
Potent Inhibition of Chikungunya Virus Entry by a Pyrazole–Benzene Derivative: A Computational Study Targeting the E1–E2 Glycoprotein Complex

 , , , and
, , , and
Abstract

1. Introduction
2. Results
2.1. Quantitative Evaluation of Ligand Bindings
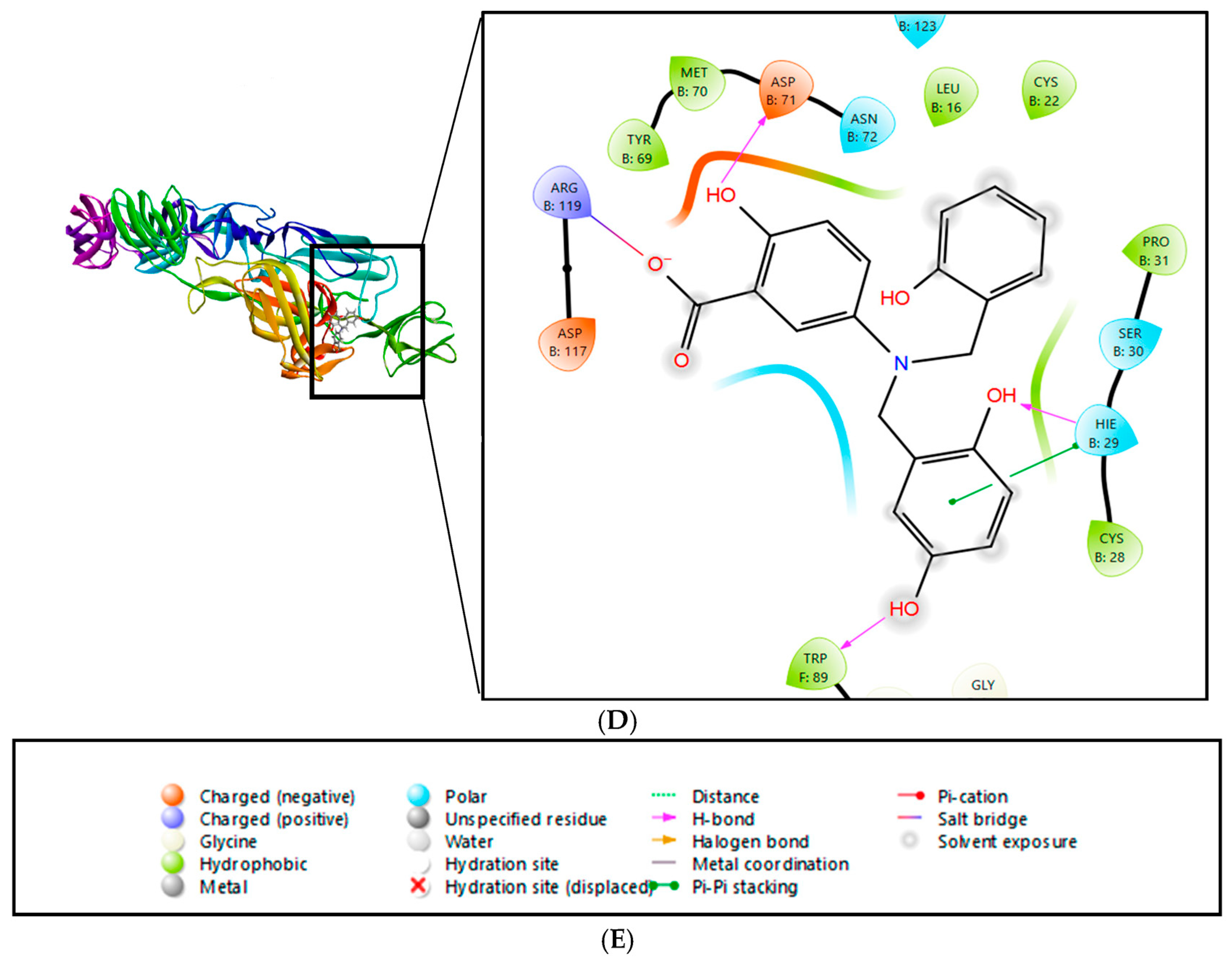
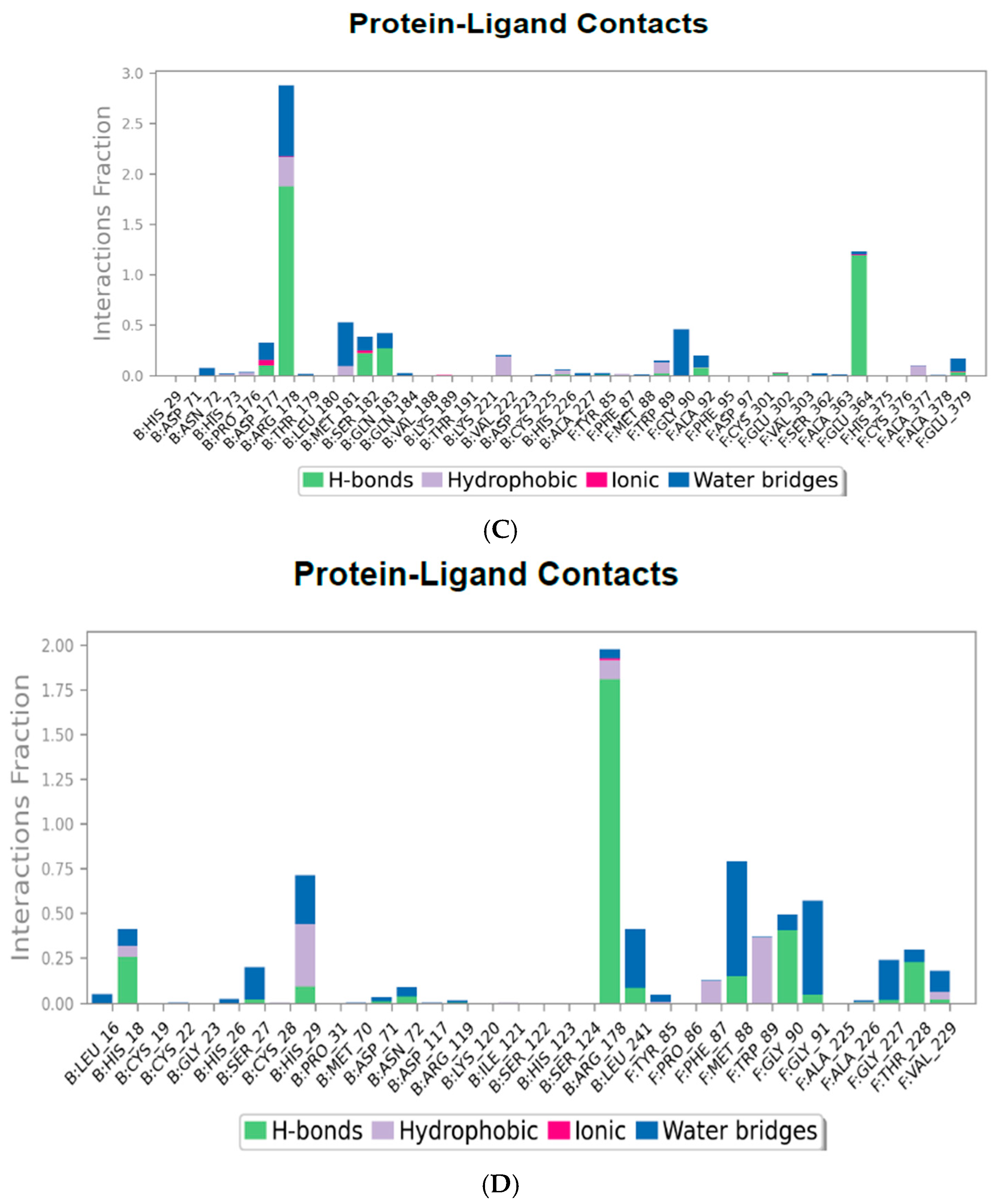
2.2. Interactions and Residue Contacts Between Protein and Ligands
2.3. ADME/T Properties Analysis
2.4. Molecular Dynamics Simulation
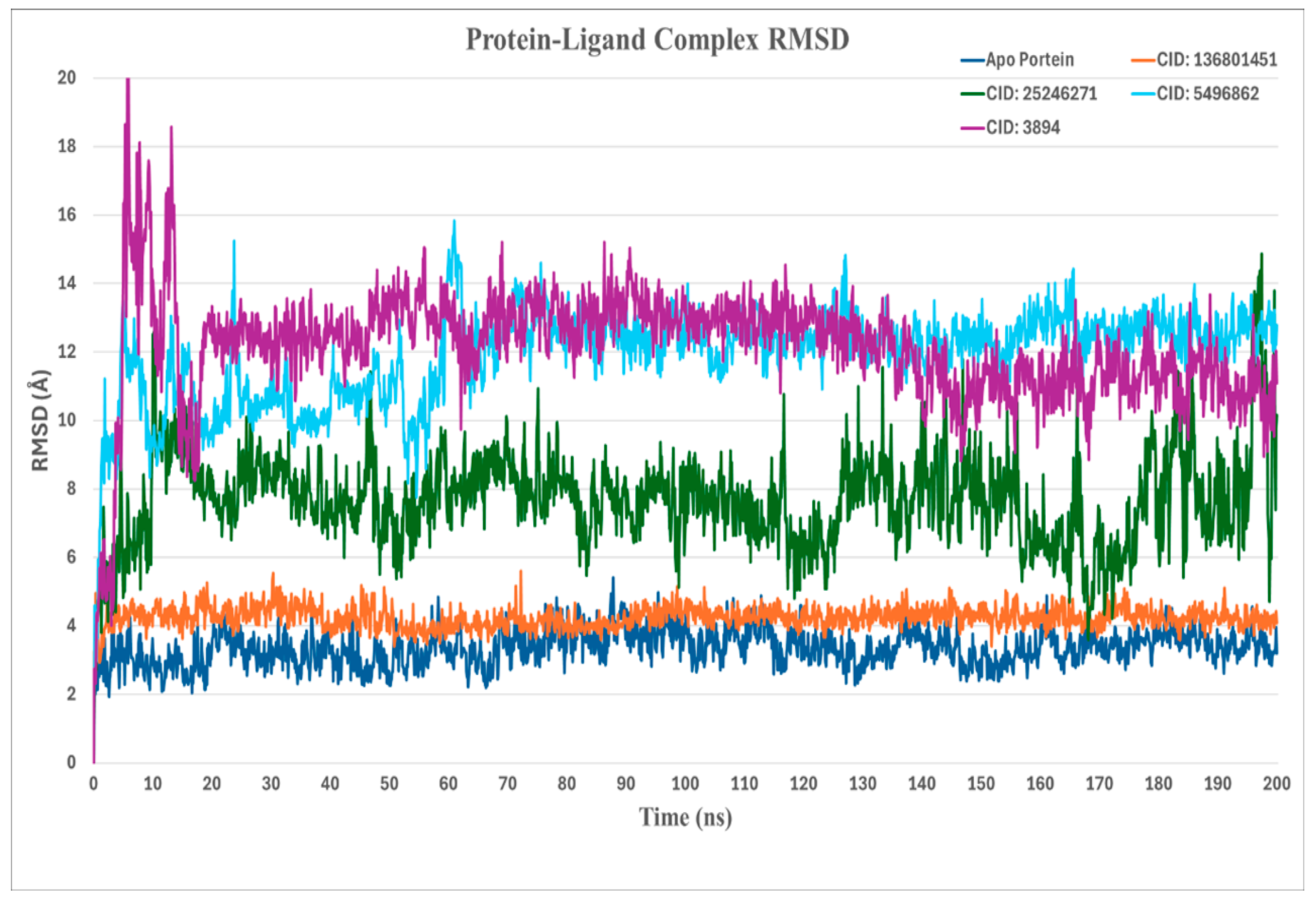
2.4.1. RMSD (Root-Mean-Square Deviation) Analysis
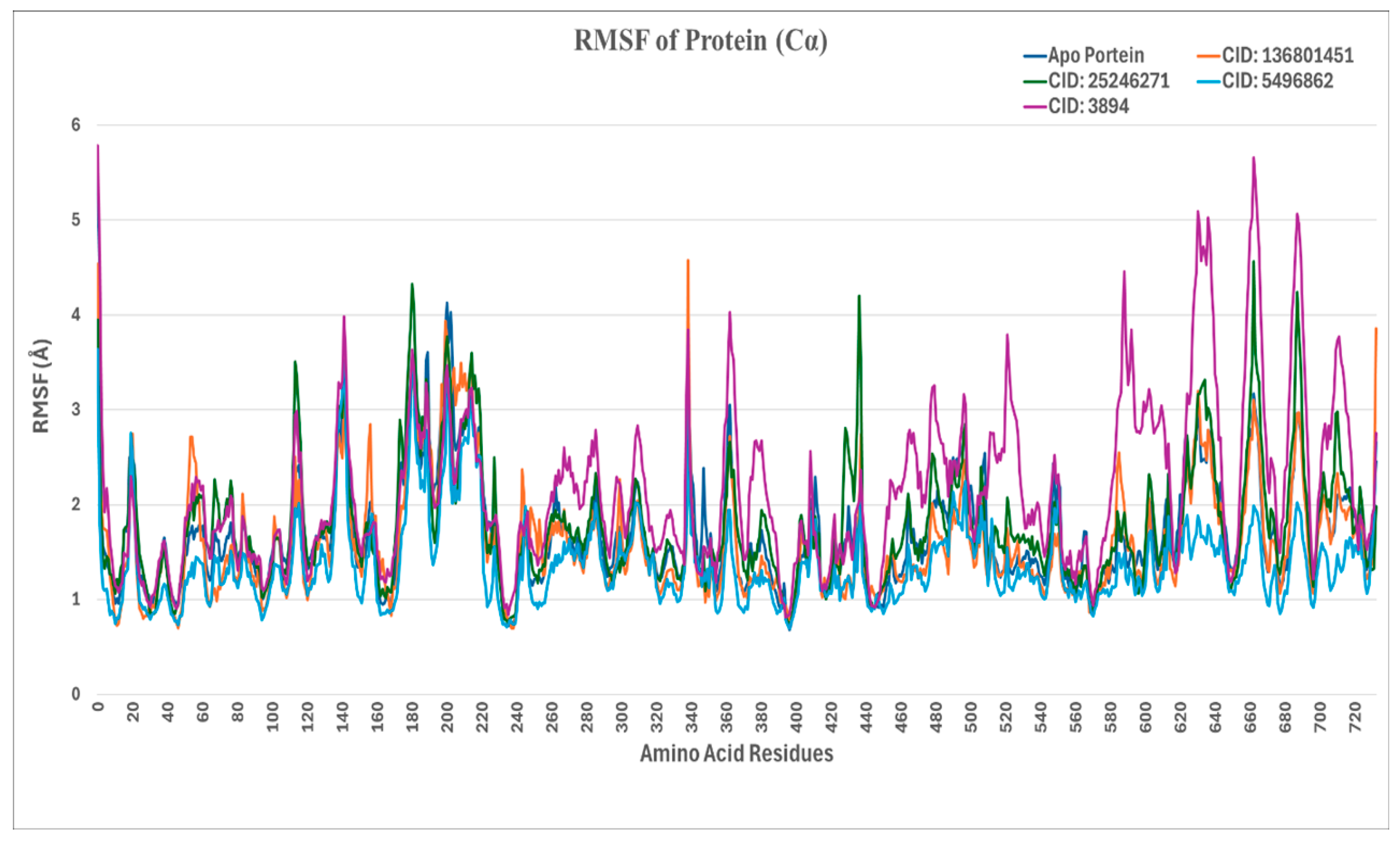
2.4.2. RMSF (Root-Mean-Square Fluctuation) Analysis
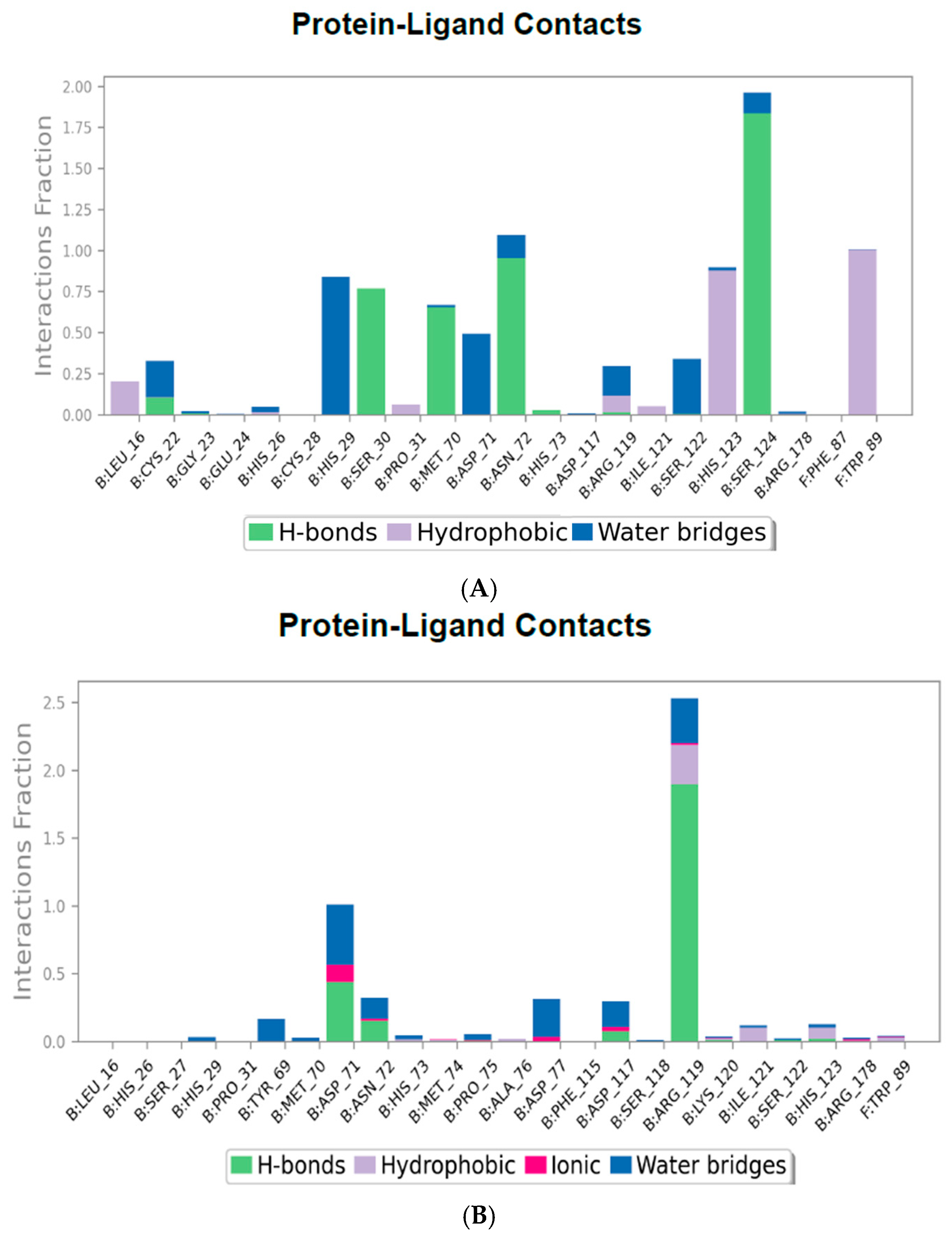
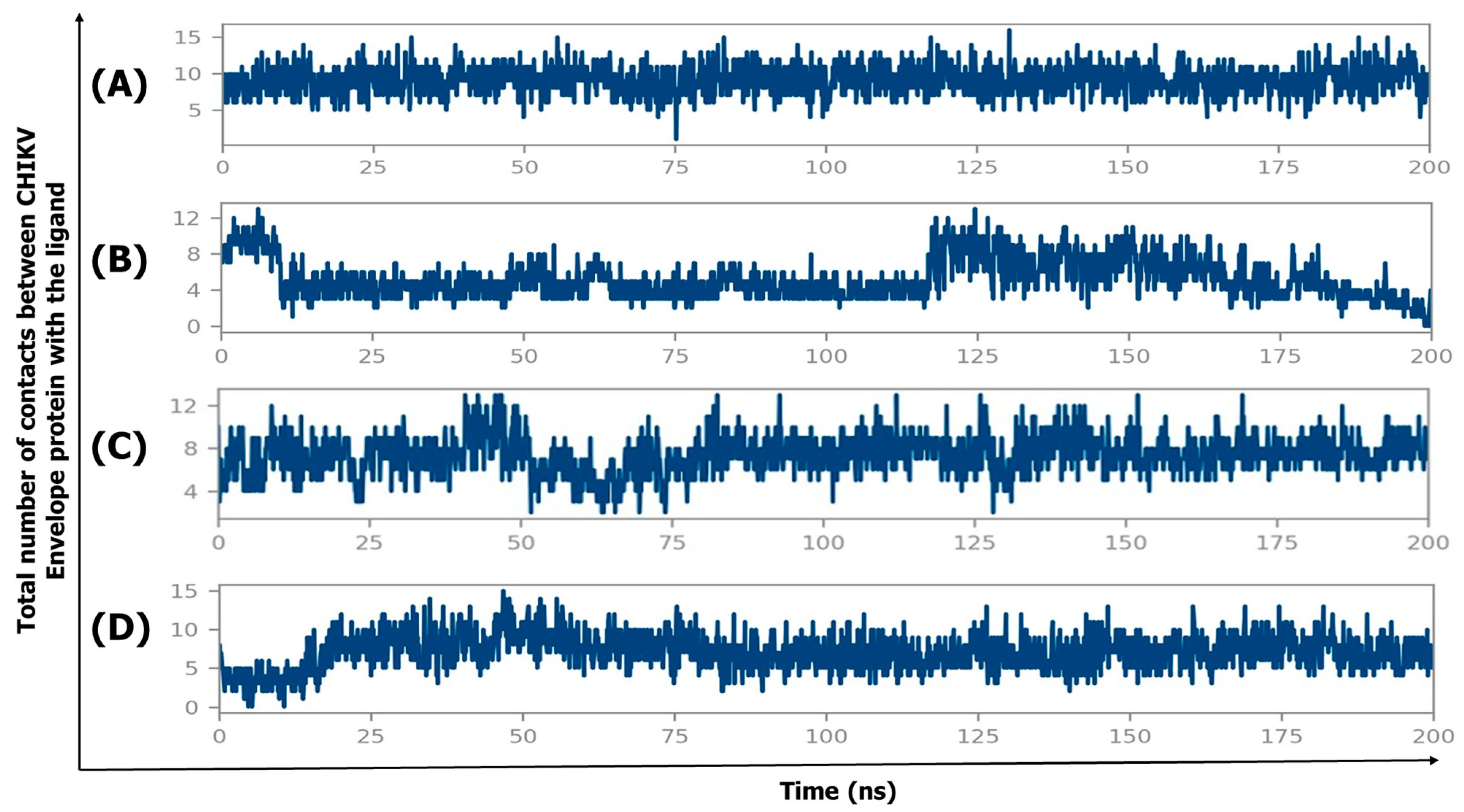
2.4.3. Protein–Ligand Contact Mapping
2.4.4. Protein–Ligand Contact Timeline
2.4.5. MM/GBSA Computations from MD Trajectories
3. Discussion
4. Materials and Methods
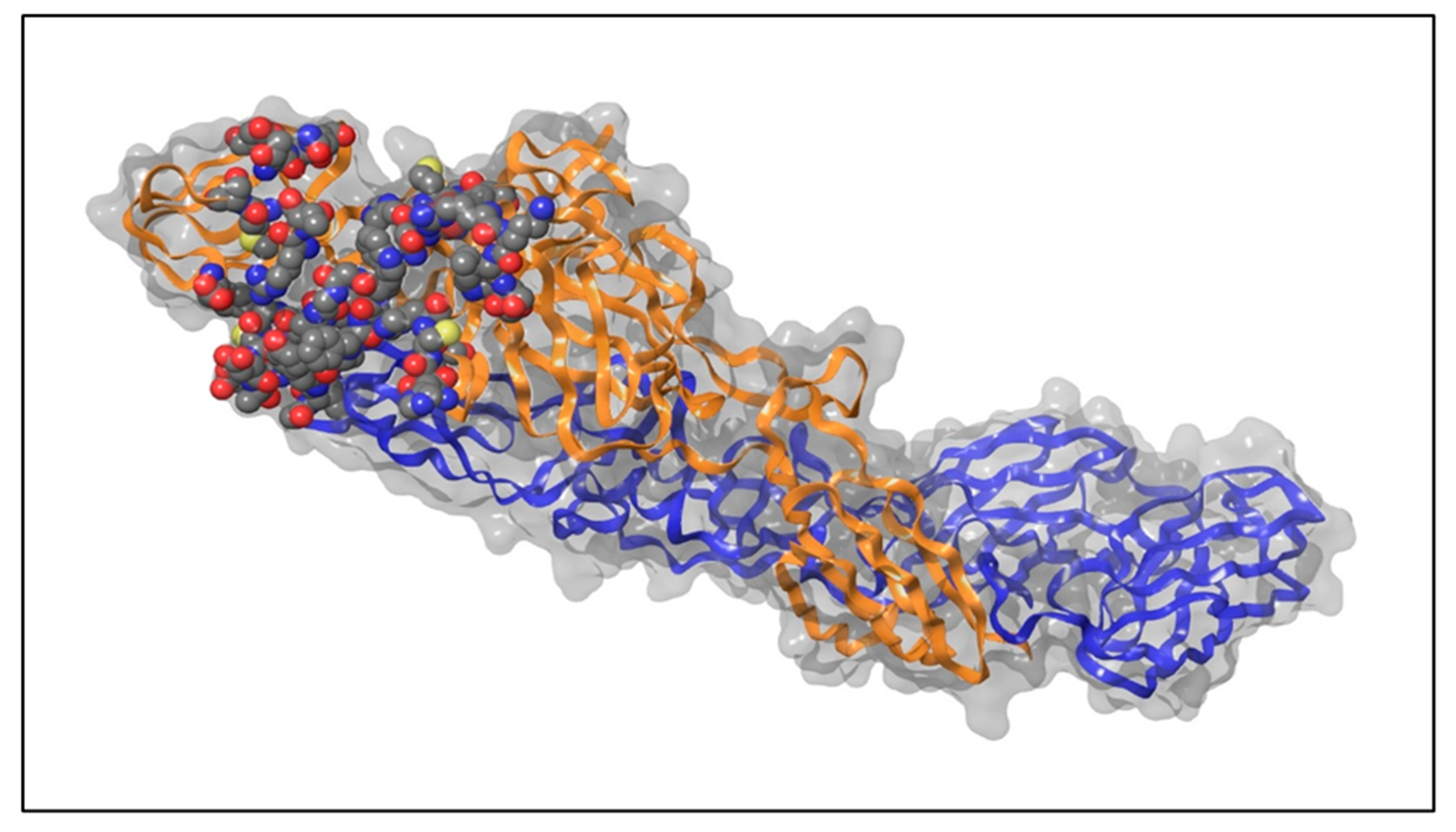
4.1. Retrieval and Preparation of Protein Structure
4.2. Ligand Preparation
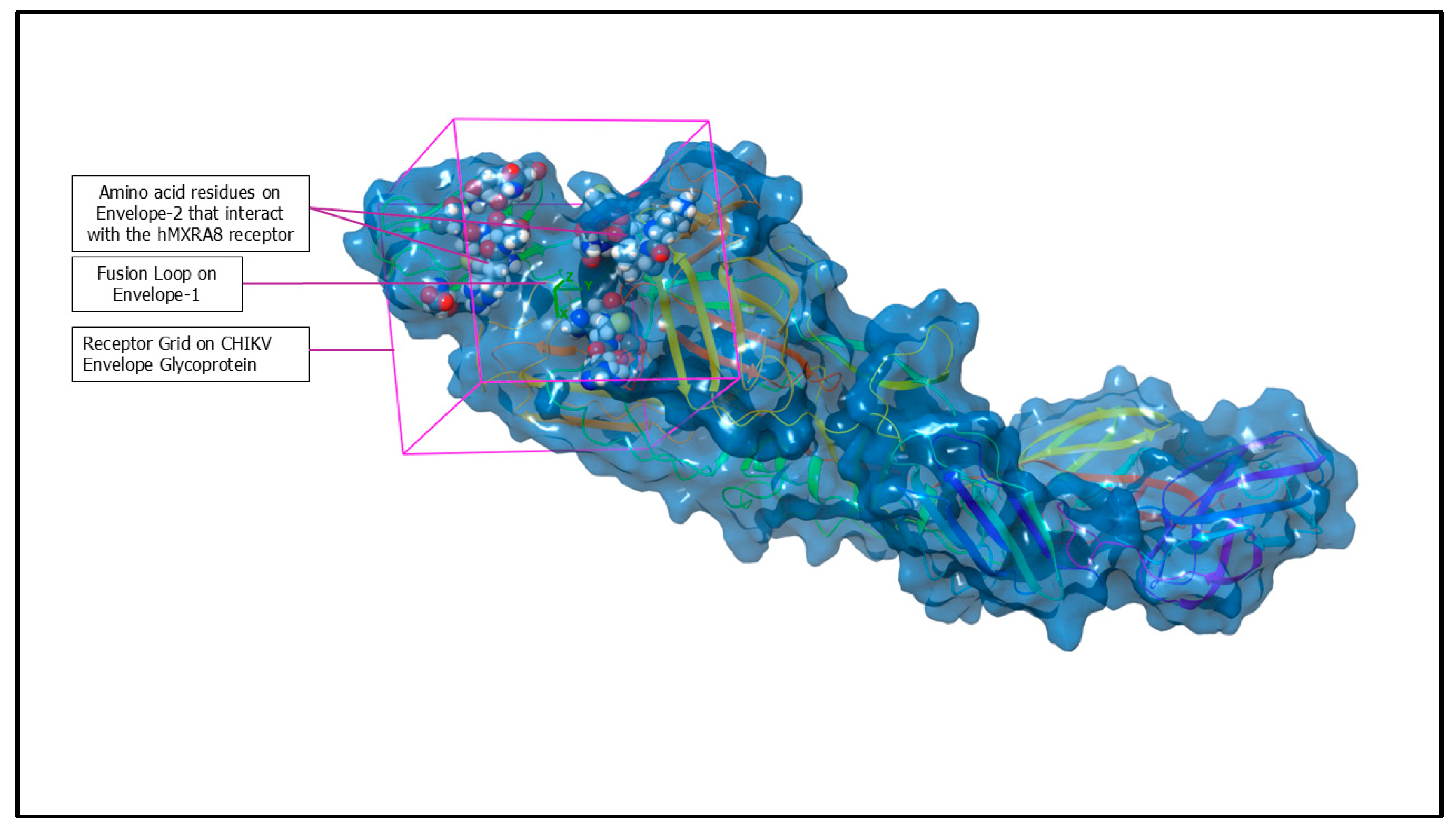
4.3. Receptor Grid Generation
4.4. Molecular Docking
4.5. Binding Free Energy Calculations (Rescoring Function)
4.6. Ligand-Based ADME/Tox Prediction
4.7. Molecular Dynamics Simulation and Binding Free Energy Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CHIKV | Chikungunya virus |
| MM-GBSA | Molecular Mechanics-Generalized Born Surface Area |
| OPLS | Optimized Potentials for Liquid Simulations |
| RMSD | Root-mean-square deviation |
| RMSF | Root-mean-square fluctuation |
| TIP3P | Transferable Intermolecular Potential 3 Points |
References
- Schwartz, O.; Albert, M.L. Biology and pathogenesis of chikungunya virus. Nat. Rev. Microbiol. 2010, 8, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Chikungunya Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/chikungunya (accessed on 6 March 2025).
- Anwar, S.; Taslem Mourosi, J.; Khan, M.F.; Ullah, M.O.; Vanakker, O.M.; Hosen, M.J. Chikungunya outbreak in Bangladesh (2017): Clinical and hematological findings. PLoS Negl. Trop. Dis. 2020, 14, e0007466. [Google Scholar] [CrossRef] [PubMed]
- Bettis, A.A.; L’Azou Jackson, M.; Yoon, I.-K.; Breugelmans, J.G.; Goios, A.; Gubler, D.J.; Powers, A.M. The global epidemiology of chikungunya from 1999 to 2020: A systematic literature review to inform the development and introduction of vaccines. PLoS Negl. Trop. Dis. 2022, 16, e0010069. [Google Scholar] [CrossRef] [PubMed]
- de Souza, W.M.; Ribeiro, G.S.; de Lima, S.T.S.; de Jesus, R.; Moreira, F.R.R.; Whittaker, C.; Sallum, M.A.M.; Carrington, C.V.F.; Sabino, E.C.; Kitron, U.; et al. Chikungunya: A decade of burden in the Americas. Lancet Reg. Health Am. 2024, 30, 100673. [Google Scholar] [CrossRef]
- Chikungunya|CDC Yellow Book. 2024. Available online: https://wwwnc.cdc.gov/travel/yellowbook/2024/infections-diseases/chikungunya (accessed on 6 March 2025).
- Chikungunya: Symptoms, Prevention and Treatments—PAHO/WHO. Available online: https://www.paho.org/en/topics/chikungunya (accessed on 6 March 2025).
- Staples, J.E.; Breiman, R.F.; Powers, A.M. Chikungunya Fever: An Epidemiological Review of a Re-Emerging Infectious Disease. Clin. Infect. Dis. 2009, 49, 942–948. [Google Scholar] [CrossRef]
- Weber, W.C.; Streblow, Z.J.; Kreklywich, C.N.; Denton, M.; Sulgey, G.; Streblow, M.M.; Marcano, D.; Flores, P.N.; Rodriguez-Santiago, R.M.; Alvarado, L.I.; et al. The Approved Live-Attenuated Chikungunya Virus Vaccine (IXCHIQ®) Elicits Cross-Neutralizing Antibody Breadth Extending to Multiple Arthritogenic Alphaviruses Similar to the Antibody Breadth Following Natural Infection. Vaccines 2024, 12, 893. [Google Scholar] [CrossRef]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 2018, 557, 570–574. [Google Scholar] [CrossRef]
- Schnierle, B.S. Cellular Attachment and Entry Factors for Chikungunya Virus. Viruses 2019, 11, 1078. [Google Scholar] [CrossRef]
- Song, H.; Zhao, Z.; Chai, Y.; Jin, X.; Li, C.; Yuan, F.; Liu, S.; Gao, Z.; Wang, H.; Song, J.; et al. Molecular Basis of Arthritogenic Alphavirus Receptor MXRA8 Binding to Chikungunya Virus Envelope Protein. Cell 2019, 177, 1714–1724.e12. [Google Scholar] [CrossRef]
- Feng, F.; Bouma, E.M.; Hu, G.; Zhu, Y.; Yu, Y.; Smit, J.M.; Diamond, M.S.; Zhang, R. Colocalization of Chikungunya Virus with Its Receptor MXRA8 during Cell Attachment, Internalization, and Membrane Fusion. J. Virol. 2023, 97, e01557-22. [Google Scholar] [CrossRef]
- Weber, C.; Sliva, K.; Von Rhein, C.; Kümmerer, B.M.; Schnierle, B.S. The green tea catechin, epigallocatechin gallate inhibits chikungunya virus infection. Antivir. Res. 2015, 113, 1–3. [Google Scholar] [CrossRef]
- Kovacikova, K.; Van Hemert, M.J. Small-Molecule Inhibitors of Chikungunya Virus: Mechanisms of Action and Antiviral Drug Resistance. Antimicrob. Agents Chemother. 2020, 64, e01788-20. [Google Scholar] [CrossRef] [PubMed]
- Battisti, V.; Urban, E.; Langer, T. Antivirals against the Chikungunya Virus. Viruses 2021, 13, 1307. [Google Scholar] [CrossRef] [PubMed]
- Hucke, F.I.L.; Bugert, J.J. Current and Promising Antivirals Against Chikungunya Virus. Front. Public Health 2020, 8, 618624. [Google Scholar] [CrossRef]
- Battini, L.; Fidalgo, D.M.; Álvarez, D.E.; Bollini, M. Discovery of a Potent and Selective Chikungunya Virus Envelope Protein Inhibitor through Computer-Aided Drug Design. ACS Infect. Dis. 2021, 7, 1503–1518. [Google Scholar] [CrossRef]
- Silva, L.R.; Rodrigues, É.E.D.S.; Taniele-Silva, J.; Anderson, L.; Araújo-Júnior, J.X.D.; Bassi, Ê.J.; Silva-Júnior, E.F.D. Targeting Chikungunya Virus Entry: Alternatives for New Inhibitorsin Drug Discovery. Curr. Med. Chem. 2022, 29, 612–634. [Google Scholar] [CrossRef] [PubMed]
- Verma, J.; Hasan, A.; Sunil, S.; Subbarao, N. In silico identification and in vitro antiviral validation of potential inhibitors against Chikungunya virus. J. Comput. Aided Mol. Des. 2022, 36, 521–536. [Google Scholar] [CrossRef]
- De Wispelaere, M.; Lian, W.; Potisopon, S.; Li, P.-C.; Jang, J.; Ficarro, S.B.; Clark, M.J.; Zhu, X.; Kaplan, J.B.; Pitts, J.D.; et al. Inhibition of Flaviviruses by Targeting a Conserved Pocket on the Viral Envelope Protein. Cell Chem. Biol. 2018, 25, 1006–1016.e8. [Google Scholar] [CrossRef]
- Helle, F.; Wychowski, C.; Vu-Dac, N.; Gustafson, K.R.; Voisset, C.; Dubuisson, J. Cyanovirin-N Inhibits Hepatitis C Virus Entry by Binding to Envelope Protein Glycans. J. Biol. Chem. 2006, 281, 25177–25183. [Google Scholar] [CrossRef]
- Albulescu, I.C.; Van Hoolwerff, M.; Wolters, L.A.; Bottaro, E.; Nastruzzi, C.; Yang, S.C.; Tsay, S.-C.; Hwu, J.R.; Snijder, E.J.; Van Hemert, M.J. Suramin inhibits chikungunya virus replication through multiple mechanisms. Antivir. Res. 2015, 121, 39–46. [Google Scholar] [CrossRef]
- Ho, Y.-J.; Wang, Y.-M.; Lu, J.; Wu, T.-Y.; Lin, L.-I.; Kuo, S.-C.; Lin, C.-C. Suramin Inhibits Chikungunya Virus Entry and Transmission. PLoS ONE 2015, 10, e0133511. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-W.; Hsieh, P.-S.; Lin, C.-C.; Hu, M.-K.; Huang, S.-M.; Wang, Y.-M.; Liang, C.-Y.; Gong, Z.; Ho, Y.-J. Synergistic effects of combination treatment using EGCG and suramin against the chikungunya virus. Biochem. Biophys. Res. Commun. 2017, 491, 595–602. [Google Scholar] [CrossRef] [PubMed]
- Oo, A.; Hassandarvish, P.; Chin, S.P.; Lee, V.S.; Abu Bakar, S.; Zandi, K. In silico study on anti-Chikungunya virus activity of hesperetin. PeerJ 2016, 4, e2602. [Google Scholar] [CrossRef]
- Mangala Prasad, V.; Blijleven, J.S.; Smit, J.M.; Lee, K.K. Visualization of conformational changes and membrane remodeling leading to genome delivery by viral class-II fusion machinery. Nat. Commun. 2022, 13, 4772. [Google Scholar] [CrossRef]
- De Paris, R.; Quevedo, C.V.; Ruiz, D.D.A.; Norberto De Souza, O. An Effective Approach for Clustering InhA Molecular Dynamics Trajectory Using Substrate-Binding Cavity Features. PLoS ONE 2015, 10, e0133172. [Google Scholar] [CrossRef]
- Puranik, N.V.; Rani, R.; Singh, V.A.; Tomar, S.; Puntambekar, H.M.; Srivastava, P. Evaluation of the Antiviral Potential of Halogenated Dihydrorugosaflavonoids and Molecular Modeling with nsP3 Protein of Chikungunya Virus (CHIKV). ACS Omega 2019, 4, 20335–20345. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.S.; Simeonov, A.; Jadhav, A.; Eidam, O.; Mott, B.T.; Keiser, M.J.; McKerrow, J.H.; Maloney, D.J.; Irwin, J.J.; Shoichet, B.K. Complementarity between docking and HTS in discovering new cruzain inhibitors. J. Med. Chem. 2010, 53, 4891–4905. [Google Scholar] [CrossRef]
- Das, P.K.; Puusepp, L.; Varghese, F.S.; Utt, A.; Ahola, T.; Kananovich, D.G.; Lopp, M.; Merits, A.; Karelson, M. Design and Validation of Novel Chikungunya Virus Protease Inhibitors. Antimicrob. Agents Chemother. 2016, 60, 7382–7395. [Google Scholar] [CrossRef]
- Voss, J.E.; Vaney, M.-C.; Duquerroy, S.; Vonrhein, C.; Girard-Blanc, C.; Crublet, E.; Thompson, A.; Bricogne, G.; Rey, F.A. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 2010, 468, 709–712. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Hymavati; Kumar, V.; Elizabeth Sobhia, M. Implication of Crystal Water Molecules in Inhibitor Binding at ALR2 Active Site. Comput. Math. Methods Med. 2012, 2012, 541594. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Dhasmana, A.; Raza, S.; Jahan, R.; Lohani, M.; Arif, J.M. High-Throughput Virtual Screening (HTVS) of Natural Compounds and Exploration of Their Biomolecular Mechanisms. In New Look to Phytomedicine; Elsevier: Amsterdam, The Netherlands, 2019; pp. 523–548. ISBN 978-0-12-814619-4. [Google Scholar]
- Subramaniam, S.; Mehrotra, M.; Gupta, D. Virtual high throughput screening (vHTS)—A perspective. Bioinformation 2008, 3, 14–17. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Rodosy, F.B.; Azad, M.A.K.; Halder, S.K.; Limon, M.B.H.; Jaman, S.; Lata, N.A.; Sarker, M.; Riya, A.I. The potential of phytochemicals against epidermal growth factor receptor tyrosine kinase (EGFRK): An insight from molecular dynamic simulations. J. Biomol. Struct. Dyn. 2024, 42, 2482–2493. [Google Scholar] [CrossRef]
- “Schrödinger Release Notes—Release 2023-2,” Schrödinger. Available online: https://www.schrodinger.com/life-science/download/release-notes/release-2023-2/ (accessed on 6 March 2025).
- Saxena, S.; Durgam, L.; Guruprasad, L. Multiple e-pharmacophore modelling pooled with high-throughput virtual screening, docking and molecular dynamics simulations to discover potential inhibitors of Plasmodium falciparum lactate dehydrogenase (PfLDH). J. Biomol. Struct. Dyn. 2019, 37, 1783–1799. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kokubo, H. Exploring the Stability of Ligand Binding Modes to Proteins by Molecular Dynamics Simulations: A Cross-docking Study. J. Chem. Inf. Model. 2017, 57, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Harder, E.; Damm, W.; Friesner, R.A.; Sherman, W. Improving the Prediction of Absolute Solvation Free Energies Using the Next Generation OPLS Force Field. J. Chem. Theory Comput. 2012, 8, 2553–2558. [Google Scholar] [CrossRef] [PubMed]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building Water Models: A Different Approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar] [CrossRef]
- Sigala, P.A.; Tsuchida, M.A.; Herschlag, D. Hydrogen bond dynamics in the active site of photoactive yellow protein. Proc. Natl. Acad. Sci. USA 2009, 106, 9232–9237. [Google Scholar] [CrossRef]
- Qiu, S.; Azofra, L.M.; MacFarlane, D.R.; Sun, C. Hydrogen bonding effect between active site and protein environment on catalysis performance in H2 -producing [NiFe] hydrogenases. Phys. Chem. Chem. Phys. 2018, 20, 6735–6743. [Google Scholar] [CrossRef]
- Azam, F.; Eid, E.E.M.; Almutairi, A. Targeting SARS-CoV-2 main protease by teicoplanin: A mechanistic insight by docking, MM/GBSA and molecular dynamics simulation. J. Mol. Struct. 2021, 1246, 131124. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial No. | Compound PubChem ID (CID) | XP GScore | Docking Score |
|---|---|---|---|
| 1 | 136801451 | −10.227 | −10.227 |
| 2 | 25246271 | −10.362 | −9.39 |
| 3 | 5496862 | −9.365 | −9.362 |
| 4 | 3894 | −9.169 | −9.169 |
| Criteria | CID 136801451 | CID 25246271 | CID 5496862 | CID 3894 | Acceptable Range |
|---|---|---|---|---|---|
| QPpolrz | 36.463 | 19.185 | 40.583 | 34.795 | 13.0 to 70.0 |
| QPlogPC16 | 13.309 | 8.563 | 14.564 | 13.336 | 4.0 to 18.0 |
| QPlogPoct | 22.937 | 18.922 | 24.058 | 21.202 | 8.0 to 35.0 |
| QPlogPw | 16.089 | 16.072 | 15.246 | 13.428 | 4.0 to 45.0 |
| QPlogPo/w | 1.764 | −2.393 | 3.336 | 2.94 | −2 to 6.5 |
| QPlogS | −4.176 | −0.618 | −4.849 | −3.281 | −6.5 to 0.5 |
| QPlogHERG | −5.794 | −0.619 | −4.103 | −3.269 | Concern below −5 |
| QPPCaco | 53.22 | 0.702 | 17.129 | 16.864 | <25 = poor, >500 = Great |
| QPlogBB | −2.477 | −1.585 | −2.519 | −2.039 | −3 to 12 |
| QPPMDCK | 20.768 | 0.345 | 7.756 | 7.627 | <25 = poor, >500 = Great |
| QPlogKp | −4.319 | −7.18 | −5.971 | −3.670 | −8 to −1 |
| QPlogKhsa | −0.113 | −1.182 | 0.024 | −0.095 | −1.5 to 1.5 |
| HOAP | 68.166 | 10.183 | 68.557 | 66.117 | <25 = poor |
| #metab | 6 | 7 | 7 | 7 | 1 to 8 |
| #rtvFG | 0 | 1 | 0 | 0 | 0 to 2 |
| CNS | −2 | −2 | −2 | −2 | −2 to +2 |
| Glob | 0.799196 | 0.895275 | 0.793301 | 34.795 | 13.0 to 70.0 |
| Rule of Five | 0 | 0 | 0 | 0 | 4.0 to 18.0 |
| Serial No | Compound PubChem ID (CID) | MMGBSA ΔG Bind Energy of Docked Conformation a (kcal/mol) | Binding Energy Calculated from Frames of the MD Simulation Trajectories b (Kcal/mol) |
|---|---|---|---|
| 1 | 136801451 | −51.53 | −62.48 ± 9.06 |
| 2 | 25246271 | −3.58 | −8.19 ± 4.84 |
| 3 | 5496862 | −20.06 | −32.43 ± 11.62 |
| 4 | 3894 | −24.18 | −38.08 ± 8.40 |
| CHIKV Envelope | Amino Acids | Positions on Envelope Proteins |
|---|---|---|
| Envelope-2 (E2) (Chain-B of 3N42) | His | 18, 26, 29 |
| Ser | 27, 182 | |
| Cys | 28 | |
| Asp | 71, 214, 223 | |
| Asn | 72, 193 | |
| Met | 74, 181 | |
| Pro | 75 | |
| Ala | 76 | |
| Arg | 119, 178 | |
| Lys | 120 | |
| Ile | 121 | |
| Thr | 179, 191 | |
| Val | 192 | |
| Envelope-1 (E1) (Chain-F of 3N42) | Gly | 83, 90, 91 |
| Val | 84 | |
| Tyr | 85, 93 | |
| Pro | 86 | |
| Phe | 87, 95 | |
| Met | 88 | |
| Trp | 89 | |
| Ala | 92 | |
| Asp | 97 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.M.; Limon, M.B.H.; Saikat, T.A.; Saha, P.; Nahid, A.H.; Alam, M.M.; Rahman, M.Z. Potent Inhibition of Chikungunya Virus Entry by a Pyrazole–Benzene Derivative: A Computational Study Targeting the E1–E2 Glycoprotein Complex. Int. J. Mol. Sci. 2025, 26, 6480. https://doi.org/10.3390/ijms26136480
Rahman MM, Limon MBH, Saikat TA, Saha P, Nahid AH, Alam MM, Rahman MZ. Potent Inhibition of Chikungunya Virus Entry by a Pyrazole–Benzene Derivative: A Computational Study Targeting the E1–E2 Glycoprotein Complex. International Journal of Molecular Sciences. 2025; 26(13):6480. https://doi.org/10.3390/ijms26136480
Chicago/Turabian StyleRahman, Md. Mohibur, Md. Belayet Hasan Limon, Tanvir Ahmed Saikat, Poulomi Saha, Abdul Hadi Nahid, Mohammad Mamun Alam, and Mohammed Ziaur Rahman. 2025. "Potent Inhibition of Chikungunya Virus Entry by a Pyrazole–Benzene Derivative: A Computational Study Targeting the E1–E2 Glycoprotein Complex" International Journal of Molecular Sciences 26, no. 13: 6480. https://doi.org/10.3390/ijms26136480
APA StyleRahman, M. M., Limon, M. B. H., Saikat, T. A., Saha, P., Nahid, A. H., Alam, M. M., & Rahman, M. Z. (2025). Potent Inhibition of Chikungunya Virus Entry by a Pyrazole–Benzene Derivative: A Computational Study Targeting the E1–E2 Glycoprotein Complex. International Journal of Molecular Sciences, 26(13), 6480. https://doi.org/10.3390/ijms26136480