
Development of FGF21 Mutant with Potent Cardioprotective Effects in T2D Mice via FGFR1–AMPK-Mediated Inhibition of Oxidative Stress


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
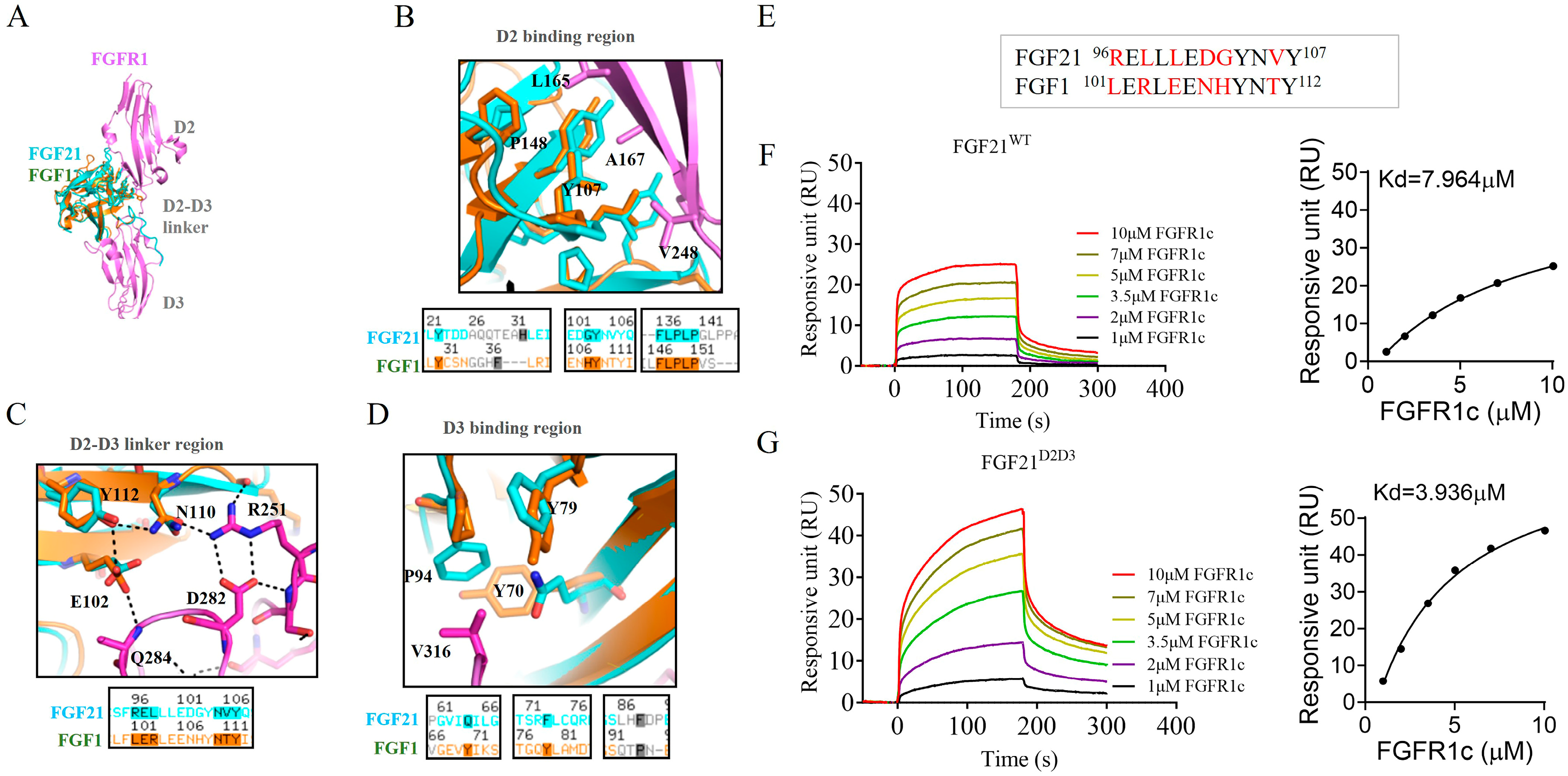
2.1. Design of FGF21 Analog with Enhanced Affinity Toward FGFR1c
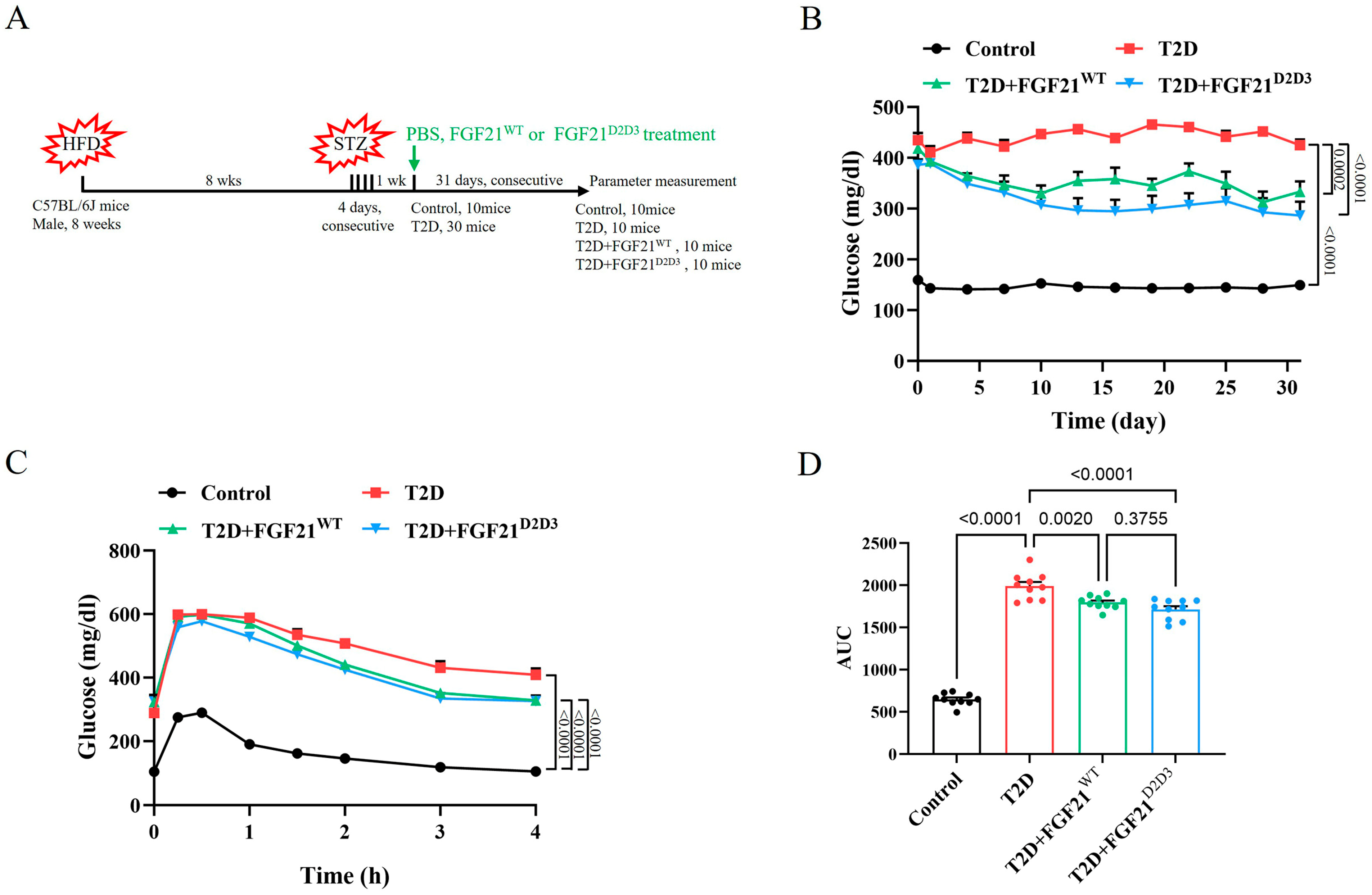
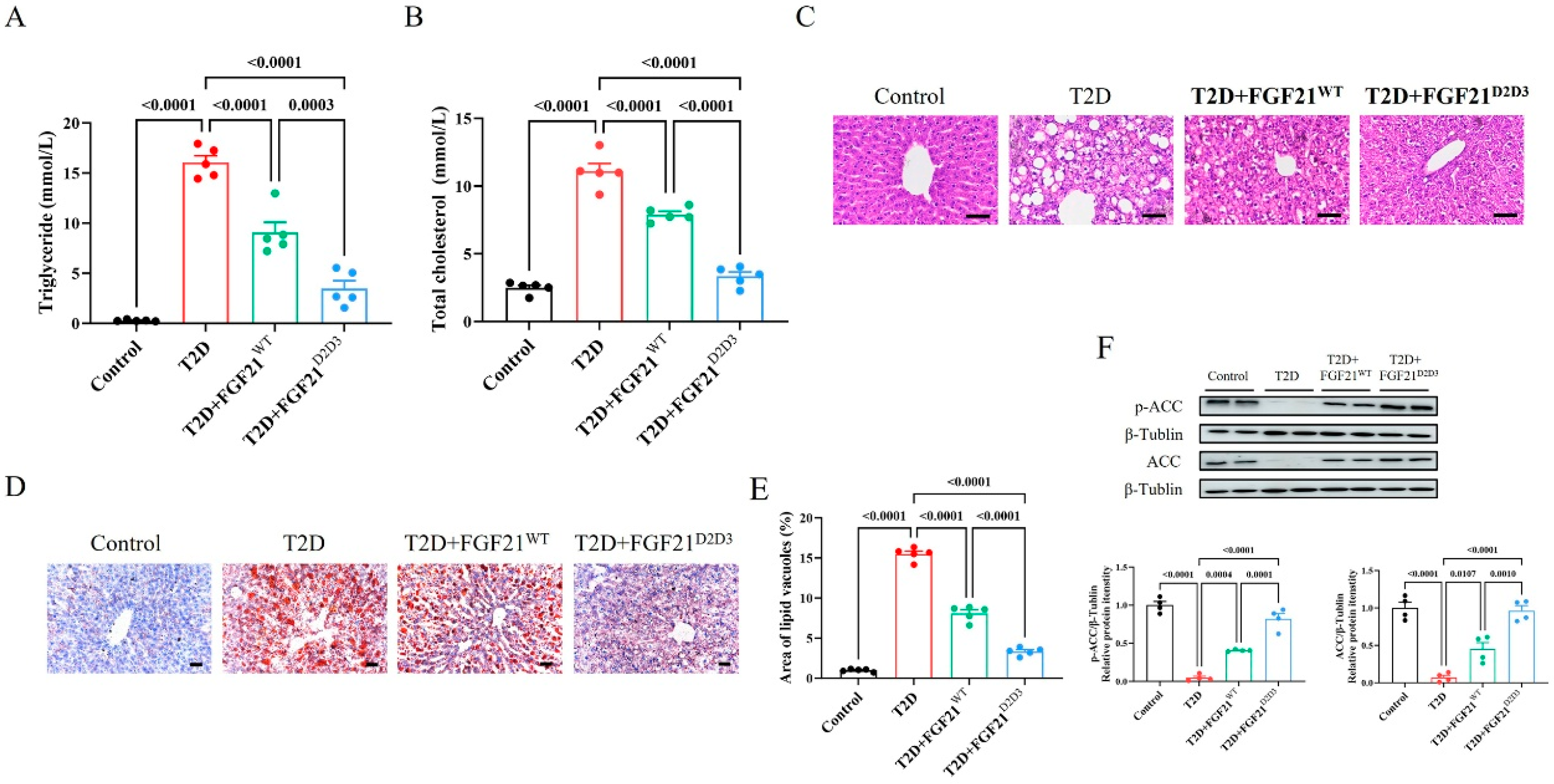
2.2. FGF21D2D3 Has a More Potent Lipid-Lowering Effect than FGF21WT in T2D Mice
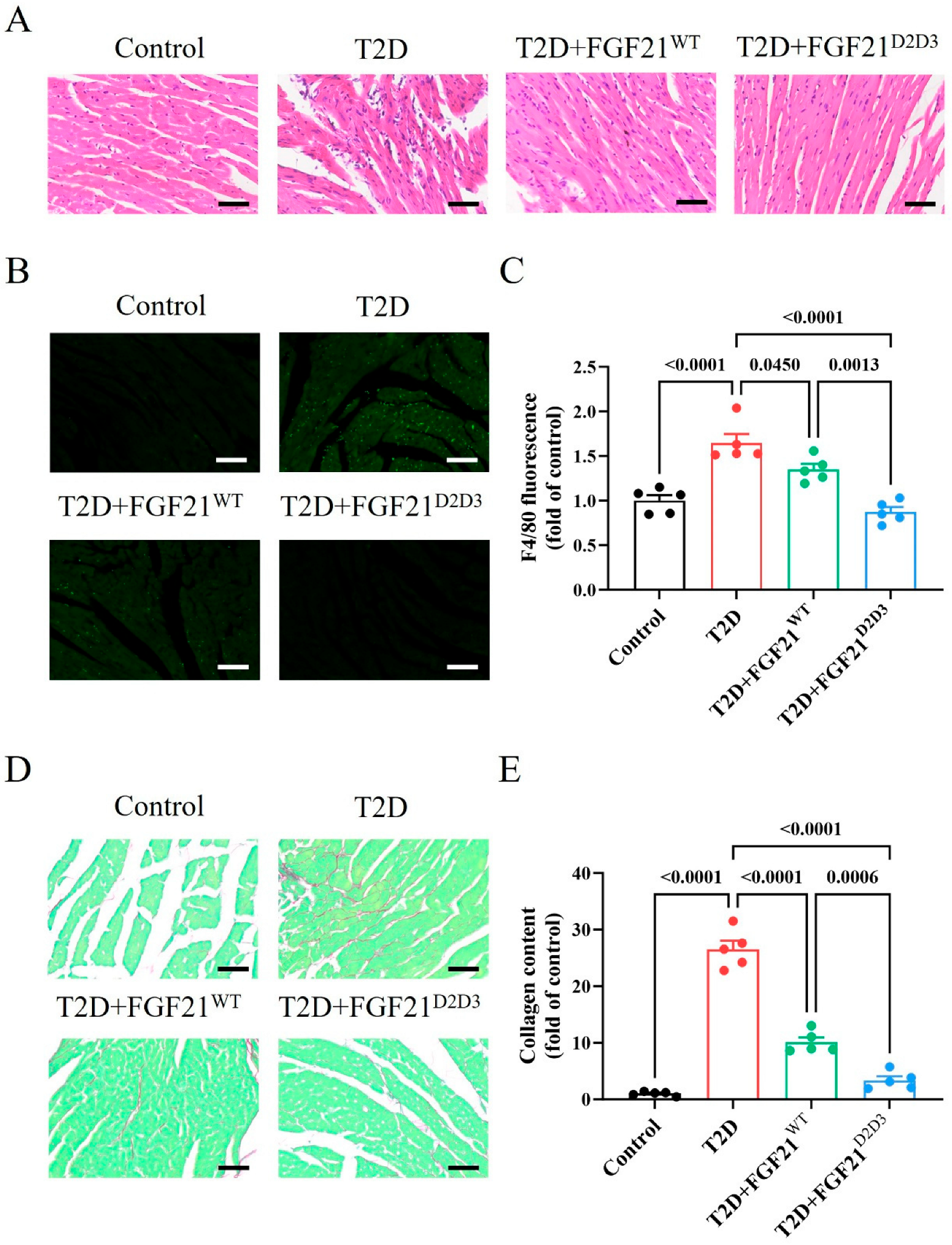
2.3. FGF21D2D3 Has a More Potent Cardio-Protective Effect than FGF21WT in T2D Mice
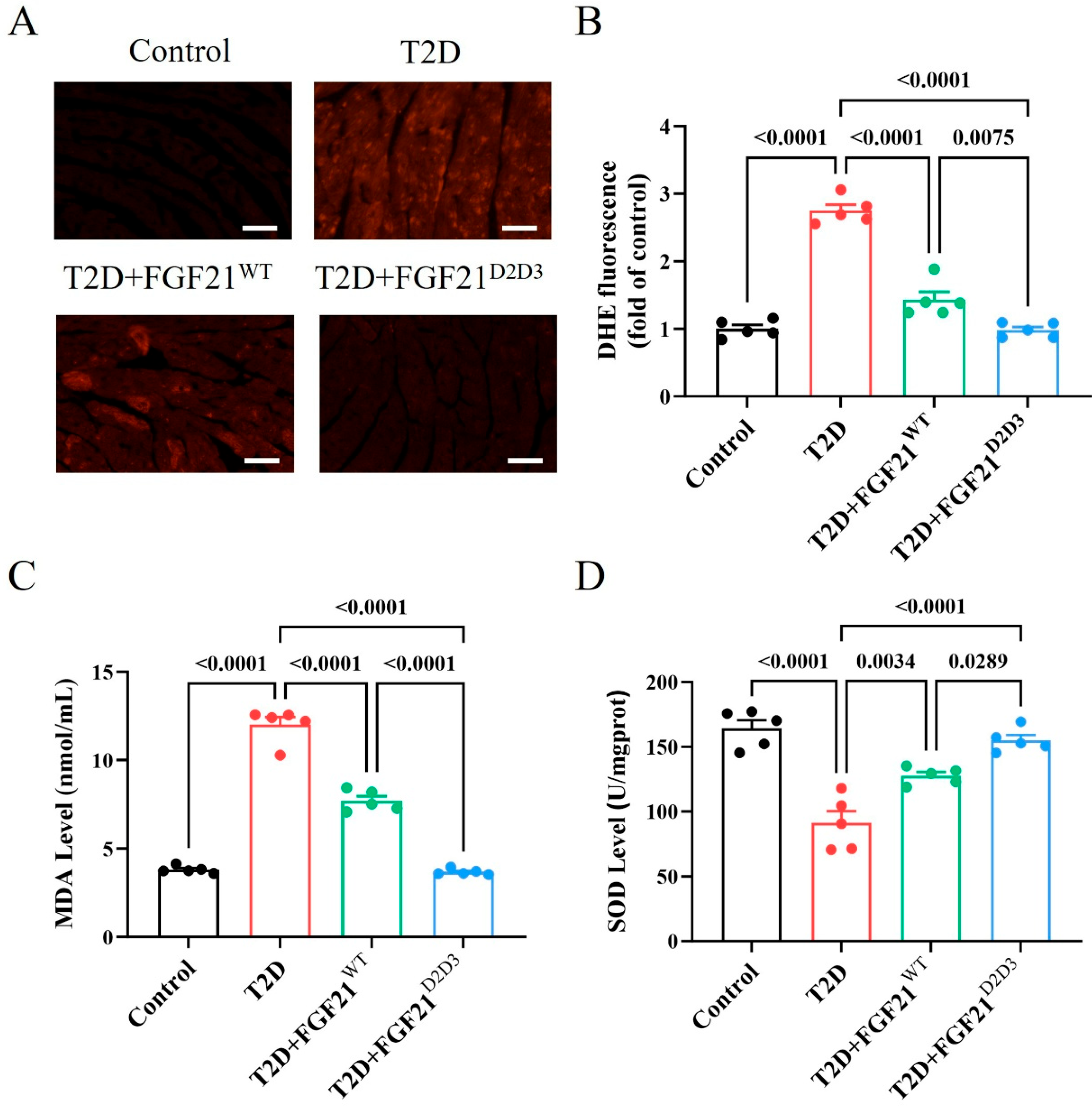
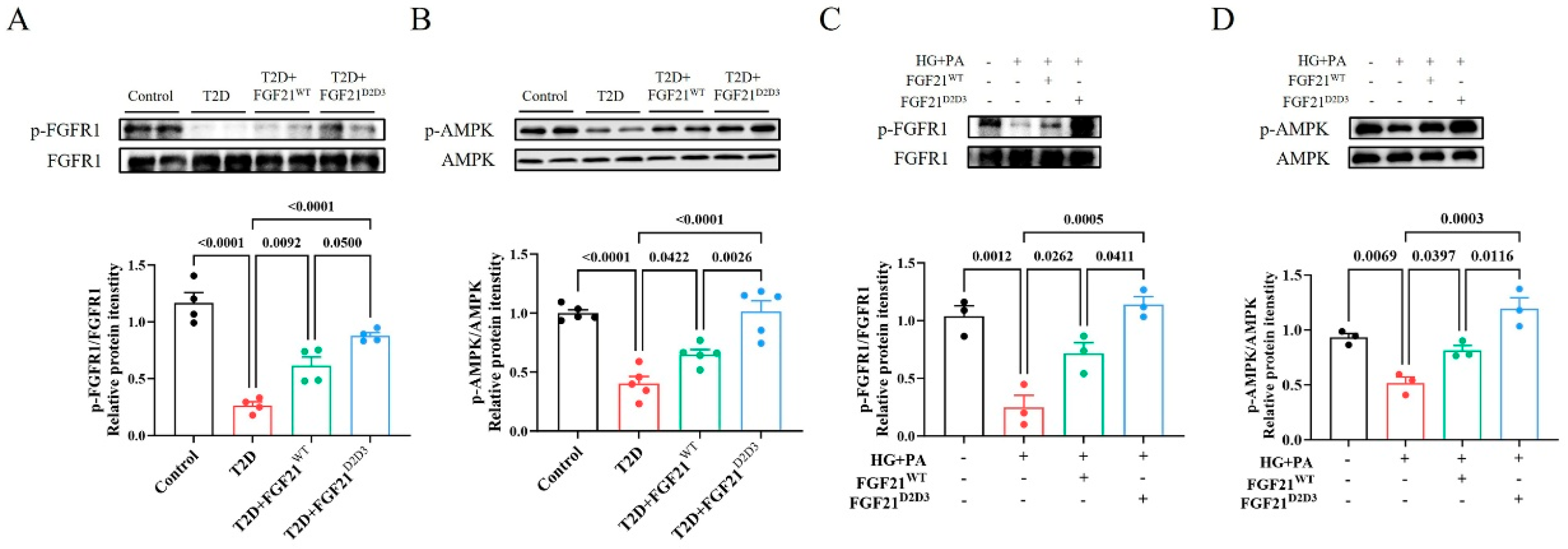
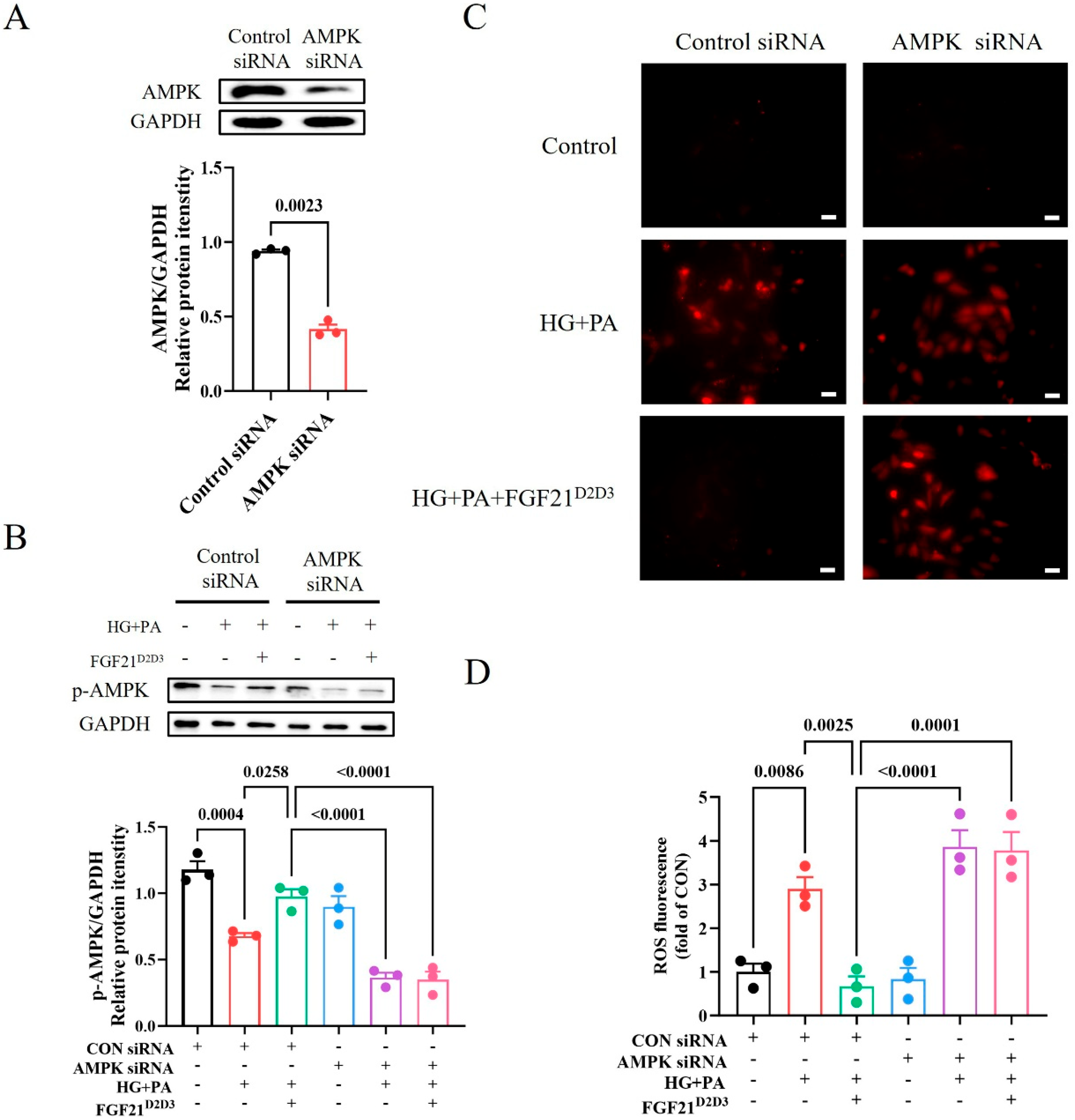
2.4. FGF21D2D3 More Potently Alleviates Cardiac Damage than FGF21WT via FGFR1–AMPKa-Mediated Inhibition of Oxidative Stress in T2D Mice
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of Wild-Type FGF21 and Its Mutant
4.2. Surface Plasmon Resonance (SPR) Spectroscopy
4.3. Animals and Animal Welfare
4.4. Functional Evaluation of FGF21WT and FGF21D2D3 in T2D Mice
4.5. Immunohistochemical and Immunofluorescent Staining of Mouse Tissues
4.6. MDA and SOD Measurements
4.7. Experiments Using H9c2 Cardiomyocytes
4.8. Western Blot Analysis
4.9. DHE Staining
4.10. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DCM | Diabetic cardiomyopathy |
| T2D | Type 2 diabetes |
| FGF21 | Fibroblast growth factor 21 |
| FGFR1c | Fibroblast growth factor receptor 1c |
| FGF21WT | Wild-type FGF21 |
| FGF | Fibroblast growth factor |
| KLB | β-klotho |
| FGFR1 | Fibroblast growth factor receptor 1 |
| HFD | High-fat diet |
| STZ | Streptozotocin |
| ACC | Acetyl-CoA carboxylase |
| DHE | Dihydroethidium |
| AMPK | Adenosine 5′-monophosphate (AMP)-activated protein kinase |
| HG | High glucose |
| PA | Palmitic acid |
| SPR | Surface plasmon resonance |
| IPGTTs | Intraperitoneal glucose tolerance tests |
| AUC | Area under the curve |
| H&E | Hematoxylin and eosin |
| ANOVA | Analysis of variance |
References
- Ritchie, R.H.; Abel, E.D. Basic Mechanisms of Diabetic Heart Disease. Circ. Res. 2020, 126, 1501–1525. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Ma, X.N.; Guan, C.H.; Li, Y.D.; Mauricio, D.; Fu, S.B. Cardiovascular disease in type 2 diabetes mellitus: Progress toward personalized management. Cardiovasc. Diabetol. 2022, 21, 74. [Google Scholar] [CrossRef]
- Zhang, P.Y. Cardiovascular disease in diabetes. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 2205–2214. [Google Scholar]
- Fuentes-Antras, J.; Picatoste, B.; Ramirez, E.; Egido, J.; Tunon, J.; Lorenzo, O. Targeting metabolic disturbance in the diabetic heart. Cardiovasc. Diabetol. 2015, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Parim, B.; Sathibabu Uddandrao, V.V.; Saravanan, G. Diabetic cardiomyopathy: Molecular mechanisms, detrimental effects of conventional treatment, and beneficial effects of natural therapy. Heart Fail. Rev. 2019, 24, 279–299. [Google Scholar] [CrossRef]
- Nishimura, T.; Nakatake, Y.; Konishi, M.; Itoh, N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim. Biophys. Acta 2000, 1492, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Degirolamo, C.; Sabba, C.; Moschetta, A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat. Rev. Drug Discov. 2016, 15, 51–69. [Google Scholar] [CrossRef]
- Flippo, K.H.; Potthoff, M.J. Metabolic Messengers: FGF21. Nat. Metab. 2021, 3, 309–317. [Google Scholar] [CrossRef]
- Fisher, F.M.; Maratos-Flier, E. Understanding the Physiology of FGF21. Annu. Rev. Physiol. 2016, 78, 223–241. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef]
- Coskun, T.; Bina, H.A.; Schneider, M.A.; Dunbar, J.D.; Hu, C.C.; Chen, Y.; Moller, D.E.; Kharitonenkov, A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 2008, 149, 6018–6027. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lloyd, D.J.; Hale, C.; Stanislaus, S.; Chen, M.; Sivits, G.; Vonderfecht, S.; Hecht, R.; Li, Y.S.; Lindberg, R.A.; et al. Fibroblast growth factor 21 reverses hepatic steatosis, increases energy expenditure, and improves insulin sensitivity in diet-induced obese mice. Diabetes 2009, 58, 250–259. [Google Scholar] [CrossRef]
- Xu, J.; Stanislaus, S.; Chinookoswong, N.; Lau, Y.Y.; Hager, T.; Patel, J.; Ge, H.; Weiszmann, J.; Lu, S.C.; Graham, M.; et al. Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models--association with liver and adipose tissue effects. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1105–E1114. [Google Scholar] [CrossRef] [PubMed]
- Kharitonenkov, A.; Wroblewski, V.J.; Koester, A.; Chen, Y.F.; Clutinger, C.K.; Tigno, X.T.; Hansen, B.C.; Shanafelt, A.B.; Etgen, G.J. The metabolic state of diabetic monkeys is regulated by fibroblast growth factor-21. Endocrinology 2007, 148, 774–781. [Google Scholar] [CrossRef]
- Planavila, A.; Redondo-Angulo, I.; Villarroya, F. FGF21 and Cardiac Physiopathology. Front. Endocrinol. 2015, 6, 133. [Google Scholar] [CrossRef]
- Itoh, N.; Ohta, H. Pathophysiological roles of FGF signaling in the heart. Front. Physiol. 2013, 4, 247. [Google Scholar] [CrossRef]
- Yang, H.; Feng, A.; Lin, S.; Yu, L.; Lin, X.; Yan, X.; Lu, X.; Zhang, C. Fibroblast growth factor-21 prevents diabetic cardiomyopathy via AMPK-mediated antioxidation and lipid-lowering effects in the heart. Cell Death Dis. 2018, 9, 227. [Google Scholar] [CrossRef]
- Suzuki, M.; Uehara, Y.; Motomura-Matsuzaka, K.; Oki, J.; Koyama, Y.; Kimura, M.; Asada, M.; Komi-Kuramochi, A.; Oka, S.; Imamura, T. betaKlotho is required for fibroblast growth factor (FGF) 21 signaling through FGF receptor (FGFR) 1c and FGFR3c. Mol. Endocrinol. 2008, 22, 1006–1014. [Google Scholar] [CrossRef]
- Cheng, P.; Zhang, F.; Yu, L.; Lin, X.; He, L.; Li, X.; Lu, X.; Yan, X.; Tan, Y.; Zhang, C. Physiological and Pharmacological Roles of FGF21 in Cardiovascular Diseases. J. Diabetes Res. 2016, 2016, 1540267. [Google Scholar] [CrossRef]
- Tucker, W.; Tucker, B.; Rye, K.A.; Ong, K.L. Fibroblast growth factor 21 in heart failure. Heart Fail. Rev. 2023, 28, 261–272. [Google Scholar] [CrossRef]
- Jin, L.; Geng, L.; Ying, L.; Shu, L.; Ye, K.; Yang, R.; Liu, Y.; Wang, Y.; Cai, Y.; Jiang, X.; et al. FGF21-Sirtuin 3 Axis Confers the Protective Effects of Exercise Against Diabetic Cardiomyopathy by Governing Mitochondrial Integrity. Circulation 2022, 146, 1537–1557. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N. FGF21 as a Hepatokine, Adipokine, and Myokine in Metabolism and Diseases. Front. Endocrinol. 2014, 5, 107. [Google Scholar] [CrossRef]
- Huang, Z.; Tan, Y.; Gu, J.; Liu, Y.; Song, L.; Niu, J.; Zhao, L.; Srinivasan, L.; Lin, Q.; Deng, J.; et al. Uncoupling the Mitogenic and Metabolic Functions of FGF1 by Tuning FGF1-FGF Receptor Dimer Stability. Cell Rep. 2017, 20, 1717–1728. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Lam, K.S.L.; Xu, A. The therapeutic potential of FGF21 in metabolic diseases: From bench to clinic. Nat. Rev. Endocrinol. 2020, 16, 654–667. [Google Scholar] [CrossRef]
- Zhao, L.; Niu, J.; Lin, H.; Zhao, J.; Liu, Y.; Song, Z.; Xiang, C.; Wang, X.; Yang, Y.; Li, X.; et al. Paracrine-endocrine FGF chimeras as potent therapeutics for metabolic diseases. EBioMedicine 2019, 48, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, G.; Fang, Q.; Zhang, M.; Hui, X.; Sheng, B.; Wu, L.; Bao, Y.; Li, P.; Xu, A.; et al. Fibroblast growth factor 21 increases insulin sensitivity through specific expansion of subcutaneous fat. Nat. Commun. 2018, 9, 272. [Google Scholar] [CrossRef]
- Emanuelli, B.; Vienberg, S.G.; Smyth, G.; Cheng, C.; Stanford, K.I.; Arumugam, M.; Michael, M.D.; Adams, A.C.; Kharitonenkov, A.; Kahn, C.R. Interplay between FGF21 and insulin action in the liver regulates metabolism. J. Clin. Investig. 2015, 125, 458. [Google Scholar] [CrossRef]
- Lin, Z.; Tian, H.; Lam, K.S.; Lin, S.; Hoo, R.C.; Konishi, M.; Itoh, N.; Wang, Y.; Bornstein, S.R.; Xu, A.; et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013, 17, 779–789. [Google Scholar] [CrossRef]
- Deprince, A.; Haas, J.T.; Staels, B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol. Metab. 2020, 42, 101092. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Lin, C.; Guo, Y.; Xia, Y.; Li, C.; Xu, X.; Qi, T.; Zhang, F.; Fan, M.; Hu, G.; Zhao, H.; et al. FNDC5/Irisin attenuates diabetic cardiomyopathy in a type 2 diabetes mouse model by activation of integrin alphaV/beta5-AKT signaling and reduction of oxidative/nitrosative stress. J. Mol. Cell. Cardiol. 2021, 160, 27–41. [Google Scholar] [CrossRef]
- Westermeier, F.; Riquelme, J.A.; Pavez, M.; Garrido, V.; Diaz, A.; Verdejo, H.E.; Castro, P.F.; Garcia, L.; Lavandero, S. New Molecular Insights of Insulin in Diabetic Cardiomyopathy. Front. Physiol. 2016, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Faria, A.; Persaud, S.J. Cardiac oxidative stress in diabetes: Mechanisms and therapeutic potential. Pharmacol. Ther. 2017, 172, 50–62. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, S.; Cai, L. Diabetic cardiomyopathy and its mechanisms: Role of oxidative stress and damage. J. Diabetes Investig. 2014, 5, 623–634. [Google Scholar] [CrossRef]
- Ying, L.; Li, N.; He, Z.; Zeng, X.; Nan, Y.; Chen, J.; Miao, P.; Ying, Y.; Lin, W.; Zhao, X.; et al. Fibroblast growth factor 21 Ameliorates diabetes-induced endothelial dysfunction in mouse aorta via activation of the CaMKK2/AMPKalpha signaling pathway. Cell Death Dis. 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed]
- Ying, L.; Wang, L.; Guo, K.; Hou, Y.; Li, N.; Wang, S.; Liu, X.; Zhao, Q.; Zhou, J.; Zhao, L.; et al. Paracrine FGFs target skeletal muscle to exert potent anti-hyperglycemic effects. Nat. Commun. 2021, 12, 7256. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Tuo, H.; Tang, N.; Liu, F.Y.; Ma, S.Q.; An, P.; Yang, D.; Wang, M.Y.; Fan, D.; Yang, Z.; et al. Neuraminidase 1 deficiency attenuates cardiac dysfunction, oxidative stress, fibrosis, inflammatory via AMPK-SIRT3 pathway in diabetic cardiomyopathy mice. Int. J. Biol. Sci. 2022, 18, 826–840. [Google Scholar] [CrossRef]
- Wang, D.; Yin, Y.; Wang, S.; Zhao, T.; Gong, F.; Zhao, Y.; Wang, B.; Huang, Y.; Cheng, Z.; Zhu, G.; et al. FGF1(DeltaHBS) prevents diabetic cardiomyopathy by maintaining mitochondrial homeostasis and reducing oxidative stress via AMPK/Nur77 suppression. Signal Transduct. Target. Ther. 2021, 6, 133. [Google Scholar] [CrossRef]
- Xie, Z.; He, C.; Zou, M.H. AMP-activated protein kinase modulates cardiac autophagy in diabetic cardiomyopathy. Autophagy 2011, 7, 1254–1255. [Google Scholar] [CrossRef]
- Panek, R.L.; Lu, G.H.; Dahring, T.K.; Batley, B.L.; Connolly, C.; Hamby, J.M.; Brown, K.J. In vitro biological characterization and antiangiogenic effects of PD 166866, a selective inhibitor of the FGF-1 receptor tyrosine kinase. J. Pharmacol. Exp. Ther. 1998, 286, 569–577. [Google Scholar] [CrossRef]
- Wong, W.T.; Tian, X.Y.; Xu, A.; Yu, J.; Lau, C.W.; Hoo, R.L.; Wang, Y.; Lee, V.W.; Lam, K.S.; Vanhoutte, P.M.; et al. Adiponectin is required for PPARgamma-mediated improvement of endothelial function in diabetic mice. Cell Metab. 2011, 14, 104–115. [Google Scholar] [CrossRef]
- Nies, V.J.; Sancar, G.; Liu, W.; van Zutphen, T.; Struik, D.; Yu, R.T.; Atkins, A.R.; Evans, R.M.; Jonker, J.W.; Downes, M.R. Fibroblast Growth Factor Signaling in Metabolic Regulation. Front. Endocrinol. 2015, 6, 193. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Kahn, C.R. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001, 414, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Klip, A.; Paquet, M.R. Glucose transport and glucose transporters in muscle and their metabolic regulation. Diabetes Care 1990, 13, 228–243. [Google Scholar] [CrossRef]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro, O.M. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef]
- Shaw, A.; Doherty, M.K.; Mutch, N.J.; MacRury, S.M.; Megson, I.L. Endothelial cell oxidative stress in diabetes: A key driver of cardiovascular complications? Biochem. Soc. Trans. 2014, 42, 928–933. [Google Scholar] [CrossRef]
- King, G.L.; Loeken, M.R. Hyperglycemia-induced oxidative stress in diabetic complications. Histochem. Cell Biol. 2004, 122, 333–338. [Google Scholar] [CrossRef]
- Penckofer, S.; Schwertz, D.; Florczak, K. Oxidative stress and cardiovascular disease in type 2 diabetes: The role of antioxidants and pro-oxidants. J. Cardiovasc. Nurs. 2002, 16, 68–85. [Google Scholar] [CrossRef]
- Robson, R.; Kundur, A.R.; Singh, I. Oxidative stress biomarkers in type 2 diabetes mellitus for assessment of cardiovascular disease risk. Diabetes Metab. Syndr. 2018, 12, 455–462. [Google Scholar] [CrossRef]
- Ceriello, A.; Motz, E. Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 816–823. [Google Scholar] [CrossRef]
- Coughlan, K.A.; Valentine, R.J.; Ruderman, N.B.; Saha, A.K. AMPK activation: A therapeutic target for type 2 diabetes? Diabetes Metab. Syndr. Obes. 2014, 7, 241–253. [Google Scholar] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Zhu, Y.; Wang, H.; Belov, A.A.; Niu, J.; Shi, L.; Xie, Y.; Ye, C.; Li, X.; Huang, Z. A solid-phase PEGylation strategy for protein therapeutics using a potent FGF21 analog. Biomaterials 2014, 35, 5206–5215. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Zhao, J.; Wu, J.; Qiao, G.; Gu, J.; Zhou, C.; Li, Q.; Ying, L.; Wang, D.; Lin, H.; et al. Curtailing FGF19's mitogenicity by suppressing its receptor dimerization ability. Proc. Natl. Acad. Sci. USA 2020, 117, 29025–29034. [Google Scholar] [CrossRef]
- Li, Q.; Zhao, Y.; Guo, H.; Li, Q.; Yan, C.; Li, Y.; He, S.; Wang, N.; Wang, Q. Impaired lipophagy induced-microglial lipid droplets accumulation contributes to the buildup of TREM1 in diabetes-associated cognitive impairment. Autophagy 2023, 19, 2639–2656. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Z.; Gao, L.; Zhang, L.; Yao, R.; Li, X.; Deng, L.; Fan, J.; Ying, L.; Wang, Y. Development of FGF21 Mutant with Potent Cardioprotective Effects in T2D Mice via FGFR1–AMPK-Mediated Inhibition of Oxidative Stress. Int. J. Mol. Sci. 2025, 26, 6577. https://doi.org/10.3390/ijms26146577
Peng Z, Gao L, Zhang L, Yao R, Li X, Deng L, Fan J, Ying L, Wang Y. Development of FGF21 Mutant with Potent Cardioprotective Effects in T2D Mice via FGFR1–AMPK-Mediated Inhibition of Oxidative Stress. International Journal of Molecular Sciences. 2025; 26(14):6577. https://doi.org/10.3390/ijms26146577
Chicago/Turabian StylePeng, Ziying, Ling Gao, Lei Zhang, Ruina Yao, Xiaoxiao Li, Long Deng, Jinxia Fan, Lei Ying, and Yang Wang. 2025. "Development of FGF21 Mutant with Potent Cardioprotective Effects in T2D Mice via FGFR1–AMPK-Mediated Inhibition of Oxidative Stress" International Journal of Molecular Sciences 26, no. 14: 6577. https://doi.org/10.3390/ijms26146577
APA StylePeng, Z., Gao, L., Zhang, L., Yao, R., Li, X., Deng, L., Fan, J., Ying, L., & Wang, Y. (2025). Development of FGF21 Mutant with Potent Cardioprotective Effects in T2D Mice via FGFR1–AMPK-Mediated Inhibition of Oxidative Stress. International Journal of Molecular Sciences, 26(14), 6577. https://doi.org/10.3390/ijms26146577