
BRCA2 Pre-mRNA Differential 5′ Splicing: A Rescue of Functional Protein Properties from Pathogenic Gene Variants and a Lifeline for Fanconi Anemia D1 Patients

 ,
,  , and
, and
Abstract
1. Introduction
2. Specific Roles and Issues of BRCA2 in Fanconi Anemia
3. BRCA2 Variant 5′ Splicing Can Rescue DNA Repair Functions
4. BRCA2 Variant 5′ Splicing—Buying Survival with Cancer Risk
5. Recurrent Cancer Signatures but Variable Organ Manifestation for the Same PVs—A Paradox of BRCA2 Variant 5′ Splicing
6. Implications of BRCA2 Differential 5′ Splicing in Non-FA Tissues
7. Summary, Conclusions, and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taylor, A.M.R.; Rothblum-Oviatt, C.; Ellis, N.A.; Hickson, I.D.; Meyer, S.; Crawford, T.O.; Smogorzewska, A.; Pietrucha, B.; Weemaes, C.; Stewart, G.S. Chromosome instability syndromes. Nat. Rev. Dis. Primers 2019, 5, 64. [Google Scholar] [CrossRef]
- Kuehl, J.; Xue, Y.; Yuan, F.; Ramanagoudr-Bhojappa, R.; Pickel, S.; Kalb, R.; Chandrasekharappa, S.C.; Wang, W.; Zhang, Y.; Schindler, D. Genetic inactivation of FAAP100 causes Fanconi anemia due to disruption of the monoubiquitin ligase core complex. J. Clin. Investig. 2025, 135, e187323. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Sarangi, P.; D’Andrea, A.D. The Fanconi anaemia pathway: New players and new functions. Nat. Rev. Mol. Cell Biol. 2016, 17, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef]
- Mehta, P.A.; Ebens, C. Fanconi Anemia. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Foulkes, W.D. Inherited susceptibility to common cancers. N. Engl. J. Med. 2008, 359, 2143–2153. [Google Scholar] [CrossRef]
- Kratz, C.P.; Smirnov, D.; Autry, R.; Jager, N.; Waszak, S.M.; Grosshennig, A.; Berutti, R.; Wendorff, M.; Hainaut, P.; Pfister, S.M.; et al. Heterozygous BRCA1 and BRCA2 and Mismatch Repair Gene Pathogenic Variants in Children and Adolescents with Cancer. J. Natl. Cancer Inst. 2022, 114, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Woodward, E.R.; Meyer, S. Fanconi Anaemia, Childhood Cancer and the BRCA Genes. Genes 2021, 12, 1520. [Google Scholar] [CrossRef]
- Howlett, N.G.; Taniguchi, T.; Olson, S.; Cox, B.; Waisfisz, Q.; De Die-Smulders, C.; Persky, N.; Grompe, M.; Joenje, H.; Pals, G.; et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science 2002, 297, 606–609. [Google Scholar] [CrossRef]
- Radulovic, I.; Schundeln, M.M.; Muller, L.; Ptok, J.; Honisch, E.; Niederacher, D.; Wiek, C.; Scheckenbach, K.; Leblanc, T.; Larcher, L.; et al. A novel cancer risk prediction score for the natural course of FA patients with biallelic BRCA2/FANCD1 mutations. Hum. Mol. Genet. 2023, 32, 1836–1849. [Google Scholar] [CrossRef]
- Meyer, S.; Tischkowitz, M.; Chandler, K.; Gillespie, A.; Birch, J.M.; Evans, D.G. Fanconi anaemia, BRCA2 mutations and childhood cancer: A developmental perspective from clinical and epidemiological observations with implications for genetic counselling. J. Med. Genet. 2014, 51, 71–75. [Google Scholar] [CrossRef]
- McReynolds, L.J.; Biswas, K.; Giri, N.; Sharan, S.K.; Alter, B.P. Genotype-cancer association in patients with Fanconi anemia due to pathogenic variants in FANCD1 (BRCA2) or FANCN (PALB2). Cancer Genet. 2021, 258–259, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Dutzmann, C.M.; Spix, C.; Popp, I.; Kaiser, M.; Erdmann, F.; Erlacher, M.; Dork, T.; Schindler, D.; Kalb, R.; Kratz, C.P. Cancer in Children with Fanconi Anemia and Ataxia-Telangiectasia-A Nationwide Register-Based Cohort Study in Germany. J. Clin. Oncol. 2022, 40, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Evers, B.; Jonkers, J. Mouse models of BRCA1 and BRCA2 deficiency: Past lessons, current understanding and future prospects. Oncogene 2006, 25, 5885–5897. [Google Scholar] [CrossRef] [PubMed]
- Sharan, S.K.; Morimatsu, M.; Albrecht, U.; Lim, D.S.; Regel, E.; Dinh, C.; Sands, A.; Eichele, G.; Hasty, P.; Bradley, A. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2. Nature 1997, 386, 804–810. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Soper, E.R.; Odgis, J.A.; Cullina, S.; Bobo, D.; Moscati, A.; Rodriguez, J.E.; Team, C.G.; Regeneron Genetics, C.; Loos, R.J.F.; et al. Exome sequencing reveals a high prevalence of BRCA1 and BRCA2 founder variants in a diverse population-based biobank. Genome Med. 2019, 12, 2. [Google Scholar] [CrossRef]
- Risch, H.A.; McLaughlin, J.R.; Cole, D.E.; Rosen, B.; Bradley, L.; Fan, I.; Tang, J.; Li, S.; Zhang, S.; Shaw, P.A.; et al. Population BRCA1 and BRCA2 mutation frequencies and cancer penetrances: A kin-cohort study in Ontario, Canada. J. Natl. Cancer Inst. 2006, 98, 1694–1706. [Google Scholar] [CrossRef]
- Gulbis, B.; Eleftheriou, A.; Angastiniotis, M.; Ball, S.; Surralles, J.; Castella, M.; Heimpel, H.; Hill, A.; Corrons, J.L. Epidemiology of rare anaemias in Europe. Adv. Exp. Med. Biol. 2010, 686, 375–396. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. BRCA2 is required for homology-directed repair of chromosomal breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef]
- Jensen, R.B.; Carreira, A.; Kowalczykowski, S.C. Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 2010, 467, 678–683. [Google Scholar] [CrossRef]
- Andreassen, P.R.; Seo, J.; Wiek, C.; Hanenberg, H. Understanding BRCA2 Function as a Tumor Suppressor Based on Domain-Specific Activities in DNA Damage Responses. Genes 2021, 12, 1034. [Google Scholar] [CrossRef]
- Shahid, T.; Soroka, J.; Kong, E.; Malivert, L.; McIlwraith, M.J.; Pape, T.; West, S.C.; Zhang, X. Structure and mechanism of action of the BRCA2 breast cancer tumor suppressor. Nat. Struct. Mol. Biol. 2014, 21, 962–968. [Google Scholar] [CrossRef] [PubMed]
- Xia, B.; Sheng, Q.; Nakanishi, K.; Ohashi, A.; Wu, J.; Christ, N.; Liu, X.; Jasin, M.; Couch, F.J.; Livingston, D.M. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006, 22, 719–729. [Google Scholar] [CrossRef]
- Oliver, A.W.; Swift, S.; Lord, C.J.; Ashworth, A.; Pearl, L.H. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009, 10, 990–996. [Google Scholar] [CrossRef]
- Marzio, A.; Kurz, E.; Sahni, J.M.; Di Feo, G.; Puccini, J.; Jiang, S.; Hirsch, C.A.; Arbini, A.A.; Wu, W.L.; Pass, H.I.; et al. EMSY inhibits homologous recombination repair and the interferon response, promoting lung cancer immune evasion. Cell 2022, 185, 169–183.e19. [Google Scholar] [CrossRef] [PubMed]
- Hughes-Davies, L.; Huntsman, D.; Ruas, M.; Fuks, F.; Bye, J.; Chin, S.F.; Milner, J.; Brown, L.A.; Hsu, F.; Gilks, B.; et al. EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer. Cell 2003, 115, 523–535. [Google Scholar] [CrossRef] [PubMed]
- Le, H.P.; Heyer, W.D.; Liu, J. Guardians of the Genome: BRCA2 and Its Partners. Genes 2021, 12, 1229. [Google Scholar] [CrossRef]
- Julien, M.; Miron, S.; Carreira, A.; Theillet, F.X.; Zinn-Justin, S. 1H, 13C and 15N backbone resonance assignment of the human BRCA2 N-terminal region. Biomol. NMR Assign. 2020, 14, 79–85. [Google Scholar] [CrossRef]
- Venkitaraman, A.R. How do mutations affecting the breast cancer genes BRCA1 and BRCA2 cause cancer susceptibility? DNA Repair 2019, 81, 102668. [Google Scholar] [CrossRef]
- Sidhu, A.; Grosbart, M.; Sanchez, H.; Verhagen, B.; van der Zon, N.L.L.; Ristic, D.; van Rossum-Fikkert, S.E.; Wyman, C. Conformational flexibility and oligomerization of BRCA2 regions induced by RAD51 interaction. Nucleic Acids Res. 2020, 48, 9649–9659. [Google Scholar] [CrossRef]
- von Nicolai, C.; Ehlen, A.; Martin, C.; Zhang, X.; Carreira, A. A second DNA binding site in human BRCA2 promotes homologous recombination. Nat. Commun. 2016, 7, 12813. [Google Scholar] [CrossRef]
- Gelli, E.; Colombo, M.; Pinto, A.M.; De Vecchi, G.; Foglia, C.; Amitrano, S.; Morbidoni, V.; Imperatore, V.; Manoukian, S.; Baldassarri, M.; et al. Usefulness and Limitations of Comprehensive Characterization of mRNA Splicing Profiles in the Definition of the Clinical Relevance of BRCA1/2 Variants of Uncertain Significance. Cancers 2019, 11, 295. [Google Scholar] [CrossRef] [PubMed]
- Friebel, T.M.; Andrulis, I.L.; Balmana, J.; Blanco, A.M.; Couch, F.J.; Daly, M.B.; Domchek, S.M.; Easton, D.F.; Foulkes, W.D.; Ganz, P.A.; et al. BRCA1 and BRCA2 pathogenic sequence variants in women of African origin or ancestry. Hum. Mutat. 2019, 40, 1781–1796. [Google Scholar] [CrossRef]
- Laitman, Y.; Friebel, T.M.; Yannoukakos, D.; Fostira, F.; Konstantopoulou, I.; Figlioli, G.; Bonanni, B.; Manoukian, S.; Zuradelli, M.; Tondini, C.; et al. The spectrum of BRCA1 and BRCA2 pathogenic sequence variants in Middle Eastern, North African, and South European countries. Hum. Mutat. 2019, 40, e1–e23. [Google Scholar] [CrossRef] [PubMed]
- Neveling, K.; Endt, D.; Hoehn, H.; Schindler, D. Genotype-phenotype correlations in Fanconi anemia. Mutat. Res. 2009, 668, 73–91. [Google Scholar] [CrossRef]
- Biswas, K.; Das, R.; Alter, B.P.; Kuznetsov, S.G.; Stauffer, S.; North, S.L.; Burkett, S.; Brody, L.C.; Meyer, S.; Byrd, R.A.; et al. A comprehensive functional characterization of BRCA2 variants associated with Fanconi anemia using mouse ES cell-based assay. Blood 2011, 118, 2430–2442. [Google Scholar] [CrossRef]
- Thirthagiri, E.; Klarmann, K.D.; Shukla, A.K.; Southon, E.; Biswas, K.; Martin, B.K.; North, S.L.; Magidson, V.; Burkett, S.; Haines, D.C.; et al. BRCA2 minor transcript lacking exons 4–7 supports viability in mice and may account for survival of humans with a pathogenic biallelic mutation. Hum. Mol. Genet. 2016, 25, 1934–1945. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Fergusson, W.D.; Oostra, A.B.; Medhurst, A.L.; Waisfisz, Q.; de Winter, J.P.; Chen, F.; Carr, T.F.; Clayton-Smith, J.; Clancy, T.; et al. A cross-linker-sensitive myeloid leukemia cell line from a 2-year-old boy with severe Fanconi anemia and biallelic FANCD1/BRCA2 mutations. Genes. Chromosomes Cancer 2005, 42, 404–415. [Google Scholar] [CrossRef]
- Meyer, S.; Stevens, A.; Paredes, R.; Schneider, M.; Walker, M.J.; Williamson, A.J.K.; Gonzalez-Sanchez, M.B.; Smetsers, S.; Dalal, V.; Teng, H.Y.; et al. Acquired cross-linker resistance associated with a novel spliced BRCA2 protein variant for molecular phenotyping of BRCA2 disruption. Cell Death Dis. 2017, 8, e2875. [Google Scholar] [CrossRef]
- Radulovic, I.; Kuechler, A.; Schundeln, M.M.; Paulussen, M.; von Neuhoff, N.; Reinhardt, D.; Hanenberg, H. A homozygous nonsense mutation early in exon 5 of BRCA2 is associated with very severe Fanconi anemia. Eur. J. Med. Genet. 2021, 64, 104260. [Google Scholar] [CrossRef]
- Nix, P.; Mundt, E.; Coffee, B.; Goossen, E.; Warf, B.M.; Brown, K.; Bowles, K.; Roa, B. Interpretation of BRCA2 Splicing Variants: A Case Series of Challenging Variant Interpretations and the Importance of Functional RNA Analysis. Fam. Cancer 2022, 21, 7–19. [Google Scholar] [CrossRef]
- Fackenthal, J.D.; Yoshimatsu, T.; Zhang, B.; de Garibay, G.R.; Colombo, M.; De Vecchi, G.; Ayoub, S.C.; Lal, K.; Olopade, O.I.; Vega, A.; et al. Naturally occurring BRCA2 alternative mRNA splicing events in clinically relevant samples. J. Med. Genet. 2016, 53, 548–558. [Google Scholar] [CrossRef]
- Mahdi, A.H.; Huo, Y.; Chen, Y.; Selenica, P.; Sharma, A.; Merritt, E.; Barnard, N.; Chan, C.; Ganesan, S.; Reis-Filho, J.S.; et al. Loss of the BRCA1-PALB2 interaction accelerates p53-associated tumor development in mice. Genes. Dis. 2022, 9, 807–813. [Google Scholar] [CrossRef]
- Lin, H.R.; Ting, N.S.; Qin, J.; Lee, W.H. M phase-specific phosphorylation of BRCA2 by Polo-like kinase 1 correlates with the dissociation of the BRCA2-P/CAF complex. J. Biol. Chem. 2003, 278, 35979–35987. [Google Scholar] [CrossRef] [PubMed]
- Ehlen, A.; Martin, C.; Miron, S.; Julien, M.; Theillet, F.X.; Ropars, V.; Sessa, G.; Beaurepere, R.; Boucherit, V.; Duchambon, P.; et al. Proper chromosome alignment depends on BRCA2 phosphorylation by PLK1. Nat. Commun. 2020, 11, 1819. [Google Scholar] [CrossRef]
- Takaoka, M.; Saito, H.; Takenaka, K.; Miki, Y.; Nakanishi, A. BRCA2 phosphorylated by PLK1 moves to the midbody to regulate cytokinesis mediated by nonmuscle myosin IIC. Cancer Res. 2014, 74, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Yata, K.; Bleuyard, J.Y.; Nakato, R.; Ralf, C.; Katou, Y.; Schwab, R.A.; Niedzwiedz, W.; Shirahige, K.; Esashi, F. BRCA2 coordinates the activities of cell-cycle kinases to promote genome stability. Cell Rep. 2014, 7, 1547–1559. [Google Scholar] [CrossRef]
- Alter, B.P.; Rosenberg, P.S.; Brody, L.C. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J. Med. Genet. 2007, 44, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bennett, L.M.; McAllister, K.A.; Blackshear, P.E.; Malphurs, J.; Goulding, G.; Collins, N.K.; Ward, T.; Bunch, D.O.; Eddy, E.M.; Davis, B.J.; et al. BRCA2-null embryonic survival is prolonged on the BALB/c genetic background. Mol. Carcinog. 2000, 28, 174–183. [Google Scholar] [CrossRef]
- Skoulidis, F.; Cassidy, L.D.; Pisupati, V.; Jonasson, J.G.; Bjarnason, H.; Eyfjord, J.E.; Karreth, F.A.; Lim, M.; Barber, L.M.; Clatworthy, S.A.; et al. Germline Brca2 heterozygosity promotes Kras(G12D)-driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell 2010, 18, 499–509. [Google Scholar] [CrossRef]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Z.; Peng, J.; Yuan, C.; Yan, S.; Yang, N.; Li, P.; Kong, B. The splicing factor SNRPB promotes ovarian cancer progression through regulating aberrant exon skipping of POLA1 and BRCA2. Oncogene 2023, 42, 2386–2401. [Google Scholar] [CrossRef] [PubMed]
- Sanz, D.J.; Acedo, A.; Infante, M.; Duran, M.; Perez-Cabornero, L.; Esteban-Cardenosa, E.; Lastra, E.; Pagani, F.; Miner, C.; Velasco, E.A. A high proportion of DNA variants of BRCA1 and BRCA2 is associated with aberrant splicing in breast/ovarian cancer patients. Clin. Cancer Res. 2010, 16, 1957–1967. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Karlan, B.Y.; Agarwal, M.K.; Higgins, J.; Friedman, C.; Villegas, E.; Jacquemont, C.; Farrugia, D.J.; Couch, F.J.; et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008, 451, 1116–1120. [Google Scholar] [CrossRef]
- Hadadi, N.; Spiljar, M.; Steinbach, K.; Colakoglu, M.; Chevalier, C.; Salinas, G.; Merkler, D.; Trajkovski, M. Comparative multi-tissue profiling reveals extensive tissue-specificity in transcriptome reprogramming during thermal adaptation. eLife 2022, 11, e78556. [Google Scholar] [CrossRef]
- Suntsova, M.; Gaifullin, N.; Allina, D.; Reshetun, A.; Li, X.; Mendeleeva, L.; Surin, V.; Sergeeva, A.; Spirin, P.; Prassolov, V.; et al. Atlas of RNA sequencing profiles for normal human tissues. Sci. Data 2019, 6, 36. [Google Scholar] [CrossRef]
- Wheeler, E.C.; Martin, B.J.E.; Doyle, W.C.; Neaher, S.; Conway, C.A.; Pitton, C.N.; Gorelov, R.A.; Donahue, M.; Jann, J.C.; Abdel-Wahab, O.; et al. Splicing modulators impair DNA damage response and induce killing of cohesin-mutant MDS and AML. Sci. Transl. Med. 2024, 16, eade2774. [Google Scholar] [CrossRef]
- Li, L.; Biswas, K.; Habib, L.A.; Kuznetsov, S.G.; Hamel, N.; Kirchhoff, T.; Wong, N.; Armel, S.; Chong, G.; Narod, S.A.; et al. Functional redundancy of exon 12 of BRCA2 revealed by a comprehensive analysis of the c.6853A>G (p.I2285V) variant. Hum. Mutat. 2009, 30, 1543–1550. [Google Scholar] [CrossRef]
- Meulemans, L.; Mesman, R.L.S.; Caputo, S.M.; Krieger, S.; Guillaud-Bataille, M.; Caux-Moncoutier, V.; Leone, M.; Boutry-Kryza, N.; Sokolowska, J.; Revillion, F.; et al. Skipping Nonsense to Maintain Function: The Paradigm of BRCA2 Exon 12. Cancer Res. 2020, 80, 1374–1386. [Google Scholar] [CrossRef]
- Mesman, R.L.S.; Calleja, F.; de la Hoya, M.; Devilee, P.; van Asperen, C.J.; Vrieling, H.; Vreeswijk, M.P.G. Alternative mRNA splicing can attenuate the pathogenicity of presumed loss-of-function variants in BRCA2. Genet. Med. 2020, 22, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Meeks, H.D.; Song, H.; Michailidou, K.; Bolla, M.K.; Dennis, J.; Wang, Q.; Barrowdale, D.; Frost, D.; Embrace; McGuffog, L.; et al. BRCA2 Polymorphic Stop Codon K3326X and the Risk of Breast, Prostate, and Ovarian Cancers. J. Natl. Cancer Inst. 2016, 108, djv315. [Google Scholar] [CrossRef]
- Sherlock, M.E.; Baquero Galvis, L.; Vicens, Q.; Kieft, J.S.; Jagannathan, S. Principles, mechanisms, and biological implications of translation termination-reinitiation. RNA 2023, 29, 865–884. [Google Scholar] [CrossRef] [PubMed]
- Varon, R.; Dutrannoy, V.; Weikert, G.; Tanzarella, C.; Antoccia, A.; Stockl, L.; Spadoni, E.; Kruger, L.A.; di Masi, A.; Sperling, K.; et al. Mild Nijmegen breakage syndrome phenotype due to alternative splicing. Hum. Mol. Genet. 2006, 15, 679–689. [Google Scholar] [CrossRef] [PubMed]
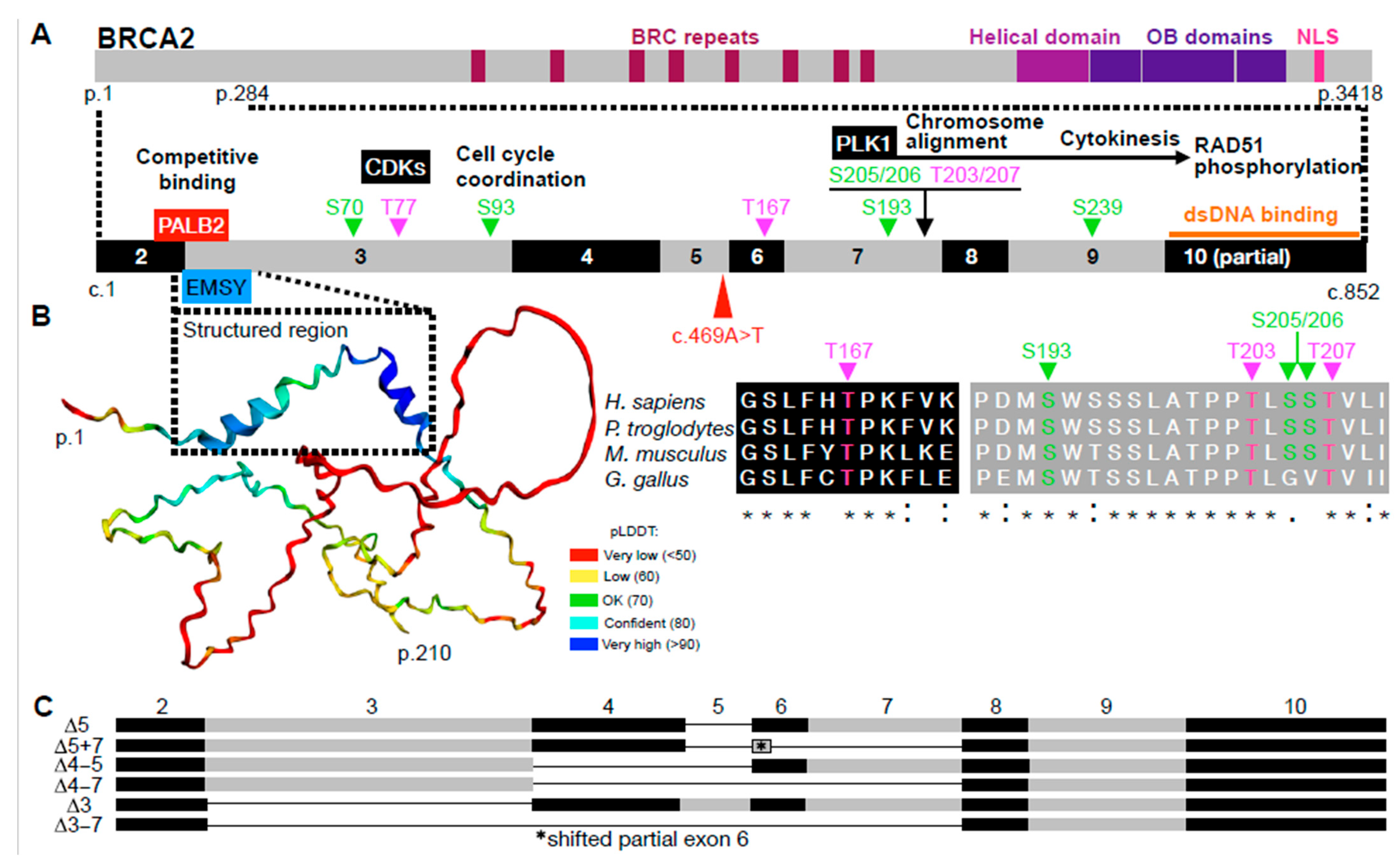
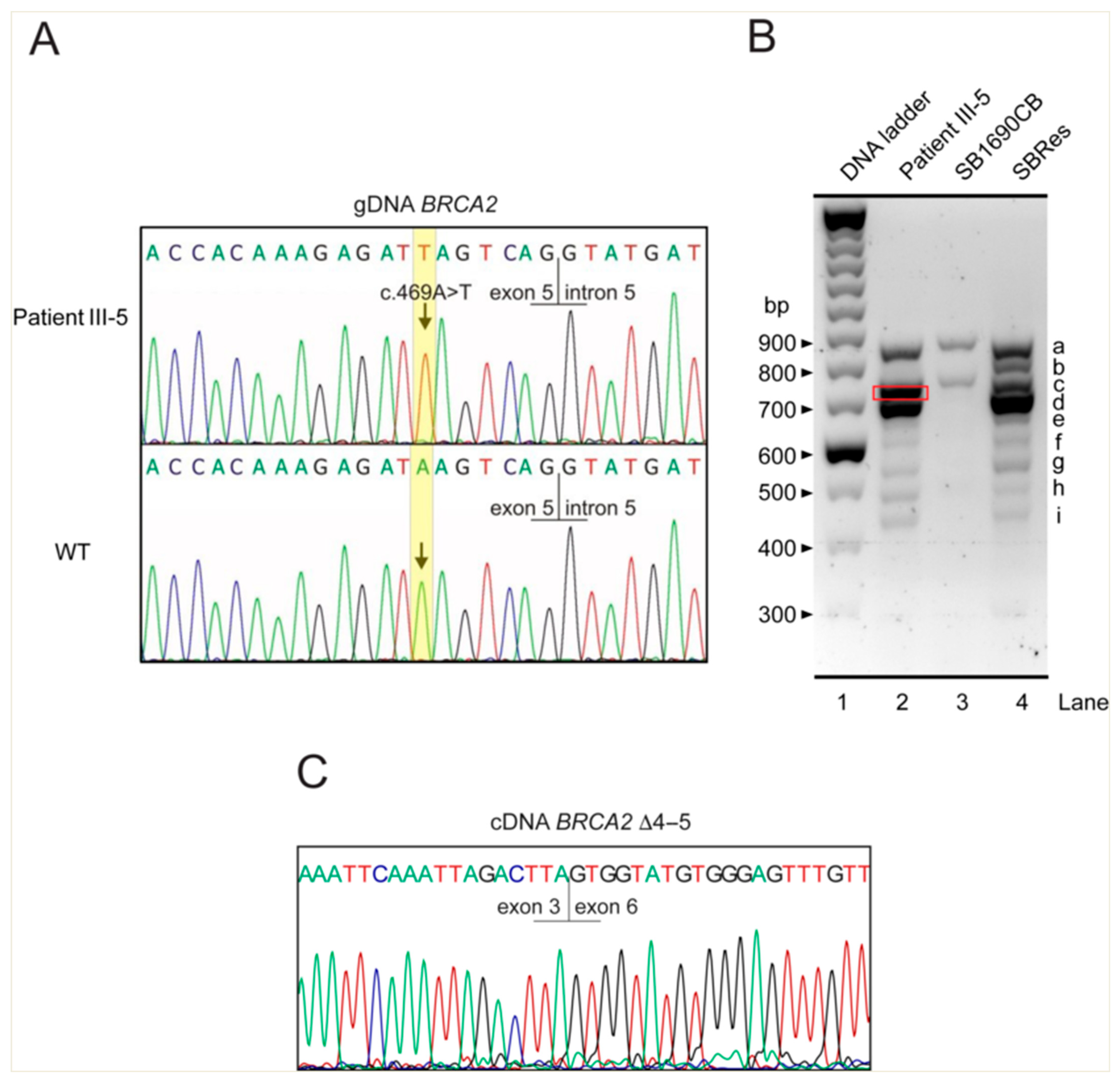
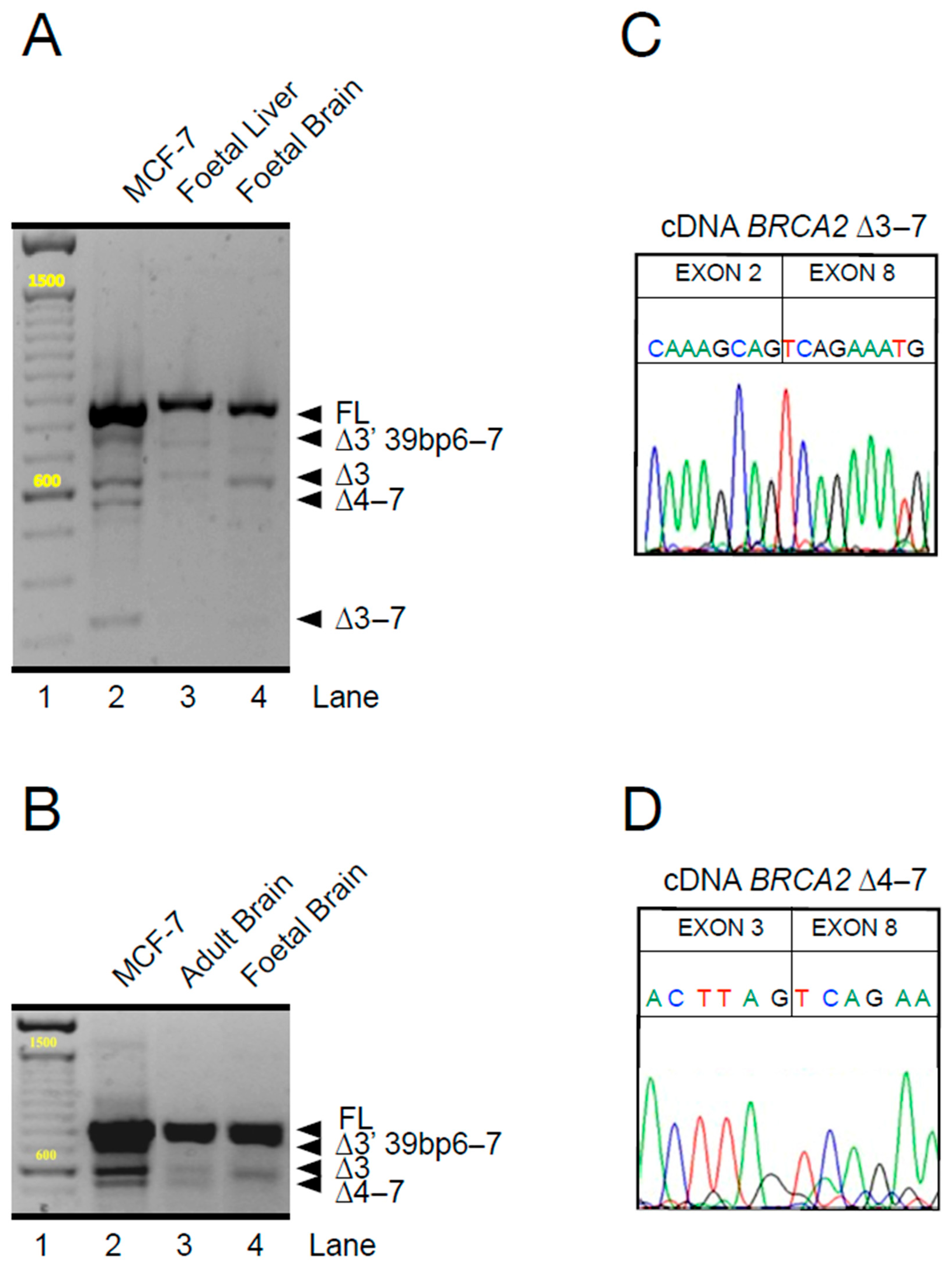

{kind=link}
{kind=link}
{kind=link}
| Cryptic Splice Site (Position) | Exon/Intron (IVS) no. | Predicted Function | Sample Count (Frequency) 1 |
|---|---|---|---|
| c.67+1254 | IVS 2 | acceptor | 3467 (3.8%) |
| c.317-419 | IVS 3 | acceptor | 120 (0.1%) |
| c.320 | 4 | acceptor | 7866 (7.7%) |
| c.425+381 | IVS 4 | acceptor | 433 (0.4%) |
| c.426-208 | IVS 4 | donor | 781 (0.7%) |
| c.426-24 | IVS 4 | acceptor | 145 (0.1%) |
| c.632-187 | IVS 7 | donor | 310 (0.3%) |
| Exon no. | Splice Variant | Length (nt) | Reduction of Transcript Length (nt) | Effect on the Reading Frame |
|---|---|---|---|---|
| 3 | canonical | 249 | − | − |
| − | ∆3 | − | −249 | in frame |
| ∆3–4 | −358 | out of frame | ||
| − | ∆3 + 7 | − | −364 | out of frame |
| − | ∆3 + 5 + 7 | − | −414 | in frame |
| − | ∆3–7 | − | −564 | in frame |
| 4 | canonical | 109 | − | − |
| ∆4 | −109 | out of frame | ||
| − | ∆4–5 | −159 | in frame | |
| − | ∆4–7 | −315 | in frame | |
| 5 | canonical | 50 | − | − |
| − | ∆5 | − | −50 | out of frame |
| − | ∆5 + 7 | − | −165 | in frame |
| − | ∆5–7, insTG | − | −204 | in frame |
| 6 | canonical | 41 | − | − |
| 7 | canonical | 115 | − | − |
| − | ∆7 | − | −115 | out of frame |
| 8 | canonical | 50 | − | − |
| TIS (CDS Position) | Exon No. | Kozak Similarity Score | Reading Frame Phase | Translation Option |
|---|---|---|---|---|
| c.1 (canonical start codon) | 2 | 0.66 | +1 | yes |
| c.323 | 4 | 0.50 | +2 | no, in-frame TGA at c.359 (exon 4) |
| c.370 | 4 | 0.71 | +1 | yes |
| c.441 | 5 | 0.62 | +3 | no, in-frame TAA at c.468 (exon 5) |
| c.574 | 7 | 0.57 | +1 | yes |
| c.638 | 8 | 0.62 | +2 | unlikely 2, TGA at c.686 (exon 9) |
| c.707 | 9 | 0.7 | +2 | no, TGA at c.719 (exon 9) |
| c.710 | 9 | 0.67 | +2 | no, TGA at c.719 (exon 9) |
| c.728 | 9 | 0.62 | +2 | no, TGA at c.749 (exon 9) |
| c.791 | 9 | 0.74 | +2 | unlikely 2, TAA at c.827 (exon 10) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paredes, R.; Batta, K.; Wiseman, D.H.; Gothbi, R.; Dalal, V.; Schmidt, C.K.; Kalb, R.; Meyer, S.; Schindler, D. BRCA2 Pre-mRNA Differential 5′ Splicing: A Rescue of Functional Protein Properties from Pathogenic Gene Variants and a Lifeline for Fanconi Anemia D1 Patients. Int. J. Mol. Sci. 2025, 26, 6694. https://doi.org/10.3390/ijms26146694
Paredes R, Batta K, Wiseman DH, Gothbi R, Dalal V, Schmidt CK, Kalb R, Meyer S, Schindler D. BRCA2 Pre-mRNA Differential 5′ Splicing: A Rescue of Functional Protein Properties from Pathogenic Gene Variants and a Lifeline for Fanconi Anemia D1 Patients. International Journal of Molecular Sciences. 2025; 26(14):6694. https://doi.org/10.3390/ijms26146694
Chicago/Turabian StyleParedes, Roberto, Kiran Batta, Daniel H. Wiseman, Reham Gothbi, Vineet Dalal, Christine K. Schmidt, Reinhard Kalb, Stefan Meyer, and Detlev Schindler. 2025. "BRCA2 Pre-mRNA Differential 5′ Splicing: A Rescue of Functional Protein Properties from Pathogenic Gene Variants and a Lifeline for Fanconi Anemia D1 Patients" International Journal of Molecular Sciences 26, no. 14: 6694. https://doi.org/10.3390/ijms26146694
APA StyleParedes, R., Batta, K., Wiseman, D. H., Gothbi, R., Dalal, V., Schmidt, C. K., Kalb, R., Meyer, S., & Schindler, D. (2025). BRCA2 Pre-mRNA Differential 5′ Splicing: A Rescue of Functional Protein Properties from Pathogenic Gene Variants and a Lifeline for Fanconi Anemia D1 Patients. International Journal of Molecular Sciences, 26(14), 6694. https://doi.org/10.3390/ijms26146694