl. 2), S10–S16. [Google Scholar] [CrossRef]
- Gingras, I.; Gebhart, G.; de Azambuja, E.; Piccart-Gebhart, M. HER2-positive breast cancer is lost in translation: Time for patient-centered research. Nat. Rev. Clin. Oncol. 2017, 14, 669–681. [Google Scholar] [CrossRef]
- Babina, I.S.; Turner, N.C. Advances and challenges in targeting FGFR signalling in cancer. Nat. Rev. Cancer 2017, 17, 318–332. [Google Scholar] [CrossRef]
- Ali, S.; Coombes, R.C. Estrogen receptor alpha in human breast cancer: Occurrence and significance. J. Mammary Gland. Biol. Neoplasia 2000, 5, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Traish, A.M.; Newton, A.W.; Styperek, K.; Beazley, R.; Kavanah, M. Estrogen receptor functional status in human breast cancer. Diagn. Mol. Pathol. 1995, 4, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Toumba, M.; Kythreotis, A.; Panayiotou, K.; Skordis, N. Estrogen receptor signaling and targets: Bones, breasts and brain (Review). Mol. Med. Rep. 2024, 30, 144. [Google Scholar] [CrossRef] [PubMed]
- Cignarella, A.; Boscaro, C.; Albiero, M.; Bolego, C.; Barton, M. Post-Transcriptional and Epigenetic Regulation of Estrogen Signaling. J. Pharmacol. Exp. Ther. 2023, 386, 288–297. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Z.; Cenciarini, M.E.; Proietti, C.J.; Amasino, M.; Hong, T.; Yang, M.; Liao, Y.; Chiang, H.C.; Kaklamani, V.G.; et al. Tamoxifen Resistance in Breast Cancer Is Regulated by the EZH2-ERα-GREB1 Transcriptional Axis. Cancer Res. 2018, 78, 671–684. [Google Scholar] [CrossRef]
- Du, F.; Yuan, P.; Wang, T.; Zhao, J.; Zhao, Z.; Luo, Y.; Xu, B. The Significance and Therapeutic Potential of GATA3 Expression and Mutation in Breast Cancer: A Systematic Review. Med. Res. Rev. 2015, 35, 1300–1315. [Google Scholar] [CrossRef]
- Carroll, J.S.; Liu, X.S.; Brodsky, A.S.; Li, W.; Meyer, C.A.; Szary, A.J.; Eeckhoute, J.; Shao, W.; Hestermann, E.V.; Geistlinger, T.R.; et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005, 122, 33–43. [Google Scholar] [CrossRef]
- Theodorou, V.; Stark, R.; Menon, S.; Carroll, J.S. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013, 23, 12–22. [Google Scholar] [CrossRef]
- Viedma-Rodríguez, R.; Baiza-Gutman, L.; Salamanca-Gómez, F.; Diaz-Zaragoza, M.; Martínez-Hernández, G.; Ruiz Esparza-Garrido, R.; Velázquez-Flores, M.A.; Arenas-Aranda, D. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer (review). Oncol. Rep. 2014, 32, 3–15. [Google Scholar] [CrossRef]
- Adams, B.D.; Guttilla, I.K.; White, B.A. Involvement of microRNAs in breast cancer. Semin. Reprod. Med. 2008, 26, 522–536. [Google Scholar] [CrossRef]
- Bird, J.K.; Troesch, B.; Warnke, I.; Calder, P.C. The effect of long chain omega-3 polyunsaturated fatty acids on muscle mass and function in sarcopenia: A scoping systematic review and meta-analysis. Clin. Nutr. ESPEN 2021, 46, 73–86. [Google Scholar] [CrossRef]
- Ferraro, G.B.; Ali, A.; Luengo, A.; Kodack, D.P.; Deik, A.; Abbott, K.L.; Bezwada, D.; Blanc, L.; Prideaux, B.; Jin, X.; et al. Fatty Acid Synthesis Is Required for Breast Cancer Brain Metastasis. Nat. Cancer 2021, 2, 414–428. [Google Scholar] [CrossRef] [PubMed]
- Rose, D.P.; Rayburn, J.; Hatala, M.A.; Connolly, J.M. Effects of dietary fish oil on fatty acids and eicosanoids in metastasizing human breast cancer cells. Nutr. Cancer 1994, 22, 131–141. [Google Scholar] [CrossRef]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef]
- Xia, L.; Zhou, Z.; Chen, X.; Luo, W.; Ding, L.; Xie, H.; Zhuang, W.; Ni, K.; Li, G. Ligand-dependent CD36 functions in cancer progression, metastasis, immune response, and drug resistance. Biomed. Pharmacother. 2023, 168, 115834. [Google Scholar] [CrossRef]
- Yang, B.; Ren, X.L.; Wang, Z.Y.; Wang, L.; Zhao, F.; Guo, X.J.; Li, D. Biomarker of long-chain n-3 fatty acid intake and breast cancer: Accumulative evidence from an updated meta-analysis of epidemiological studies. Crit. Rev. Food Sci. Nutr. 2019, 59, 3152–3164. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.M.; Handa, P.; Tateya, S.; Schwartz, J.; Tang, C.; Mitra, P.; Oram, J.F.; Chait, A.; Kim, F. Apolipoprotein A-I attenuates palmitate-mediated NF-κB activation by reducing Toll-like receptor-4 recruitment into lipid rafts. PLoS ONE 2012, 7, e33917. [Google Scholar] [CrossRef] [PubMed]
- Rayaprolu, S.J.; Hettiarachchy, N.S.; Horax, R.; Phillips, G.K.; Mahendran, M.; Chen, P. Soybean peptide fractions inhibit human blood, breast and prostate cancer cell proliferation. J. Food Sci. Technol. 2017, 54, 38–44. [Google Scholar] [CrossRef]
- Huang, L.H.; Chung, H.Y.; Su, H.M. Docosahexaenoic acid reduces sterol regulatory element binding protein-1 and fatty acid synthase expression and inhibits cell proliferation by inhibiting pAkt signaling in a human breast cancer MCF-7 cell line. BMC Cancer 2017, 17, 890. [Google Scholar] [CrossRef]
- Rose, D.P.; Goldman, M.; Connolly, J.M.; Strong, L.E. High-fiber diet reduces serum estrogen concentrations in premenopausal women. Am. J. Clin. Nutr. 1991, 54, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Edin, M.L.; Duval, C.; Zhang, G.; Zeldin, D.C. Role of linoleic acid-derived oxylipins in cancer. Cancer Metastasis Rev. 2020, 39, 581–582. [Google Scholar] [CrossRef] [PubMed]
- Ogata, R.; Mori, S.; Kishi, S.; Sasaki, R.; Iwata, N.; Ohmori, H.; Sasaki, T.; Nishiguchi, Y.; Nakashima, C.; Goto, K.; et al. Linoleic Acid Upregulates Microrna-494 to Induce Quiescence in Colorectal Cancer. Int. J. Mol. Sci. 2021, 23, 225. [Google Scholar] [CrossRef] [PubMed]
- Ogata, R.; Mori, S.; Ohmori, H.; Kishi, S.; Fujiwara-Tani, R.; Sasaki, T.; Nishiguchi, Y.; Nakashima, C.; Goto, K.; Kawahara, I.; et al. Suppressive GLI2 fragment enhances liver metastasis in colorectal cancer. Oncotarget 2022, 13, 122–135. [Google Scholar] [CrossRef]
- Ohmori, H.; Sasahira, T.; Fujii, K.; Yi, L.; Shimomoto, T.; Kuniyasu, H. Linoleic-acid-induced growth suppression induces quiescent cancer cell nests in nude mice. Pathobiology 2008, 75, 226–232. [Google Scholar] [CrossRef]
- Kawahara, I.; Mori, T.; Goto, K.; Fujii, K.; Ohmori, H.; Kishi, S.; Fujiwara-Tani, R.; Kuniyasu, H. Fatty Acids Induce Stemness in the Stromal Cells of a CT26 Mouse Tumor Model. Pathobiology 2017, 84, 237–242. [Google Scholar] [CrossRef]
- Tanabe, E.; Kitayoshi, M.; Fujii, K.; Ohmori, H.; Luo, Y.; Kadochi, Y.; Mori, S.; Fujiwara, R.; Nishiguchi, Y.; Sasaki, T.; et al. Fatty acids inhibit anticancer effects of 5-fluorouracil in mouse cancer cell lines. Oncol. Lett. 2017, 14, 681–686. [Google Scholar] [CrossRef]
- Shimomoto, T.; Luo, Y.; Ohmori, H.; Chihara, Y.; Fujii, K.; Sasahira, T.; Denda, A.; Kuniyasu, H. Advanced glycation end products (AGE) induce the receptor for AGE in the colonic mucosa of azoxymethane-injected Fischer 344 rats fed with a high-linoleic acid and high-glucose diet. J. Gastroenterol. 2012, 47, 1073–1083. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Cheng, C.F.; Ku, H.C.; Lin, H. PGC-1α as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef]
- He, Q.; Chen, Y.; Wang, Z.; He, H.; Yu, P. Cellular Uptake, Metabolism and Sensing of Long-Chain Fatty Acids. Front. Biosci. (Landmark Ed.) 2023, 28, 10. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, H.; Ogura, H.; Arai, Y.; Baba, S.; Kosugi, I.; Tsutsui, Y.; Iwashita, T. Aggressive progression of breast cancer with microscopic pulmonary emboli possessing a stem cell-like phenotype independent of its origin. Pathol. Int. 2010, 60, 228–234. [Google Scholar] [CrossRef]
- Xiong, J.; Yu, D.; Wei, N.; Fu, H.; Cai, T.; Huang, Y.; Wu, C.; Zheng, X.; Du, Q.; Lin, D.; et al. An estrogen receptor alpha suppressor, microRNA-22, is downregulated in estrogen receptor alpha-positive human breast cancer cell lines and clinical samples. FEBS J. 2010, 277, 1684–1694. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Gasparini, P.; Piovan, C.; Ngankeu, A.; Garofalo, M.; Taccioli, C.; Iorio, M.V.; Li, M.; Volinia, S.; Alder, H.; et al. MicroRNA cluster 221-222 and estrogen receptor alpha interactions in breast cancer. J. Natl. Cancer Inst. 2010, 102, 706–721. [Google Scholar] [CrossRef]
- Vesuna, F.; Lisok, A.; van Diest, P.; Raman, V. Twist activates miR-22 to suppress estrogen receptor alpha in breast cancer. Mol. Cell Biochem. 2021, 476, 2295–2306. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Toyama, T.; Sugiura, H.; Fujii, Y.; Yamashita, H. miR-206 Expression is down-regulated in estrogen receptor alpha-positive human breast cancer. Cancer Res. 2008, 68, 5004–5008. [Google Scholar] [CrossRef]
- Chakkaravarthi, K.; Ramesh, R.; Palaniyandi, T.; Baskar, G.; Viswanathan, S.; Wahab, M.R.A.; Surendran, H.; Ravi, M.; Sivaji, A. Prospectives of mirna gene signaling pathway in triple-negative breast cancer. Pathol. Res. Pract. 2023, 248, 154658. [Google Scholar] [CrossRef]
- Shao, X.; Huang, P.; Shi, L.; Lei, L.; Cao, W.; Chen, Z.; Wang, X.; Zheng, Y. MicroRNA and LncRNA Expression Profiles in Human Estrogen Receptor Positive Breast Cancer. Clin. Lab. 2019, 65, 7. [Google Scholar] [CrossRef]
- Adams, B.D.; Furneaux, H.; White, B.A. The micro-ribonucleic acid (miRNA) miR-206 targets the human estrogen receptor-alpha (ERalpha) and represses ERalpha messenger RNA and protein expression in breast cancer cell lines. Mol. Endocrinol. 2007, 21, 1132–1147. [Google Scholar] [CrossRef]
- Endo, Y.; Toyama, T.; Takahashi, S.; Yoshimoto, N.; Iwasa, M.; Asano, T.; Fujii, Y.; Yamashita, H. miR-1290 and its potential targets are associated with characteristics of estrogen receptor α-positive breast cancer. Endocr. Relat. Cancer 2013, 20, 91–102. [Google Scholar] [CrossRef]
- Tang, Y.; Tian, W.; Xie, J.; Zou, Y.; Wang, Z.; Li, N.; Zeng, Y.; Wu, L.; Zhang, Y.; Wu, S.; et al. Prognosis and Dissection of Immunosuppressive Microenvironment in Breast Cancer Based on Fatty Acid Metabolism-Related Signature. Front. Immunol. 2022, 31, 843515. [Google Scholar] [CrossRef]
- Palomer, X.; Pizarro-Delgado, J.; Barroso, E.; Vázquez-Carrera, M. Palmitic and Oleic Acid: The Yin and Yang of Fatty Acids in Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2018, 29, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Khoi, C.S.; Lin, T.Y.; Chiang, C.K. Targeting Insulin Resistance, Reactive Oxygen Species, Inflammation, Programmed Cell Death, ER Stress, and Mitochondrial Dysfunction for the Therapeutic Prevention of Free Fatty Acid-Induced Vascular Endothelial Lipotoxicity. Antioxidants 2024, 13, 1486. [Google Scholar] [CrossRef]
- Mondal, A.; Burchat, N.; Sampath, H. Palmitate exacerbates bisphenol A toxicity via induction of ER stress and mitochondrial dysfunction. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158816. [Google Scholar] [CrossRef]
- González-Becerra, K.; Ramos-Lopez, O.; Barrón-Cabrera, E.; Riezu-Boj, J.I.; Milagro, F.I.; Martínez-López, E.; Martínez, J.A. Fatty acids, epigenetic mechanisms and chronic diseases: A systematic review. Lipids Health Dis. 2019, 18, 178. [Google Scholar] [CrossRef]
- Al-Bader, M.; Ford, C.; Al-Ayadhy, B.; Francis, I. Analysis of estrogen receptor isoforms and variants in breast cancer cell lines. Exp. Ther. Med. 2011, 2, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, S.A.; Fitzgerald, S.D.; Allred, D.C.; Elledge, R.M.; Nawaz, Z.; McDonnell, D.P.; O’Malley, B.W.; Greene, G.L.; McGuire, W.L. Inhibition of estrogen receptor action by a naturally occurring variant in human breast tumors. Cancer Res. 1992, 65, 483–486. [Google Scholar]
- Ge, Y.; Jin, J.; Chen, G.; Li, J.; Ye, M.; Jin, X. Endometrial cancer (EC) derived G3BP1 overexpression and mutant promote EC tumorigenesis and metastasis via SPOP/ERα axis. Cell Commun. Signal 2023, 21, 303. [Google Scholar] [CrossRef]
- He, X.; Zheng, Z.; Song, T.; Wei, C.; Ma, H.; Ma, Q.; Zhang, Y.; Xu, Y.; Shi, W.; Ye, Q.; et al. c-Abl regulates estrogen receptor alpha transcription activity through its stabilization by phosphorylation. Oncogene 2010, 29, 2238–2251. [Google Scholar] [CrossRef]
- Karim, A.M.; Eun Kwon, J.; Ali, T.; Jang, J.; Ullah, I.; Lee, Y.G.; Park, D.W.; Park, J.; Jeang, J.W.; Kang, S.C. Triple-negative breast cancer: Epidemiology, molecular mechanisms, and modern vaccine-based treatment strategies. Biochem. Pharmacol. 2023, 212, 115545. [Google Scholar] [CrossRef]
- Shi, M.; Guo, N. MicroRNA expression and its implications for the diagnosis and therapeutic strategies of breast cancer. Cancer Treat. Rev. 2009, 35, 328–334. [Google Scholar] [CrossRef]
- da Paixão, A.O.; Bolin, A.P.; Silvestre, J.G.; Rodrigues, A.C. Palmitic Acid Impairs Myogenesis and Alters Temporal Expression of miR-133a and miR-206 in C2C12 Myoblasts. Int. J. Mol. Sci. 2021, 22, 2748. [Google Scholar] [CrossRef] [PubMed]
- Murakami, J.; Hu, Y.; Fan, Y.; Zhang, C.; Wang, C. Palmitic acid inhibits vascular smooth muscle cell switch to synthetic phenotype via upregulation of miR-22 expression. Aging 2022, 14, 8046–8060. [Google Scholar]
- Huang, F.; Chen, J.; Wang, J.; Zhu, P.; Lin, W. Palmitic Acid Induces MicroRNA-221 Expression to Decrease Glucose Uptake in HepG2 Cells via the PI3K/AKT/GLUT4 Pathway. Biomed. Res. Int. 2019, 2019, 8171989. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Lei, C.; Gong, K.; Bai, S.; Sun, L. Palmitic acid promotes human retinal pigment epithelial cells migration by upregulating miR-222 expression and inhibiting NUMB. Aging 2023, 15, 9341–9357. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Guan, H.; Huang, Z.; Liu, J.; Li, H.; Wei, G.; Cao, X.; Li, Y. Downregulation of Bcl-2 expression by miR-34a mediates palmitate-induced Min6 cells apoptosis. J. Diabetes Res. 2014, 2014, 258695. [Google Scholar] [CrossRef]
- Dian, B.; Wang, Y.; Liao, J.; Kadeer, A.; Wang, J.; Wibuer, P. Downregulation of miR-34a represses insulin resistance and mediates glucose metabolism by upregulating IGF2 to activate PI3K/AKT signaling pathway. Trop. J. Pharm. Res. 2021, 20, 2481–2487. [Google Scholar]
- Weng, Y.S.; Tseng, H.Y.; Chen, Y.A.; Shen, P.C.; Al Haq, A.T.; Chen, L.M.; Tung, Y.C.; Hsu, H.L. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol. Cancer 2019, 18, 42. [Google Scholar] [CrossRef]
- Chung, S.; Hwang, J.T.; Park, J.H.; Choi, H.K. Free fatty acid-induced histone acetyltransferase activity accelerates lipid accumulation in HepG2 cells. Nutr. Res. Pract. 2019, 13, 196–204. [Google Scholar] [CrossRef]
- Xie, X.; Gao, M.; Zhao, W.; Li, C.; Zhang, W.; Yang, J.; Zhang, Y.; Chen, E.; Guo, Y.; Guo, Z.; et al. LncRNA Snhg3 aggravates hepatic steatosis via PPARγ signaling. eLife 2024, 13, RP96988. [Google Scholar] [CrossRef]
- Madak-Erdogan, Z.; Lupien, M.; Stossi, F.; Brown, M.; Katzenellenbogen, B.S. Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol. Cell Biol. 2011, 31, 226–236. [Google Scholar] [CrossRef]
- Tang, Y.; Tian, W.; Zheng, S.; Zou, Y.; Xie, J.; Zhang, J.; Li, X.; Sun, Y.; Lan, J.; Li, N.; et al. Dissection of FOXO1-Induced LYPLAL1-DT Impeding Triple-Negative Breast Cancer Progression via Mediating hnRNPK/β-Catenin Complex. Research 2023, 15, 0289. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhu, R.; Sun, X.; Guo, Q.; Zhang, Y.; Zhang, N.; Oh, Y.; Fan, L.; Wang, C.; Gu, N. Fatty acid palmitate suppresses FoxO1 expression via PERK and IRE1 unfolded protein response in C2C12 myotubes. Toxicol. In Vitro 2022, 85, 105459. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Li, Y.; Wang, J.; Wen, X.; Marcus, M.T.; Daniels, G.; Zhang, D.Y.; Ye, F.; Wang, L.H.; Du, X.; et al. Long chain fatty Acyl-CoA synthetase 4 is a biomarker for and mediator of hormone resistance in human breast cancer. PLoS ONE 2013, 8, e77060. [Google Scholar] [CrossRef] [PubMed]
- Brechbuhl, H.M.; Han, A.; Paul, K.V.; Nemkov, T.; Ramachandran, S.; Ward, A.; Jacobsen, B.M.; Hansen, K.; Sartorius, C.A.; D’Alessandro, A.; et al. Metabolic Switch in Endocrine Resistant Estrogen Receptor Positive Breast Cancer. bioRxiv 2024. [Google Scholar] [CrossRef]
- Radde, B.N.; Alizadeh-Rad, N.; Price, S.M.; Schultz, D.J.; Klinge, C.M. Anacardic Acid, Salicylic Acid, and Oleic Acid Differentially Alter Cellular Bioenergetic Function in Breast Cancer Cells. J. Cell Biochem. 2016, 117, 2521–2532. [Google Scholar] [CrossRef]
- Bian, C.; Sun, J.; Huang, X.; Ji, S.; Ji, H. Endoplasmic reticulum stress is involved in lipid accumulation induced by oleic acid in adipocytes of grass carp (Ctenopharyngodon idella): Focusing on the transcriptional level. Fish. Physiol. Biochem. 2022, 48, 275–284. [Google Scholar] [CrossRef]
- Halder, S.; Macatangay, N.J.; Zerfas, B.L.; Salazar-Chaparro, A.F.; Trader, D.J. Oleic amide derivatives as small molecule stimulators of the human proteasome’s core particle. RSC Med. Chem. 2022, 13, 1077–1081. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Q.; Meng, Y.; Zhao, T.; Mu, C.; Fu, C.; Deng, C.; Feng, J.; Du, S.; Liu, W.; et al. Saturated fatty acids increase LPI to reduce FUNDC1 dimerization and stability and mitochondrial function. EMBO Rep. 2023, 24, e54731. [Google Scholar] [CrossRef]
- Cochrane, D.R.; Cittelly, D.M.; Howe, E.N.; Spoelstra, N.S.; McKinsey, E.L.; LaPara, K.; Elias, A.; Yee, D.; Richer, J.K. MicroRNAs link estrogen receptor alpha status and Dicer levels in breast cancer. Horm. Cancer 2010, 1, 306–319. [Google Scholar] [CrossRef]
- Pandey, D.P.; Picard, D. miR-22 inhibits estrogen signaling by directly targeting the estrogen receptor alpha mRNA. Mol. Cell Biol. 2009, 29, 3783–3790. [Google Scholar] [CrossRef] [PubMed]
- Leivonen, S.K.; Mäkelä, R.; Ostling, P.; Kohonen, P.; Haapa-Paananen, S.; Kleivi, K.; Enerly, E.; Aakula, A.; Hellström, K.; Sahlberg, N.; et al. Protein lysate microarray analysis to identify microRNAs regulating estrogen receptor signaling in breast cancer cell lines. Oncogene 2009, 28, 3926–3936. [Google Scholar] [CrossRef]
- Dunbier, A.K.; Hong, Y.; Masri, S.; Brown, K.A.; Sabnis, G.J.; Palomares, M.R. Progress in aromatase research and identification of key future directions. J. Steroid Biochem. Mol. Biol. 2010, 118, 311–315. [Google Scholar] [CrossRef]
- Fujiwara-Tani, R.; Sasaki, T.; Takagi, T.; Mori, S.; Kishi, S.; Nishiguchi, Y.; Ohmori, H.; Fujii, K.; Kuniyasu, H. Gemcitabine Resistance in Pancreatic Ductal Carcinoma Cell Lines Stems from Reprogramming of Energy Metabolism. Int. J. Mol. Sci. 2022, 23, 7824. [Google Scholar] [CrossRef]
- Takagi, T.; Fujiwara-Tani, R.; Mori, S.; Kishi, S.; Nishiguchi, Y.; Sasaki, T.; Ogata, R.; Ikemoto, A.; Sasaki, R.; Ohmori, H.; et al. Lauric Acid Overcomes Hypoxia-Induced Gemcitabine Chemoresistance in Pancreatic Ductal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 7506. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, M.; Xu, F.; Jiang, S. Wnt signaling in breast cancer: Biological mechanisms, challenges and opportunities. Mol. Cancer 2020, 19, 165. [Google Scholar] [CrossRef]
- Hinder, L.M.; Figueroa-Romero, C.; Pacut, C.; Hong, Y.; Vivekanandan-Giri, A.; Pennathur, S.; Feldman, E.L. Long-chain acyl coenzyme A synthetase 1 overexpression in primary cultured Schwann cells prevents long chain fatty acid-induced oxidative stress and mitochondrial dysfunction. Antioxid. Redox Signal 2014, 21, 588–600. [Google Scholar] [CrossRef]
- Holloway, G.P.; Fajardo, V.A.; McMeekin, L.; LeBlanc, P.J. Unsaturation of mitochondrial membrane lipids is related to palmitate oxidation in subsarcolemmal and intermyofibrillar mitochondria. J. Membr. Biol. 2012, 245, 165–176. [Google Scholar] [CrossRef]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Santa-María, C.; López-Enríquez, S.; Montserrat-de la Paz, S.; Geniz, I.; Reyes-Quiroz, M.E.; Moreno, M.; Palomares, F.; Sobrino, F.; Alba, G. Update on Anti-Inflammatory Molecular Mechanisms Induced by Oleic Acid. Nutrients 2023, 15, 224. [Google Scholar] [CrossRef]
- Tosti, V.; Bertozzi, B.; Fontana, L. Health Benefits of the Mediterranean Diet: Metabolic and Molecular Mechanisms. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 318–326. [Google Scholar] [CrossRef]
- Berrino, F.; Villarini, A.; Gargano, G.; Krogh, V.; Grioni, S.; Bellegotti, M.; Venturelli, E.; Raimondi, M.; Traina, A.; Zarcone, M.; et al. The Effect of Diet on Breast Cancer Recurrence: The DIANA-5 Randomized Trial. Clin. Cancer Res. 2024, 30, 965–974. [Google Scholar] [CrossRef]
- Creighton, C.J.; Massarweh, S.; Huang, S.; Tsimelzon, A.; Hilsenbeck, S.G.; Osborne, C.K.; Shou, J.; Malorni, L.; Schiff, R. Development of resistance to targeted therapies transforms the clinically associated molecular profile subtype of breast tumor xenografts. Cancer Res. 2008, 68, 7493–7501. [Google Scholar] [CrossRef] [PubMed]
- Khaled, N.; Bidet, Y. New Insights into the Implication of Epigenetic Alterations in the EMT of Triple Negative Breast Cancer. Cancers 2019, 11, 559. [Google Scholar] [CrossRef] [PubMed]
- Kouros-Mehr, H.; Kim, J.W.; Bechis, S.K.; Werb, Z. GATA-3 and the regulation of the mammary luminal cell fate. Curr. Opin. Cell Biol. 2008, 20, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Asselin-Labat, M.L.; Sutherland, K.D.; Barker, H.; Thomas, R.; Shackleton, M.; Forrest, N.C.; Hartley, L.; Robb, L.; Grosveld, F.G.; van der Wees, J.; et al. Gata-3 is an essential regulator of mammary-gland morphogenesis and luminal-cell differentiation. Nat. Cell Biol. 2007, 9, 201–209. [Google Scholar] [CrossRef]
- Yoon, E.C.; Wang, G.; Parkinson, B.; Huo, L.; Peng, Y.; Wang, J.; Salisbury, T.; Wu, Y.; Chen, H.; Albarracin, C.T.; et al. TRPS1, GATA3, and SOX10 expression in triple-negative breast carcinoma. Hum. Pathol. 2022, 125, 97–107. [Google Scholar] [CrossRef]
- Byrne, D.J.; Deb, S.; Takano, E.A.; Fox, S.B. GATA3 expression in triple-negative breast cancers. Histopathology 2017, 71, 63–71. [Google Scholar] [CrossRef]
- Choi, J.; Choi, J.H.; Lee, H.W.; Seo, D.; Lkhagvasuren, G.; Kim, J.W.; Seo, S.B.; Lee, K.; Lee, K.H. KPNA3 promotes epithelial-mesenchymal transition by regulating TGF-β and AKT signaling pathways in MDA-MB-231, a triple-negative breast cancer cell line. BMB Rep. 2023, 56, 120–125. [Google Scholar] [CrossRef]
- Chu, I.M.; Lai, W.C.; Aprelikova, O.; El Touny, L.H.; Kouros-Mehr, H.; Green, J.E. Expression of GATA3 in MDA-MB-231 triple-negative breast cancer cells induces a growth inhibitory response to TGFß. PLoS ONE 2013, 8, e61125. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Altea-Manzano, P.; Doglioni, G.; Liu, Y.; Cuadros, A.M.; Nolan, E.; Fernández-García, J.; Wu, Q.; Planque, M.; Laue, K.J.; Cidre-Aranaz, F.; et al. A palmitate-rich metastatic niche enables metastasis growth via p65 acetylation resulting in pro-metastatic NF-κB signaling. Nat. Cancer 2023, 4, 344–364. [Google Scholar] [CrossRef] [PubMed]
- Terry, A.R.; Nogueira, V.; Rho, H.; Ramakrishnan, G.; Li, J.; Kang, S.; Pathmasiri, K.C.; Bhat, S.A.; Jiang, L.; Kuchay, S.; et al. CD36 maintains lipid homeostasis via selective uptake of monounsaturated fatty acids during matrix detachment and tumor progression. Cell Metab. 2023, 35, 2060–2076.e9. [Google Scholar] [CrossRef]
- Kuniyasu, H.; Luo, Y.; Fujii, K.; Sasahira, T.; Moriwaka, Y.; Tatsumoto, N.; Sasaki, T.; Yamashita, Y.; Ohmori, H. CD10 enhances metastasis of colorectal cancer by abrogating the anti-tumoural effect of methionine-enkephalin in the liver. Gut 2010, 59, 348–356. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LCFA | IC50 (μM) | MMP (%) * | MtVol (%) * | MtROS (%) * | Basal OCR (%) * | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| MCF7 | MB231 | MCF7 | MB231 | MCF7 | MB231 | MCF7 | MB231 | MCF7 | MB231 | |
| LA | 78 | 129 | 77 | 42 | 65 | 21 | 175 | 140 | NT | NT |
| EA | 198 | NA | 76 | 40 | 37 | 20 | 156 | 50 | NT | NT |
| OA | 192 | 198 | 108 | 60 | 52 | 22 | 130 | 83 | 35 | 67 |
| PA | 224 | 58 | 82 | 102 | 17 | 34 | 138.5 | 105 | 30 | 61 |
| LNA | 138 | 176 | 92 | 80 | 76 | 140 | 137.5 | 95 | NT | NT |
| RT-PCR Primers | |||
|---|---|---|---|
| Gene | ID | Upper (5′ to 3′) | Lower (5′ to 3′) |
| ACTB | NM_001101.3 | ggacttcgagcaagagatgg | agcactgtgttggcgtacag |
| CD24 | BC064619.1 | atgggcagagcaatggtgg | ccacgaagagactggctgtt |
| OCT3 | BC117437.1 | gaaggatgtggtccgagtgt | gtgaagtgagggctcccata |
| CDH1 | BC146662.1 | cgtcctgggcagagtgaatt | gctctgtcaccttcagccat |
| CK18 | NM_000224.3 | ccgcatcgttctgcagattg | tctgactcaaggtgcagcag |
| c-Myc | NM_002467.4 | ttcgggtagtggaaaaccag | cagcagctcgaatttcttcc |
| PGC-1α | BC156323.1 | gtgaagaccagcctctttgc | aatccgtcttcatccacagg |
| ESR1 | JF810888.1 | gctccgcaaatgctacgaag | agatctccaccatgccctct |
| Antibodies | |||
| Target | RRID | Company | |
| ERα | AB_2617128 | Cell Signaling Technology, Danvers, MA, USA | |
| GAPDH | AB_2107448 | Abcam, Waltham, MA, USA | |
| TBP | − * | Abcam, Waltham, MA, USA | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogata, R.; Luo, Y.; Fujiwara-Tani, R.; Sasaki, R.; Ikemoto, A.; Maehana, K.; Sasaki, A.; Sasaki, T.; Fujii, K.; Ohmori, H.; et al. Long-Chain Fatty Acids Alter Estrogen Receptor Expression in Breast Cancer Cells. Int. J. Mol. Sci. 2025, 26, 6722. https://doi.org/10.3390/ijms26146722
Ogata R, Luo Y, Fujiwara-Tani R, Sasaki R, Ikemoto A, Maehana K, Sasaki A, Sasaki T, Fujii K, Ohmori H, et al. Long-Chain Fatty Acids Alter Estrogen Receptor Expression in Breast Cancer Cells. International Journal of Molecular Sciences. 2025; 26(14):6722. https://doi.org/10.3390/ijms26146722
Chicago/Turabian StyleOgata, Ruiko, Yi Luo, Rina Fujiwara-Tani, Rika Sasaki, Ayaka Ikemoto, Kaho Maehana, Ayaka Sasaki, Takamitsu Sasaki, Kiyomu Fujii, Hitoshi Ohmori, and et al. 2025. "Long-Chain Fatty Acids Alter Estrogen Receptor Expression in Breast Cancer Cells" International Journal of Molecular Sciences 26, no. 14: 6722. https://doi.org/10.3390/ijms26146722
APA StyleOgata, R., Luo, Y., Fujiwara-Tani, R., Sasaki, R., Ikemoto, A., Maehana, K., Sasaki, A., Sasaki, T., Fujii, K., Ohmori, H., & Kuniyasu, H. (2025). Long-Chain Fatty Acids Alter Estrogen Receptor Expression in Breast Cancer Cells. International Journal of Molecular Sciences, 26(14), 6722. https://doi.org/10.3390/ijms26146722