
Adult-Onset Deletion of CDKL5 in Forebrain Glutamatergic Neurons Impairs Synaptic Integrity and Behavior in Mice
 , ,
, ,  ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Postnatal Cdkl5 Deletion in Forebrain Glutamatergic Neurons, and Body Weight and General Health in Cdkl5flox/Y(Cre+) Mice
2.2. Motor Coordination, Repetitive and Autistic-like Behaviors in Cdkl5flox/Y(Cre+) Mice
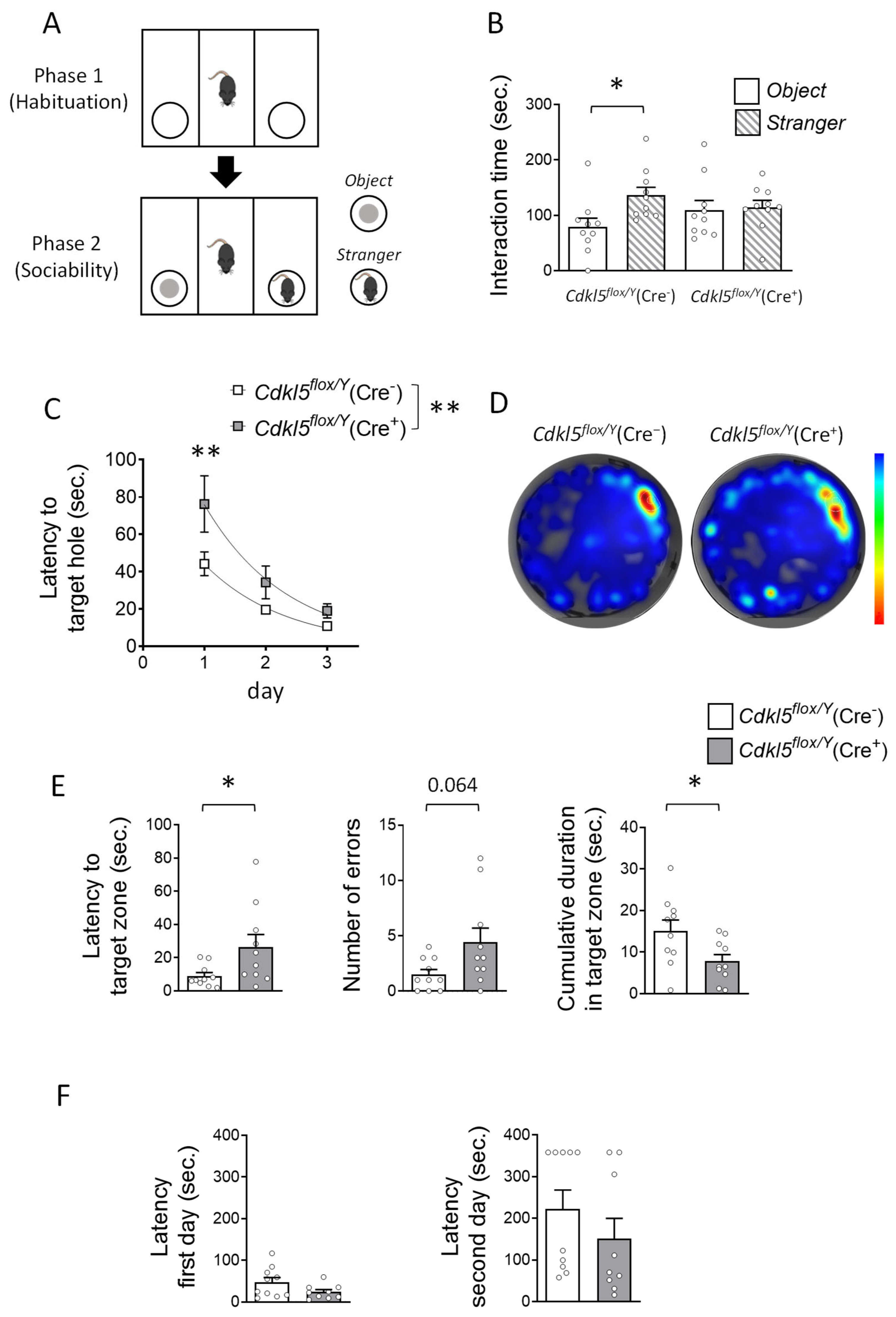
2.3. Social Interaction and Memory Impairments in Cdkl5flox/Y(Cre+) Mice
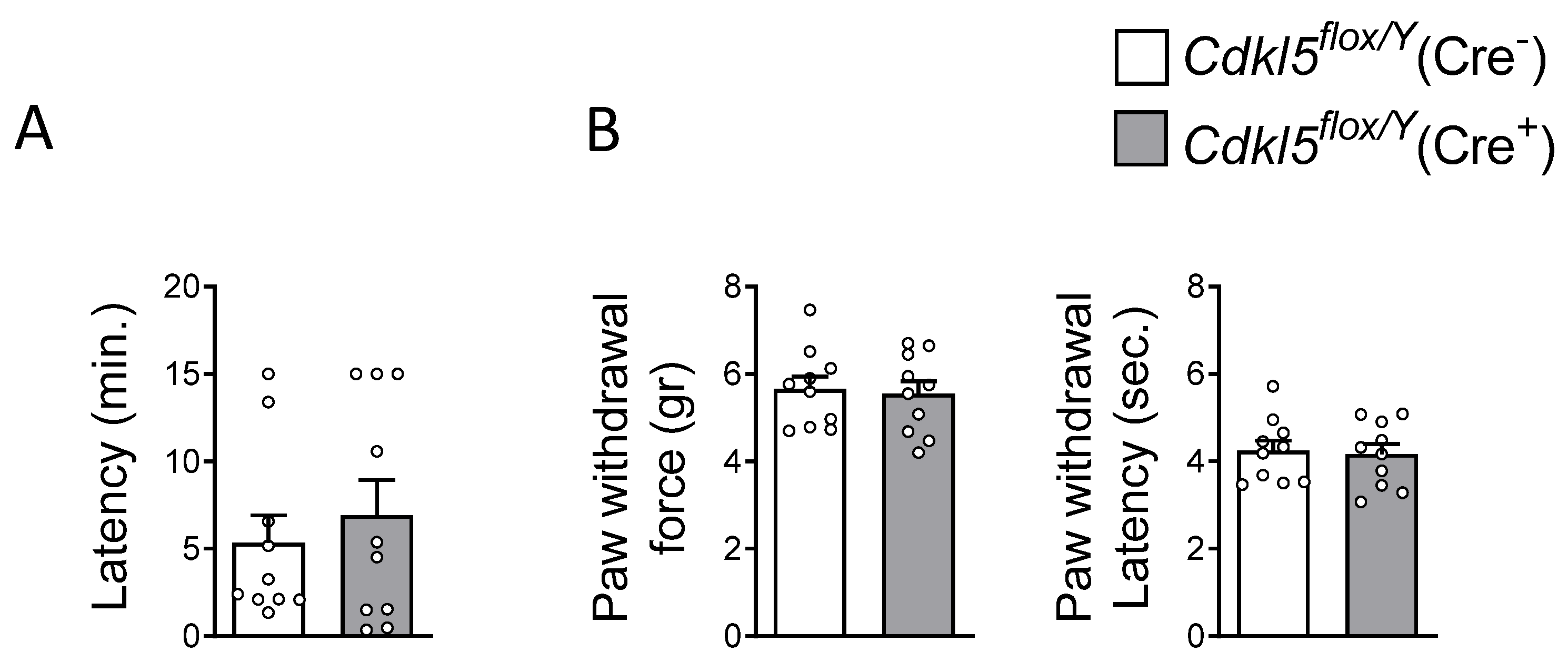
2.4. Olfaction and Pain Perception in Cdkl5flox/Y(Cre+) Mice
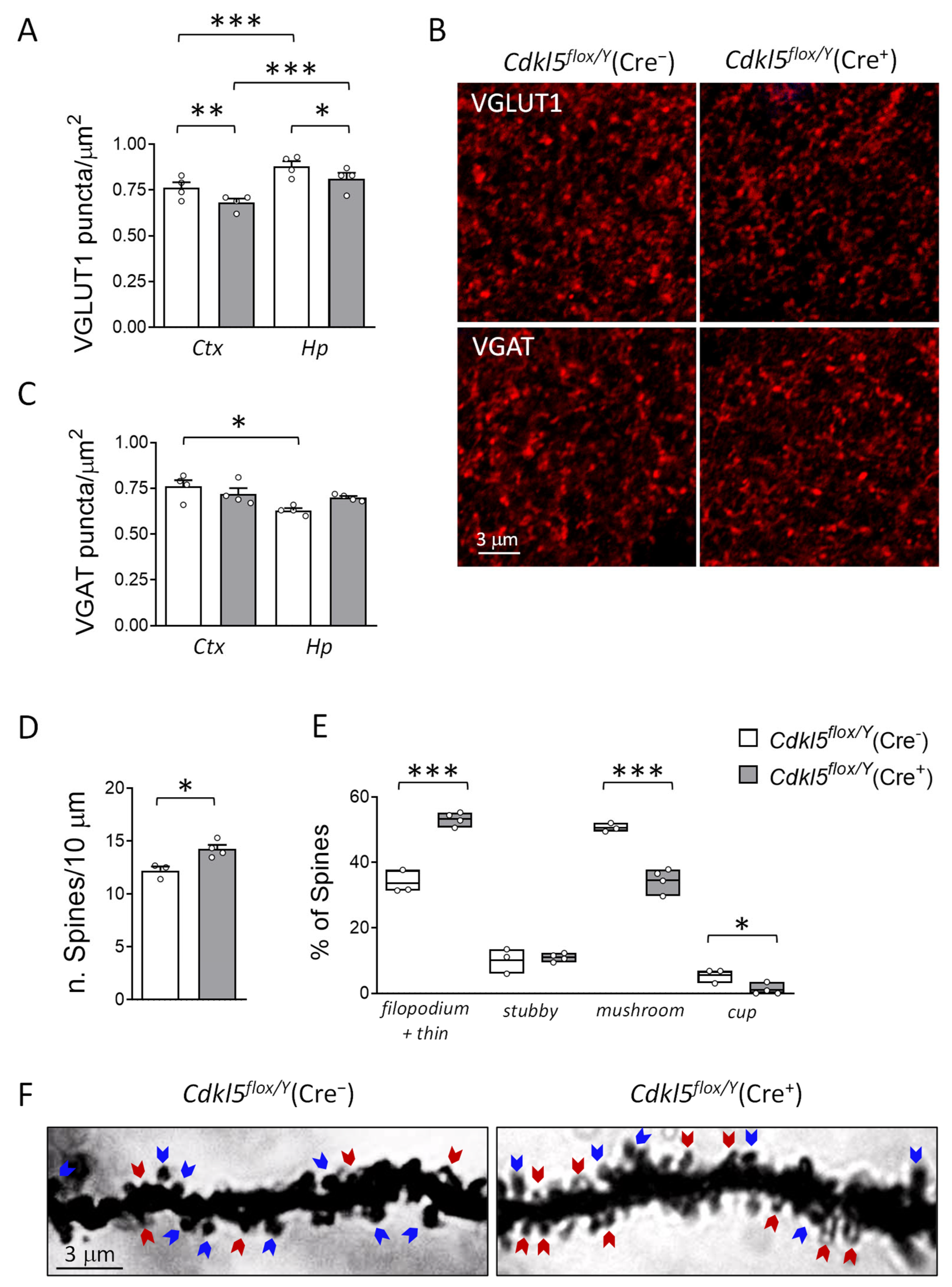
2.5. Brain Connectivity and Hippocampal Dendritic Spine Maturation in Cdkl5flox/Y(Cre+) Mice
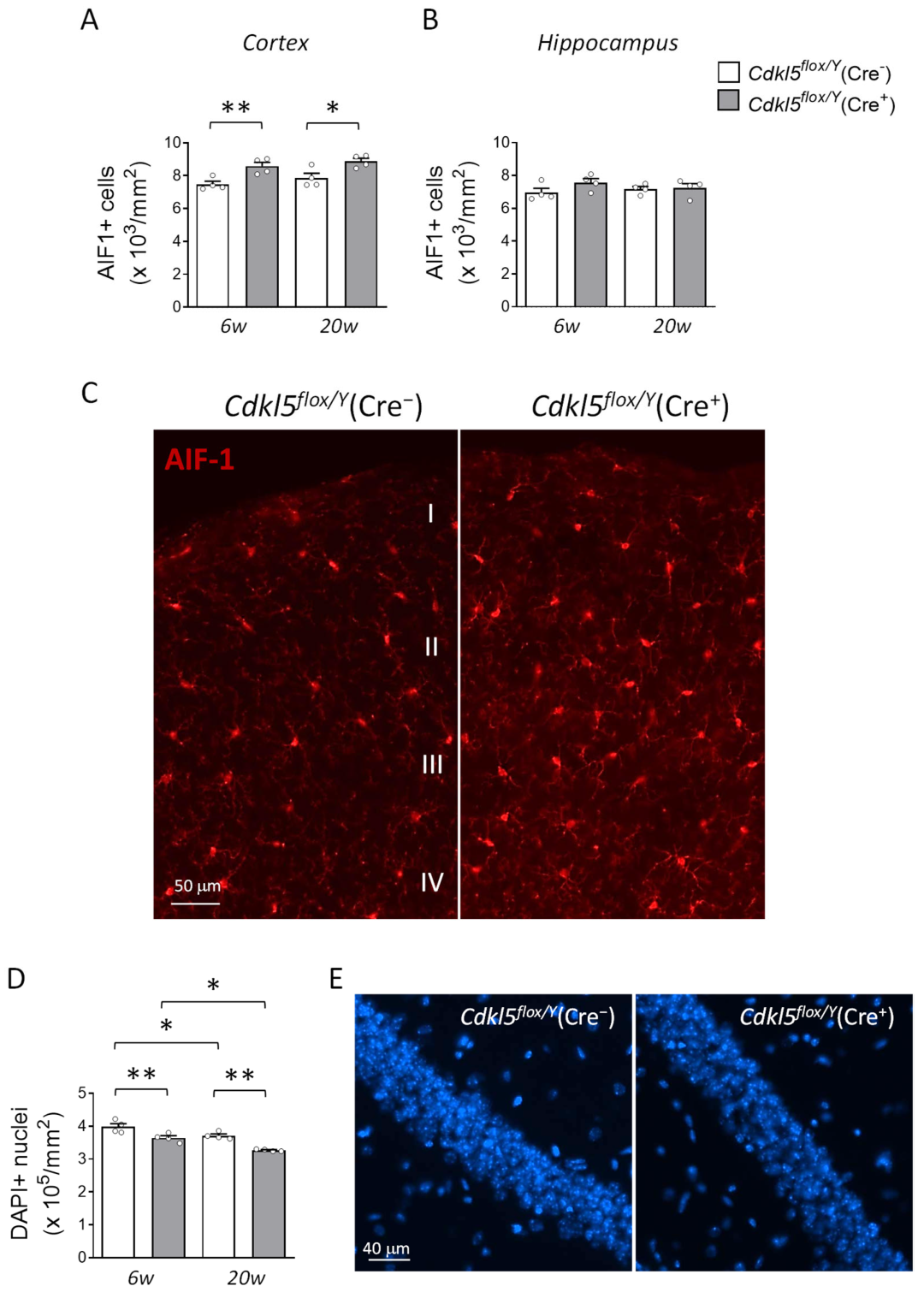
2.6. Microglial Cell Density and Hippocampal Neuronal Survival in the Brains of Cdkl5flox/Y(Cre+) Mice
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Tamoxifen Treatment
4.3. Behavioral Assays
4.3.1. Marble Burying
4.3.2. Hind-Limb Clasping
4.3.3. Accelerating Rotarod Assay
4.3.4. Open Field
4.3.5. Three-Chamber Social Interaction Test
4.3.6. Barnes Maze
4.3.7. Passive Avoidance
4.3.8. Buried-Food Test
4.3.9. Von Frey Filament Test
4.4. Histological and Immunohistochemistry Procedures
4.4.1. Immunofluorescence Staining
4.4.2. Golgi Impregnation Method
4.4.3. In Situ Hybridization (ISH)
4.5. Image Acquisition and Measurements
4.5.1. Quantification of VGAT and VGLUT1 Immunoreactive Puncta
4.5.2. Dendritic Spine Number and Morphology
4.5.3. Cell Density
4.6. Western Blotting
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Evans, J.C.; Archer, H.L.; Colley, J.P.; Ravn, K.; Nielsen, J.B.; Kerr, A.; Williams, E.; Christodoulou, J.; Gecz, J.; Jardine, P.E.; et al. Early onset seizures and Rett-like features associated with mutations in CDKL5. Eur. J. Hum. Genet. 2005, 13, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Scala, E.; Ariani, F.; Mari, F.; Caselli, R.; Pescucci, C.; Longo, I.; Meloni, I.; Giachino, D.; Bruttini, M.; Hayek, G.; et al. CDKL5/STK9 is mutated in Rett syndrome variant with infantile spasms. J. Med. Genet. 2005, 42, 103–107. [Google Scholar] [CrossRef]
- Tao, J.; Van Esch, H.; Hagedorn-Greiwe, M.; Hoffmann, K.; Moser, B.; Raynaud, M.; Sperner, J.; Fryns, J.P.; Schwinger, E.; Gecz, J.; et al. Mutations in the X-linked cyclin-dependent kinase-like 5 (CDKL5/STK9) gene are associated with severe neurodevelopmental retardation. Am. J. Hum. Genet. 2004, 75, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Weaving, L.S.; Christodoulou, J.; Williamson, S.L.; Friend, K.L.; McKenzie, O.L.; Archer, H.; Evans, J.; Clarke, A.; Pelka, G.J.; Tam, P.P.; et al. Mutations of CDKL5 cause a severe neurodevelopmental disorder with infantile spasms and mental retardation. Am. J. Hum. Genet. 2004, 75, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Bahi-Buisson, N.; Villeneuve, N.; Caietta, E.; Jacquette, A.; Maurey, H.; Matthijs, G.; Van Esch, H.; Delahaye, A.; Moncla, A.; Milh, M.; et al. Recurrent mutations in the CDKL5 gene: Genotype-phenotype relationships. Am. J. Med. Genet. A 2012, 158A, 1612–1619. [Google Scholar] [CrossRef]
- Fehr, S.; Wilson, M.; Downs, J.; Williams, S.; Murgia, A.; Sartori, S.; Vecchi, M.; Ho, G.; Polli, R.; Psoni, S.; et al. The CDKL5 disorder is an independent clinical entity associated with early-onset encephalopathy. Eur. J. Hum. Genet. 2013, 21, 266–273. [Google Scholar] [CrossRef]
- MacKay, C.I.; Wong, K.; Demarest, S.T.; Benke, T.A.; Downs, J.; Leonard, H. Exploring genotype-phenotype relationships in the CDKL5 deficiency disorder using an international dataset. Clin. Genet. 2021, 99, 157–165. [Google Scholar] [CrossRef]
- Siri, B.; Varesio, C.; Freri, E.; Darra, F.; Gana, S.; Mei, D.; Porta, F.; Fontana, E.; Galati, G.; Solazzi, R.; et al. CDKL5 deficiency disorder in males: Five new variants and review of the literature. Eur. J. Paediatr. Neurol. 2021, 33, 9–20. [Google Scholar] [CrossRef]
- Haviland, I.; Hector, R.D.; Swanson, L.C.; Verran, A.S.; Sherrill, E.; Frazier, Z.; Denny, A.M.; Lucash, J.; Zhang, B.; Dubbs, H.A.; et al. Deletions in the CDKL5 5′ untranslated region lead to CDKL5 deficiency disorder. Am. J. Med. Genet. A 2025, 197, e63843. [Google Scholar] [CrossRef]
- Montini, E.; Andolfi, G.; Caruso, A.; Buchner, G.; Walpole, S.M.; Mariani, M.; Consalez, G.; Trump, D.; Ballabio, A.; Franco, B. Identification and characterization of a novel serine-threonine kinase gene from the Xp22 region. Genomics 1998, 51, 427–433. [Google Scholar] [CrossRef]
- Kalscheuer, V.M.; Tao, J.; Donnelly, A.; Hollway, G.; Schwinger, E.; Kubart, S.; Menzel, C.; Hoeltzenbein, M.; Tommerup, N.; Eyre, H.; et al. Disruption of the serine/threonine kinase 9 gene causes severe X-linked infantile spasms and mental retardation. Am. J. Hum. Genet. 2003, 72, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.C.; Xiong, Z.Q. Molecular and Synaptic Bases of CDKL5 Disorder. Dev. Neurobiol. 2019, 79, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S.; Sueyoshi, N.; Inazu, T.; Kameshita, I. Cyclin-Dependent Kinase-Like 5 (CDKL5): Possible Cellular Signalling Targets and Involvement in CDKL5 Deficiency Disorder. Neural Plast. 2020, 2020, 6970190. [Google Scholar] [CrossRef]
- Rusconi, L.; Salvatoni, L.; Giudici, L.; Bertani, I.; Kilstrup-Nielsen, C.; Broccoli, V.; Landsberger, N. CDKL5 expression is modulated during neuronal development and its subcellular distribution is tightly regulated by the C-terminal tail. J. Biol. Chem. 2008, 283, 30101–30111. [Google Scholar] [CrossRef] [PubMed]
- Zito, A.; Lee, J.T. Variable expression of MECP2, CDKL5, and FMR1 in the human brain: Implications for gene restorative therapies. Proc. Natl. Acad. Sci. USA 2024, 121, e2312757121. [Google Scholar] [CrossRef] [PubMed]
- Kilstrup-Nielsen, C.; Rusconi, L.; La Montanara, P.; Ciceri, D.; Bergo, A.; Bedogni, F.; Landsberger, N. What we know and would like to know about CDKL5 and its involvement in epileptic encephalopathy. Neural Plast. 2012, 2012, 728267. [Google Scholar] [CrossRef]
- Barbiero, I.; De Rosa, R.; Kilstrup-Nielsen, C. Microtubules: A Key to Understand and Correct Neuronal Defects in CDKL5 Deficiency Disorder? Int. J. Mol. Sci. 2019, 20, 4075. [Google Scholar] [CrossRef]
- De Rosa, R.; Valastro, S.; Cambria, C.; Barbiero, I.; Puricelli, C.; Tramarin, M.; Randi, S.; Bianchi, M.; Antonucci, F.; Kilstrup-Nielsen, C. Loss of CDKL5 Causes Synaptic GABAergic Defects That Can Be Restored with the Neuroactive Steroid Pregnenolone-Methyl-Ether. Int. J. Mol. Sci. 2023, 24, 68. [Google Scholar] [CrossRef]
- Van Bergen, N.J.; Massey, S.; Quigley, A.; Rollo, B.; Harris, A.R.; Kapsa, R.M.I.; Christodoulou, J. CDKL5 deficiency disorder: Molecular insights and mechanisms of pathogenicity to fast-track therapeutic development. Biochem. Soc. Trans. 2022, 50, 1207–1224. [Google Scholar] [CrossRef]
- Kontaxi, C.; Ivanova, D.; Davenport, E.C.; Kind, P.C.; Cousin, M.A. Epilepsy-Related CDKL5 Deficiency Slows Synaptic Vesicle Endocytosis in Central Nerve Terminals. J. Neurosci. 2023, 43, 2002–2020. [Google Scholar] [CrossRef]
- Sun, X.; Wang, T. Research progress on the pathogenesis of CDKL5 pathogenic variants and related encephalopathy. Eur. J. Pediatr. 2023, 182, 3049–3056. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Guo, X.; Xu, C. Revealing the complex role of CDKL5 in developmental epilepsy through a calcium channel related vision. Acta Epileptol. 2024, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Bahi-Buisson, N.; Nectoux, J.; Rosas-Vargas, H.; Milh, M.; Boddaert, N.; Girard, B.; Cances, C.; Ville, D.; Afenjar, A.; Rio, M.; et al. Key clinical features to identify girls with CDKL5 mutations. Brain 2008, 131, 2647–2661. [Google Scholar] [CrossRef] [PubMed]
- Stalpers, X.L.; Spruijt, L.; Yntema, H.G.; Verrips, A. Clinical phenotype of 5 females with a CDKL5 mutation. J. Child Neurol. 2012, 27, 90–93. [Google Scholar] [CrossRef]
- Liang, J.S.; Huang, H.; Wang, J.S.; Lu, J.F. Phenotypic manifestations between male and female children with CDKL5 mutations. Brain Dev. 2019, 41, 783–789. [Google Scholar] [CrossRef]
- Kadam, S.D.; Sullivan, B.J.; Goyal, A.; Blue, M.E.; Smith-Hicks, C. Rett Syndrome and CDKL5 Deficiency Disorder: From Bench to Clinic. Int. J. Mol. Sci. 2019, 20, 5098. [Google Scholar] [CrossRef]
- Demarest, S.; Pestana-Knight, E.M.; Olson, H.E.; Downs, J.; Marsh, E.D.; Kaufmann, W.E.; Partridge, C.A.; Leonard, H.; Gwadry-Sridhar, F.; Frame, K.E.; et al. Severity Assessment in CDKL5 Deficiency Disorder. Pediatr. Neurol. 2019, 97, 38–42. [Google Scholar] [CrossRef]
- Olson, H.E.; Demarest, S.T.; Pestana-Knight, E.M.; Swanson, L.C.; Iqbal, S.; Lal, D.; Leonard, H.; Cross, J.H.; Devinsky, O.; Benke, T.A. Cyclin-Dependent Kinase-Like 5 Deficiency Disorder: Clinical Review. Pediatr. Neurol. 2019, 97, 18–25. [Google Scholar] [CrossRef]
- Jakimiec, M.; Paprocka, J.; Śmigiel, R. CDKL5 Deficiency Disorder—A Complex Epileptic Encephalopathy. Brain Sci. 2020, 10, 107. [Google Scholar] [CrossRef]
- Olson, H.E.; Daniels, C.I.; Haviland, I.; Swanson, L.C.; Greene, C.A.; Denny, A.M.M.; Demarest, S.T.; Pestana-Knight, E.; Zhang, X.; Moosa, A.N.; et al. Current neurologic treatment and emerging therapies in CDKL5 deficiency disorder. J. Neurodev. Disord. 2021, 13, 40. [Google Scholar] [CrossRef]
- Hong, W.; Haviland, I.; Pestana-Knight, E.; Weisenberg, J.L.; Demarest, S.; Marsh, E.D.; Olson, H.E. CDKL5 Deficiency Disorder-Related Epilepsy: A Review of Current and Emerging Treatment. CNS Drugs 2022, 36, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Benke, T.A.; Demarest, S.; Angione, K.; Downs, J.; Leonard, H.; Saldaris, J.; Marsh, E.D.; Olson, H.; Haviland, I. CDKL5 Deficiency Disorder. In GeneReviews; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993; Available online: https://www.ncbi.nlm.nih.gov/books/NBK602610/ (accessed on 7 July 2025).
- Fehr, S.; Downs, J.; Ho, G.; de Klerk, N.; Forbes, D.; Christodoulou, J.; Williams, S.; Leonard, H. Functional abilities in children and adults with the CDKL5 disorder. Am. J. Med. Genet. A 2016, 170, 2860–2869. [Google Scholar] [CrossRef] [PubMed]
- Mangatt, M.; Wong, K.; Anderson, B.; Epstein, A.; Hodgetts, S.; Leonard, H.; Downs, J. Prevalence and onset of comorbidities in the CDKL5 disorder differ from Rett syndrome. Orphanet J. Rare Dis. 2016, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Demarest, S.T.; Olson, H.E.; Moss, A.; Pestana-Knight, E.; Zhang, X.; Parikh, S.; Swanson, L.C.; Riley, K.D.; Bazin, G.A.; Angione, K.; et al. CDKL5 deficiency disorder: Relationship between genotype, epilepsy, cortical visual impairment, and development. Epilepsia 2019, 60, 1733–1742. [Google Scholar] [CrossRef]
- Quintiliani, M.; Ricci, D.; Petrianni, M.; Leone, S.; Orazi, L.; Amore, F.; Gambardella, M.L.; Contaldo, I.; Veredice, C.; Perulli, M.; et al. Cortical Visual Impairment in CDKL5 Deficiency Disorder. Front. Neurol. 2021, 12, 805745. [Google Scholar] [CrossRef]
- Tascini, G.; Dell’Isola, G.B.; Mencaroni, E.; Di Cara, G.; Striano, P.; Verrotti, A. Sleep Disorders in Rett Syndrome and Rett-Related Disorders: A Narrative Review. Front. Neurol. 2022, 13, 817195. [Google Scholar] [CrossRef]
- Leonard, H.; Downs, J.; Benke, T.A.; Swanson, L.; Olson, H.; Demarest, S. CDKL5 deficiency disorder: Clinical features, diagnosis, and management. Lancet Neurol. 2022, 21, 563–576. [Google Scholar] [CrossRef]
- Hagebeuk, E.E.O.; Smits, A.; de Weerd, A. Long time polysomnographic sleep and breathing evaluations in children with CDKL5 deficiency disorder. Sleep Med. 2023, 103, 173–179. [Google Scholar] [CrossRef]
- Borghi, E.; Xynomilakis, O.; Ottaviano, E.; Ceccarani, C.; Vigano, I.; Tognini, P.; Vignoli, A. Gut microbiota profile in CDKL5 deficiency disorder patients. Sci. Rep. 2024, 14, 7376. [Google Scholar] [CrossRef]
- Dell’Isola, G.B.; Perinelli, M.G.; Frulli, A.; D’Onofrio, G.; Fattorusso, A.; Siciliano, M.; Ferrara, P.; Striano, P.; Verrotti, A. Exploring neurodevelopment in CDKL5 deficiency disorder: Current insights and future directions. Epilepsy Behav. 2025, 171, 110504. [Google Scholar] [CrossRef]
- Wang, I.T.; Allen, M.; Goffin, D.; Zhu, X.; Fairless, A.H.; Brodkin, E.S.; Siegel, S.J.; Marsh, E.D.; Blendy, J.A.; Zhou, Z. Loss of CDKL5 disrupts kinome profile and event-related potentials leading to autistic-like phenotypes in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 21516–21521. [Google Scholar] [CrossRef] [PubMed]
- Amendola, E.; Zhan, Y.; Mattucci, C.; Castroflorio, E.; Calcagno, E.; Fuchs, C.; Lonetti, G.; Silingardi, D.; Vyssotski, A.L.; Farley, D.; et al. Mapping pathological phenotypes in a mouse model of CDKL5 disorder. PLoS ONE 2014, 9, e91613. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Wang, I.J.; Yue, C.; Takano, H.; Terzic, B.; Pance, K.; Lee, J.Y.; Cui, Y.; Coulter, D.A.; Zhou, Z. Loss of CDKL5 in Glutamatergic Neurons Disrupts Hippocampal Microcircuitry and Leads to Memory Impairment in Mice. J. Neurosci. 2017, 37, 7420–7437. [Google Scholar] [CrossRef] [PubMed]
- Okuda, K.; Kobayashi, S.; Fukaya, M.; Watanabe, A.; Murakami, T.; Hagiwara, M.; Sato, T.; Ueno, H.; Ogonuki, N.; Komano-Inoue, S.; et al. CDKL5 controls postsynaptic localization of GluN2B-containing NMDA receptors in the hippocampus and regulates seizure susceptibility. Neurobiol. Dis. 2017, 106, 158–170. [Google Scholar] [CrossRef]
- Tang, S.; Terzic, B.; Wang, I.J.; Sarmiento, N.; Sizov, K.; Cui, Y.; Takano, H.; Marsh, E.D.; Zhou, Z.; Coulter, D.A. Altered NMDAR signaling underlies autistic-like features in mouse models of CDKL5 deficiency disorder. Nat. Commun. 2019, 10, 2655. [Google Scholar] [CrossRef]
- Quadalti, C.; Sannia, M.; Humphreys, N.E.; Baldassarro, V.A.; Gurgone, A.; Ascolani, M.; Zanella, L.; Giardino, L.; Gross, C.T.; Croci, S.; et al. A new knockin mouse carrying the E364X patient mutation for CDKL5 deficiency disorder: Neurological, behavioral and molecular profiling. Heliyon 2024, 10, e40165. [Google Scholar] [CrossRef]
- Fuchs, C.; Trazzi, S.; Roberta, T.; Viggiano, R.; De Franceschi, M.; Amendola, E.; Gross, C.T.; Calzà, L.; Bartesaghi, R.; Ciani, E. Loss of Cdkl5 impairs survival and dendritic growth of newborn neurons by altering AKT/GSK-3beta signaling. Neurobiol. Dis. 2014, 70, 53–68. [Google Scholar] [CrossRef]
- Sivilia, S.; Mangano, C.; Beggiato, S.; Giuliani, A.; Torricella, R.; Baldassarro, V.A.; Fernandez, M.; Lorenzini, L.; Giardino, L.; Borelli, A.C.; et al. CDKL5 knockout leads to altered inhibitory transmission in the cerebellum of adult mice. Genes Brain Behav. 2016, 15, 491–502. [Google Scholar] [CrossRef]
- Fuchs, C.; Gennaccaro, L.; Trazzi, S.; Bastianini, S.; Bettini, S.; Martire, V.L.; Ren, E.; Medici, G.; Zoccoli, G.; Rimondini, R.; et al. Heterozygous CDKL5 Knockout Female Mice Are a Valuable Animal Model for CDKL5 Disorder. Neural Plast. 2018, 2018, 9726950. [Google Scholar] [CrossRef]
- Lee, K.Z.; Liao, W. Loss of CDKL5 disrupts respiratory function in mice. Respir. Physiol. Neurobiol. 2018, 248, 48–54. [Google Scholar] [CrossRef]
- Vigli, D.; Rusconi, L.; Valenti, D.; La Montanara, P.; Cosentino, L.; Lacivita, E.; Leopoldo, M.; Amendola, E.; Gross, C.; Landsberger, N.; et al. Rescue of prepulse inhibition deficit and brain mitochondrial dysfunction by pharmacological stimulation of the central serotonin receptor 7 in a mouse model of CDKL5 Deficiency Disorder. Neuropharmacology 2019, 144, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Gurgone, A.; Pizzo, R.; Raspanti, A.; Chiantia, G.; Devi, S.; Comai, D.; Morello, N.; Pilotto, F.; Gnavi, S.; Lupori, L.; et al. mGluR5 PAMs rescue cortical and behavioural defects in a mouse model of CDKL5 deficiency disorder. Neuropsychopharmacology 2023, 48, 877–886. [Google Scholar] [CrossRef]
- Della Sala, G.; Putignano, E.; Chelini, G.; Melani, R.; Calcagno, E.; Michele Ratto, G.; Amendola, E.; Gross, C.T.; Giustetto, M.; Pizzorusso, T. Dendritic Spine Instability in a Mouse Model of CDKL5 Disorder Is Rescued by Insulin-like Growth Factor 1. Biol. Psychiatry 2016, 80, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, R.; Gurgone, A.; Castroflorio, E.; Amendola, E.; Gross, C.; Sassoe-Pognetto, M.; Giustetto, M. Lack of Cdkl5 Disrupts the Organization of Excitatory and Inhibitory Synapses and Parvalbumin Interneurons in the Primary Visual Cortex. Front. Cell. Neurosci. 2016, 10, 261. [Google Scholar] [CrossRef]
- Ren, E.; Roncace, V.; Trazzi, S.; Fuchs, C.; Medici, G.; Gennaccaro, L.; Loi, M.; Galvani, G.; Ye, K.; Rimondini, R.; et al. Functional and Structural Impairments in the Perirhinal Cortex of a Mouse Model of CDKL5 Deficiency Disorder Are Rescued by a TrkB Agonist. Front. Cell Neurosci. 2019, 13, 169. [Google Scholar] [CrossRef]
- Tassinari, M.; Uguagliati, B.; Trazzi, S.; Cerchier, C.B.; Cavina, O.V.; Mottolese, N.; Loi, M.; Candini, G.; Medici, G.; Ciani, E. Early-onset brain alterations during postnatal development in a mouse model of CDKL5 deficiency disorder. Neurobiol. Dis. 2023, 182, 106146. [Google Scholar] [CrossRef]
- Fuchs, C.; Medici, G.; Trazzi, S.; Gennaccaro, L.; Galvani, G.; Berteotti, C.; Ren, E.; Loi, M.; Ciani, E. CDKL5 deficiency predisposes neurons to cell death through the deregulation of SMAD3 signaling. Brain Pathol. 2019, 29, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Galvani, G.; Mottolese, N.; Gennaccaro, L.; Loi, M.; Medici, G.; Tassinari, M.; Fuchs, C.; Ciani, E.; Trazzi, S. Inhibition of microglia overactivation restores neuronal survival in a mouse model of CDKL5 deficiency disorder. J. Neuroinflamm. 2021, 18, 155. [Google Scholar] [CrossRef]
- Terzic, B.; Davatolhagh, M.F.; Ho, Y.; Tang, S.; Liu, Y.T.; Xia, Z.; Cui, Y.; Fuccillo, M.V.; Zhou, Z. Temporal manipulation of Cdkl5 reveals essential postdevelopmental functions and reversible CDKL5 deficiency disorder-related deficits. J. Clin. Investig. 2021, 131, e143655. [Google Scholar] [CrossRef]
- Wang, H.T.; Zhu, Z.A.; Li, Y.Y.; Lou, S.S.; Yang, G.; Feng, X.; Xu, W.; Huang, Z.L.; Cheng, X.; Xiong, Z.Q. CDKL5 deficiency in forebrain glutamatergic neurons results in recurrent spontaneous seizures. Epilepsia 2021, 62, 517–528. [Google Scholar] [CrossRef]
- Silvestre, M.; Dempster, K.; Mihaylov, S.R.; Claxton, S.; Ultanir, S.K. Cell type-specific expression, regulation and compensation of CDKL5 activity in mouse brain. Mol. Psychiatry 2024, 29, 1844–1856. [Google Scholar] [CrossRef] [PubMed]
- Lupori, L.; Sagona, G.; Fuchs, C.; Mazziotti, R.; Stefanov, A.; Putignano, E.; Napoli, D.; Strettoi, E.; Ciani, E.; Pizzorusso, T. Site-specific abnormalities in the visual system of a mouse model of CDKL5 deficiency disorder. Hum. Mol. Genet. 2019, 28, 2851–2861. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.A.; Li, Y.Y.; Xu, J.; Xue, H.; Feng, X.; Zhu, Y.C.; Xiong, Z.Q. CDKL5 deficiency in adult glutamatergic neurons alters synaptic activity and causes spontaneous seizures via TrkB signaling. Cell Rep. 2023, 42, 113202. [Google Scholar] [CrossRef] [PubMed]
- Baltussen, L.L.; Negraes, P.D.; Silvestre, M.; Claxton, S.; Moeskops, M.; Christodoulou, E.; Flynn, H.R.; Snijders, A.P.; Muotri, A.R.; Ultanir, S.K. Chemical genetic identification of CDKL5 substrates reveals its role in neuronal microtubule dynamics. EMBO J. 2018, 37, 1–18. [Google Scholar] [CrossRef]
- DeFelipe, J.; Alonso-Nanclares, L.; Arellano, J.I. Microstructure of the neocortex: Comparative aspects. J. Neurocytol. 2002, 31, 299–316. [Google Scholar] [CrossRef]
- Sukenik, N.; Vinogradov, O.; Weinreb, E.; Segal, M.; Levina, A.; Moses, E. Neuronal circuits overcome imbalance in excitation and inhibition by adjusting connection numbers. Proc. Natl. Acad. Sci. USA 2021, 118, 1–9. [Google Scholar] [CrossRef]
- Fuchs, C.; Rimondini, R.; Viggiano, R.; Trazzi, S.; De Franceschi, M.; Bartesaghi, R.; Ciani, E. Inhibition of GSK3beta rescues hippocampal development and learning in a mouse model of CDKL5 disorder. Neurobiol. Dis. 2015, 82, 298–310. [Google Scholar] [CrossRef]
- Okuda, K.; Takao, K.; Watanabe, A.; Miyakawa, T.; Mizuguchi, M.; Tanaka, T. Comprehensive behavioral analysis of the Cdkl5 knockout mice revealed significant enhancement in anxiety- and fear-related behaviors and impairment in both acquisition and long-term retention of spatial reference memory. PLoS ONE 2018, 13, e0196587. [Google Scholar] [CrossRef]
- Gennaccaro, L.; Fuchs, C.; Loi, M.; Pizzo, R.; Alvente, S.; Berteotti, C.; Lupori, L.; Sagona, G.; Galvani, G.; Gurgone, A.; et al. Age-Related Cognitive and Motor Decline in a Mouse Model of CDKL5 Deficiency Disorder is Associated with Increased Neuronal Senescence and Death. Aging Dis. 2021, 12, 764–785. [Google Scholar] [CrossRef]
- La Montanara, P.; Hervera, A.; Baltussen, L.L.; Hutson, T.H.; Palmisano, I.; De Virgiliis, F.; Kong, G.; Chadwick, J.; Gao, Y.; Bartus, K.; et al. Cyclin-dependent-like kinase 5 is required for pain signaling in human sensory neurons and mouse models. Sci. Transl. Med. 2020, 12, 1–36. [Google Scholar] [CrossRef]
- Wojcik, S.M.; Rhee, J.S.; Herzog, E.; Sigler, A.; Jahn, R.; Takamori, S.; Brose, N.; Rosenmund, C. An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc. Natl. Acad. Sci. USA 2004, 101, 7158–7163. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, F.A.; Reimer, R.J.; Bellocchio, E.E.; Danbolt, N.C.; Osen, K.K.; Edwards, R.H.; Storm-Mathisen, J. The vesicular GABA transporter, VGAT, localizes to synaptic vesicles in sets of glycinergic as well as GABAergic neurons. J. Neurosci. 1998, 18, 9733–9750. [Google Scholar] [CrossRef]
- Risher, W.C.; Ustunkaya, T.; Singh Alvarado, J.; Eroglu, C. Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS ONE 2014, 9, e107591. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, S.; Ungaro, F.; Hambrock, M.; Rademacher, N.; Stefanelli, G.; Brambilla, D.; Sessa, A.; Magagnotti, C.; Bachi, A.; Giarda, E.; et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol. 2012, 14, 911–923. [Google Scholar] [CrossRef] [PubMed]
- Bourne, J.N.; Harris, K.M. Balancing structure and function at hippocampal dendritic spines. Annu. Rev. Neurosci. 2008, 31, 47–67. [Google Scholar] [CrossRef]
- Caire, M.J.; Reddy, V.; Varacallo, M.A. Physiology, Synapse. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. [Google Scholar]
- Gennaccaro, L.; Fuchs, C.; Loi, M.; Roncace, V.; Trazzi, S.; Ait-Bali, Y.; Galvani, G.; Berardi, A.C.; Medici, G.; Tassinari, M.; et al. A GABA(B) receptor antagonist rescues functional and structural impairments in the perirhinal cortex of a mouse model of CDKL5 deficiency disorder. Neurobiol. Dis. 2021, 153, 105304. [Google Scholar] [CrossRef]
- Hao, S.; Wang, Q.; Tang, B.; Wu, Z.; Yang, T.; Tang, J. CDKL5 Deficiency Augments Inhibitory Input into the Dentate Gyrus That Can Be Reversed by Deep Brain Stimulation. J. Neurosci. 2021, 41, 9031–9046. [Google Scholar] [CrossRef]
- Melrose, J. Keratan sulfate (KS)-proteoglycans and neuronal regulation in health and disease: The importance of KS-glycodynamics and interactive capability with neuroregulatory ligands. J. Neurochem. 2019, 149, 170–194. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, P.; Yan, F.; Luo, Y.; Zhao, G. Animal Models of Epilepsy: A Phenotype-oriented Review. Aging Dis. 2022, 13, 215–231. [Google Scholar] [CrossRef]
- Takashima, S.; Becker, L.E.; Armstrong, D.L.; Chan, F. Abnormal neuronal development in the visual cortex of the human fetus and infant with down’s syndrome. A quantitative and qualitative golgi study. Brain Res. 1981, 225, 1–21. [Google Scholar] [CrossRef]
- Eyo, U.B.; Murugan, M.; Wu, L.J. Microglia-Neuron Communication in Epilepsy. Glia 2017, 65, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.; Deng, X.J.; Xu, D. Microglia in epilepsy. Neurobiol. Dis. 2023, 185, 106249. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jia, N.; Tang, C.; Long, H.; Wang, J. Microglia in Microbiota-Gut-Brain Axis: A Hub in Epilepsy. Mol. Neurobiol. 2024, 61, 7109–7126. [Google Scholar] [CrossRef] [PubMed]
- Jhang, C.L.; Huang, T.N.; Hsueh, Y.P.; Liao, W. Mice lacking cyclin-dependent kinase-like 5 manifest autistic and ADHD-like behaviors. Hum. Mol. Genet. 2017, 26, 3922–3934. [Google Scholar] [CrossRef]
- Erdmann, G.; Schutz, G.; Berger, S. Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci. 2007, 8, 63. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M. Digging and marble burying in mice: Simple methods for in vivo identification of biological impacts. Nat. Protoc. 2006, 1, 122–124. [Google Scholar] [CrossRef]
- Shiotsuki, H.; Yoshimi, K.; Shimo, Y.; Funayama, M.; Takamatsu, Y.; Ikeda, K.; Takahashi, R.; Kitazawa, S.; Hattori, N. A rotarod test for evaluation of motor skill learning. J. Neurosci. Methods 2010, 189, 180–185. [Google Scholar] [CrossRef]
- Kraeuter, A.K.; Guest, P.C.; Sarnyai, Z. The Open Field Test for Measuring Locomotor Activity and Anxiety-Like Behavior. Methods Mol. Biol. 2019, 1916, 99–103. [Google Scholar] [CrossRef]
- Szabo, J.; Renczes, E.; Borbelyova, V.; Ostatnikova, D.; Celec, P. Assessing sociability using the Three-Chamber Social Interaction Test and the Reciprocal Interaction Test in a genetic mouse model of ASD. Behav. Brain Funct. 2024, 20, 24. [Google Scholar] [CrossRef]
- Gawel, K.; Gibula, E.; Marszalek-Grabska, M.; Filarowska, J.; Kotlinska, J.H. Assessment of spatial learning and memory in the Barnes maze task in rodents-methodological consideration. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 392, 1–18. [Google Scholar] [CrossRef]
- Ogren, S.O. Evidence for a role of brain serotonergic neurotransmission in avoidance learning. Acta Physiol. Scand. Suppl. 1985, 544, 1–71. [Google Scholar] [PubMed]
- Yang, M.; Crawley, J.N. Simple behavioral assessment of mouse olfaction. Curr. Protoc. Neurosci. 2009, 48, 8.24.1-8.24.12. [Google Scholar] [CrossRef] [PubMed]
- Deuis, J.R.; Dvorakova, L.S.; Vetter, I. Methods Used to Evaluate Pain Behaviors in Rodents. Front. Mol. Neurosci. 2017, 10, 284. [Google Scholar] [CrossRef]
- Medici, G.; Tassinari, M.; Galvani, G.; Bastianini, S.; Gennaccaro, L.; Loi, M.; Mottolese, N.; Alvente, S.; Berteotti, C.; Sagona, G.; et al. Expression of a Secretable, Cell-Penetrating CDKL5 Protein Enhances the Efficacy of Gene Therapy for CDKL5 Deficiency Disorder. Neurotherapeutics 2022, 19, 1886–1904. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Specchio, N.; Trivisano, M.; Lenge, M.; Ferretti, A.; Mei, D.; Parrini, E.; Napolitano, A.; Rossi-Espagnet, C.; Talenti, G.; Longo, D.; et al. CDKL5 deficiency disorder: Progressive brain atrophy may be part of the syndrome. Cereb. Cortex 2023, 33, 9709–9717. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mottolese, N.; Iannibelli, F.; Candini, G.; Trebbi, F.; Loi, M.; Bove, A.M.; Medici, G.; Xiong, Z.-Q.; Ciani, E.; Trazzi, S. Adult-Onset Deletion of CDKL5 in Forebrain Glutamatergic Neurons Impairs Synaptic Integrity and Behavior in Mice. Int. J. Mol. Sci. 2025, 26, 6626. https://doi.org/10.3390/ijms26146626
Mottolese N, Iannibelli F, Candini G, Trebbi F, Loi M, Bove AM, Medici G, Xiong Z-Q, Ciani E, Trazzi S. Adult-Onset Deletion of CDKL5 in Forebrain Glutamatergic Neurons Impairs Synaptic Integrity and Behavior in Mice. International Journal of Molecular Sciences. 2025; 26(14):6626. https://doi.org/10.3390/ijms26146626
Chicago/Turabian StyleMottolese, Nicola, Feliciana Iannibelli, Giulia Candini, Federica Trebbi, Manuela Loi, Angelica Marina Bove, Giorgio Medici, Zhi-Qi Xiong, Elisabetta Ciani, and Stefania Trazzi. 2025. "Adult-Onset Deletion of CDKL5 in Forebrain Glutamatergic Neurons Impairs Synaptic Integrity and Behavior in Mice" International Journal of Molecular Sciences 26, no. 14: 6626. https://doi.org/10.3390/ijms26146626
APA StyleMottolese, N., Iannibelli, F., Candini, G., Trebbi, F., Loi, M., Bove, A. M., Medici, G., Xiong, Z.-Q., Ciani, E., & Trazzi, S. (2025). Adult-Onset Deletion of CDKL5 in Forebrain Glutamatergic Neurons Impairs Synaptic Integrity and Behavior in Mice. International Journal of Molecular Sciences, 26(14), 6626. https://doi.org/10.3390/ijms26146626