
Novel Derivatives of 3-Amino-4-hydroxy-benzenesulfonamide: Synthesis, Binding to Carbonic Anhydrases, and Activity in Cancer Cell 2D and 3D Cultures


 , , , ,
, , , ,  ,
,  , and
, and
Abstract
1. Introduction
2. Results and Discussion
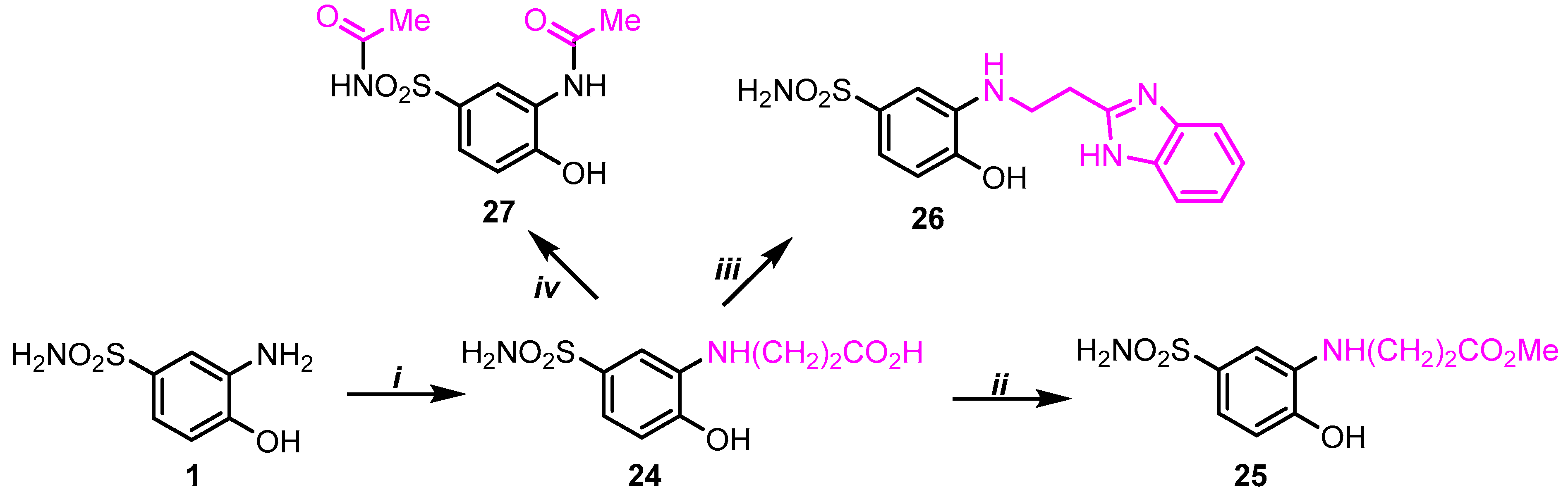
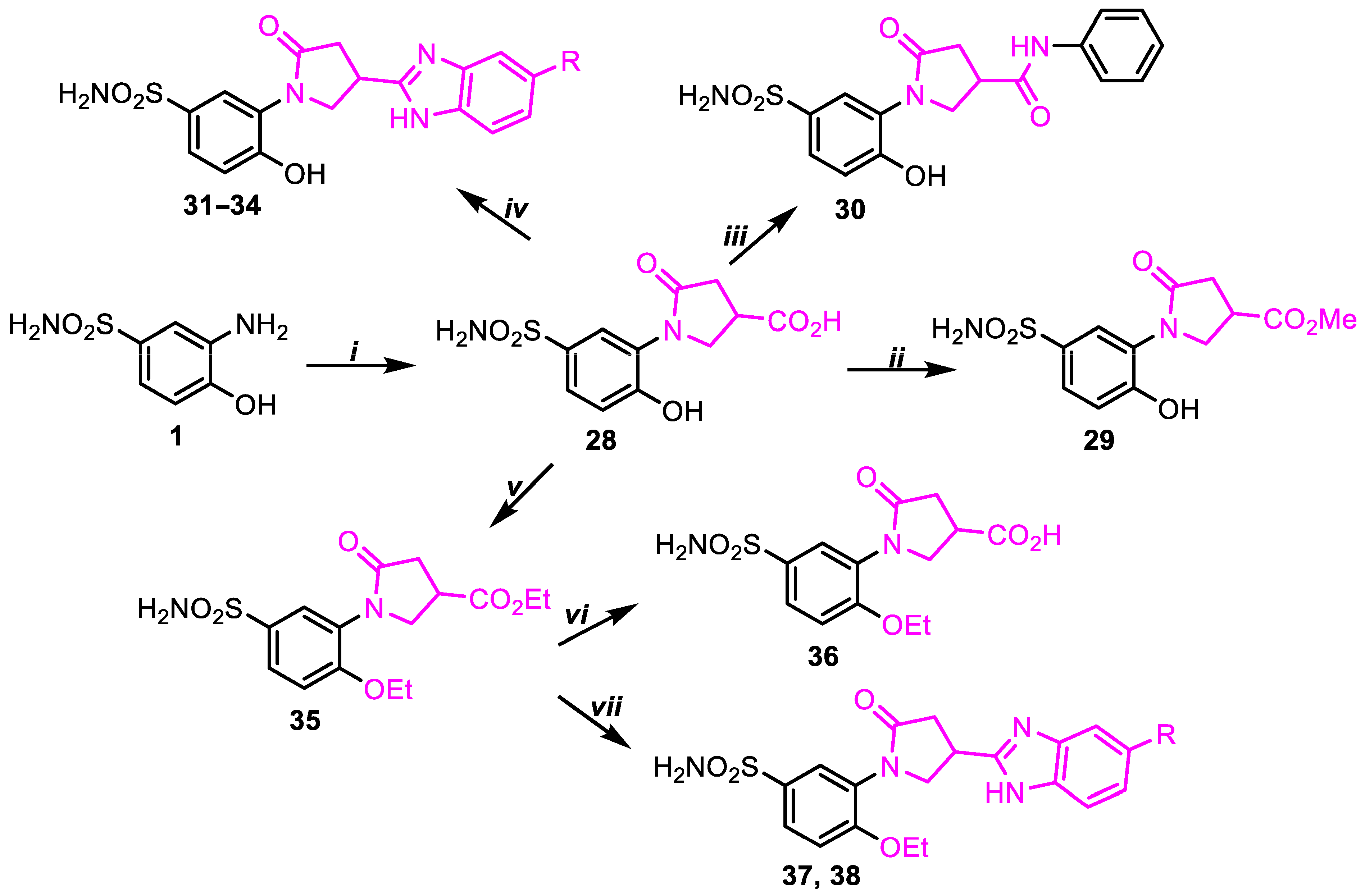
2.1. Chemistry
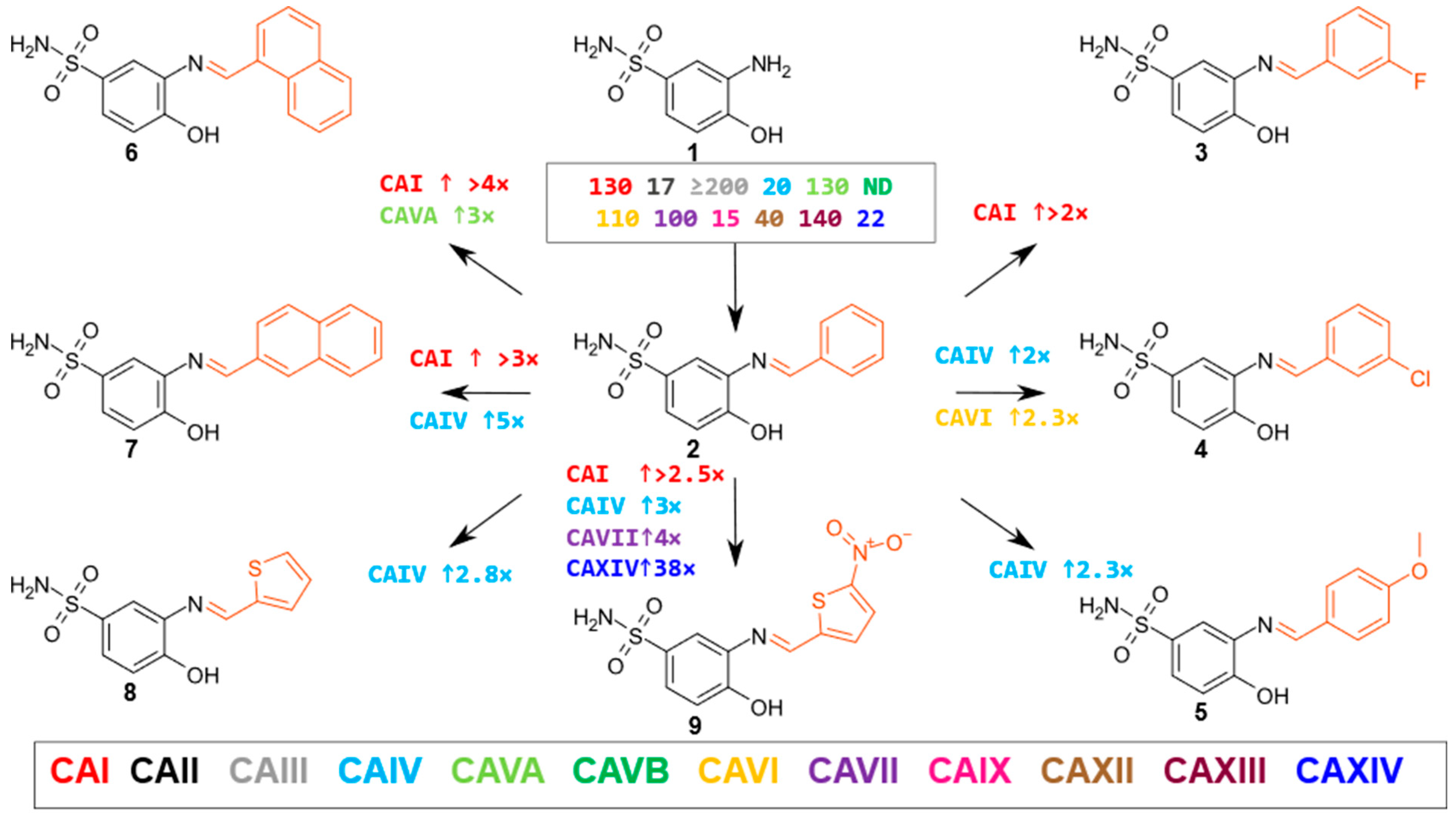
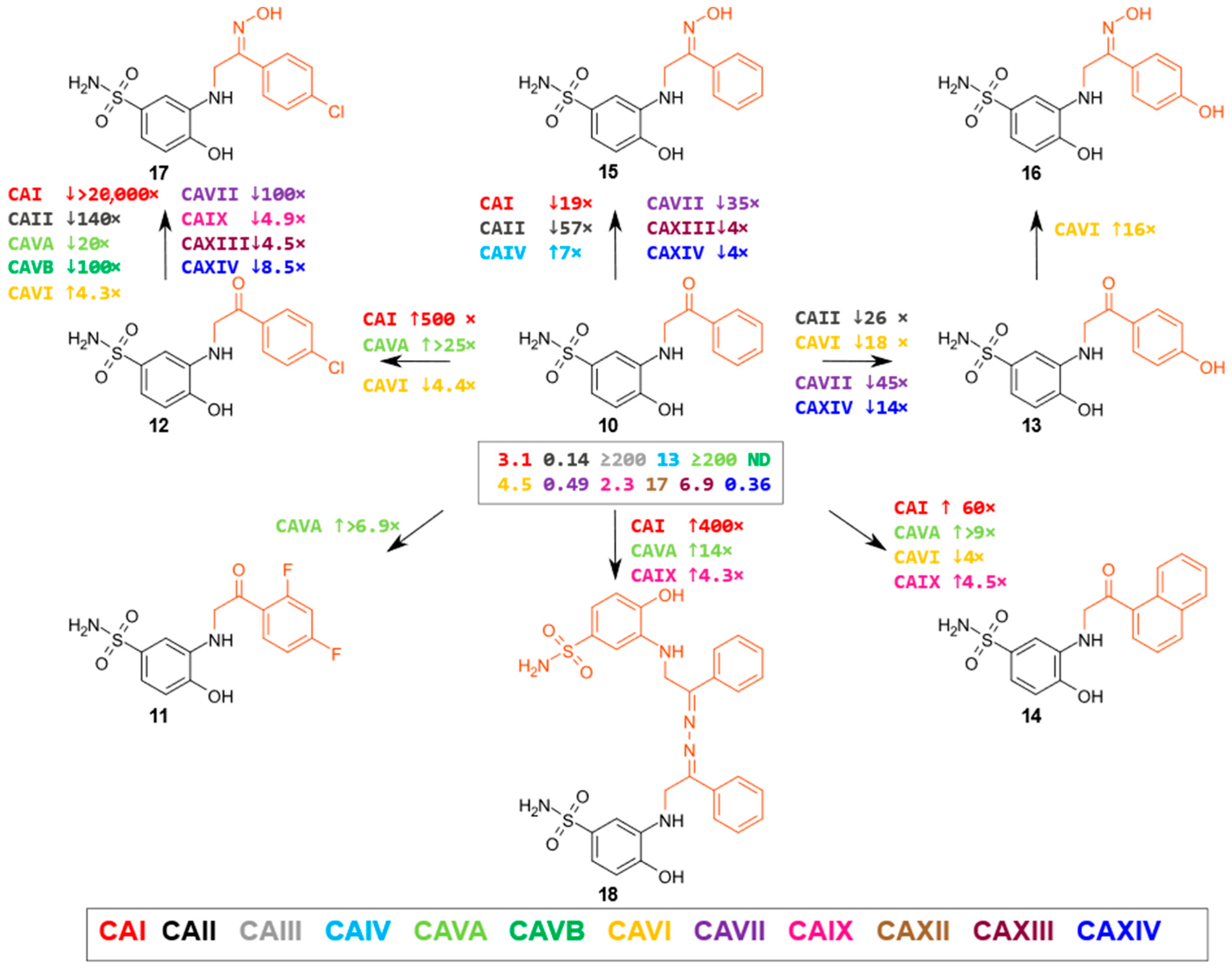
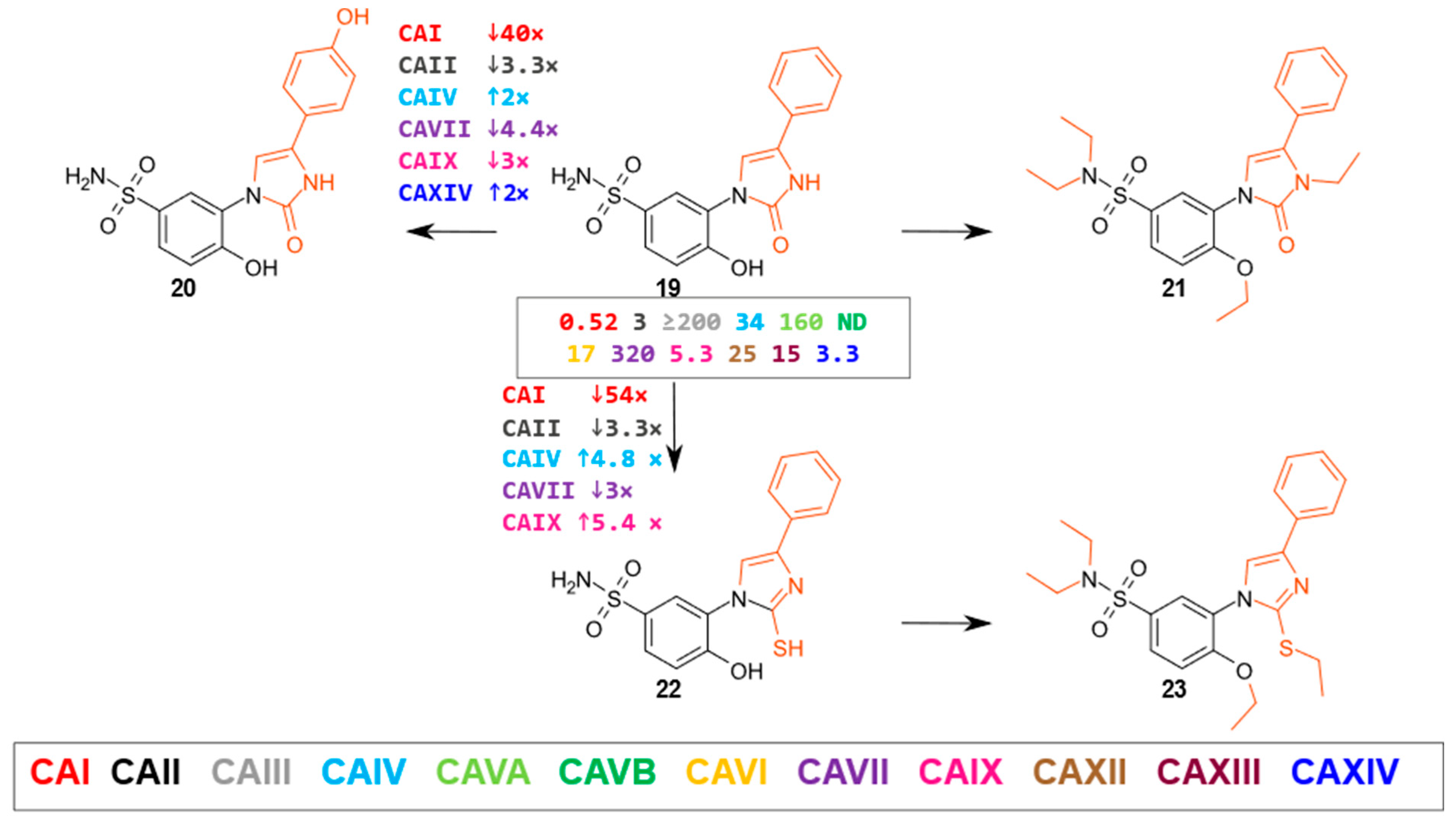
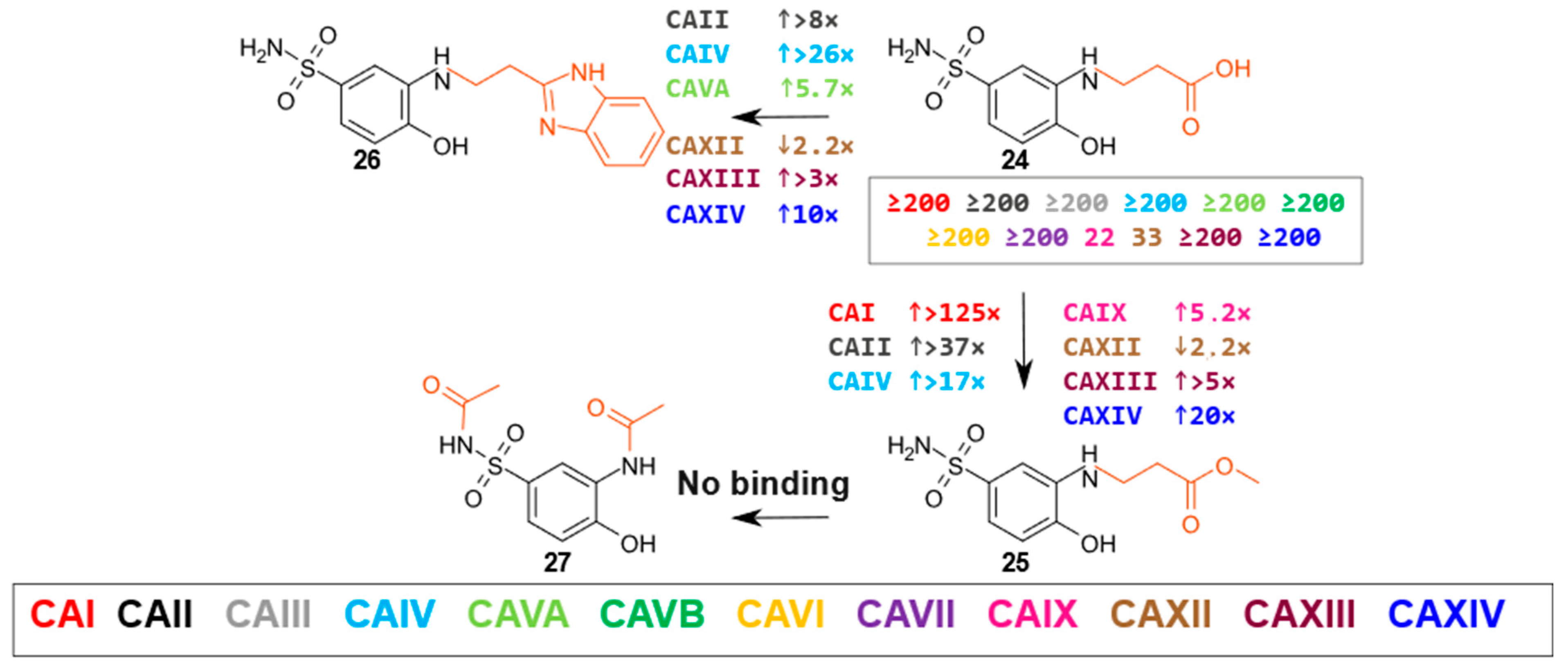
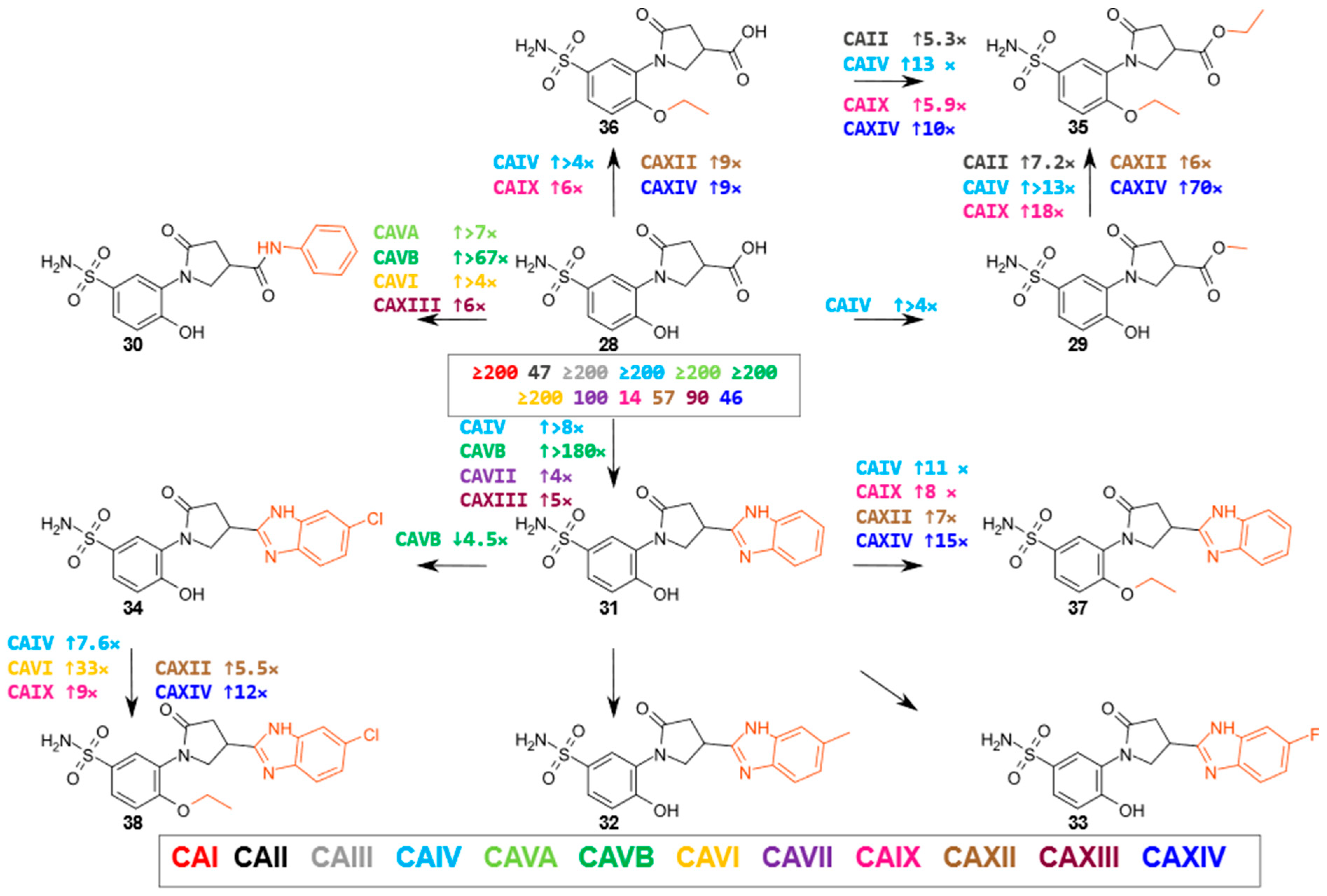
2.2. Compound Binding to Human Carbonic Anhydrases
- Oxoimidazoles
- β-Amino acids and their derivatives
- 3-substituted-5-oxopyrrolidine derivatives
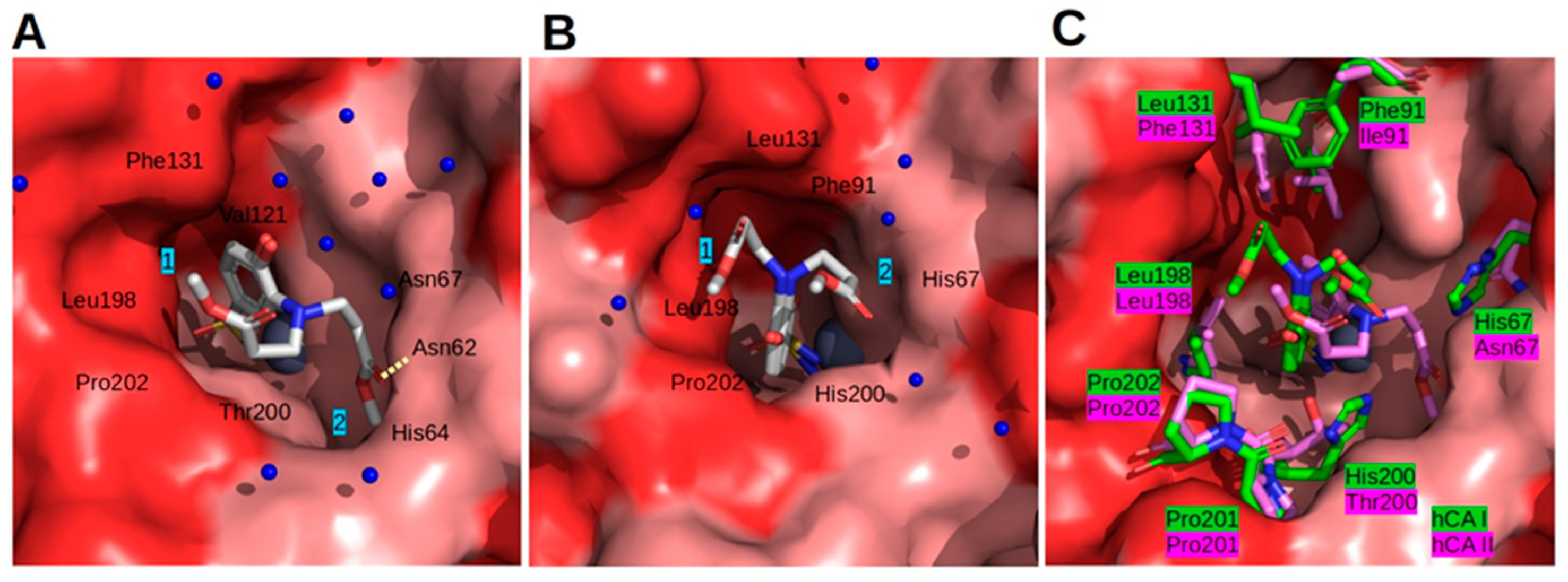
2.3. Crystal Structures
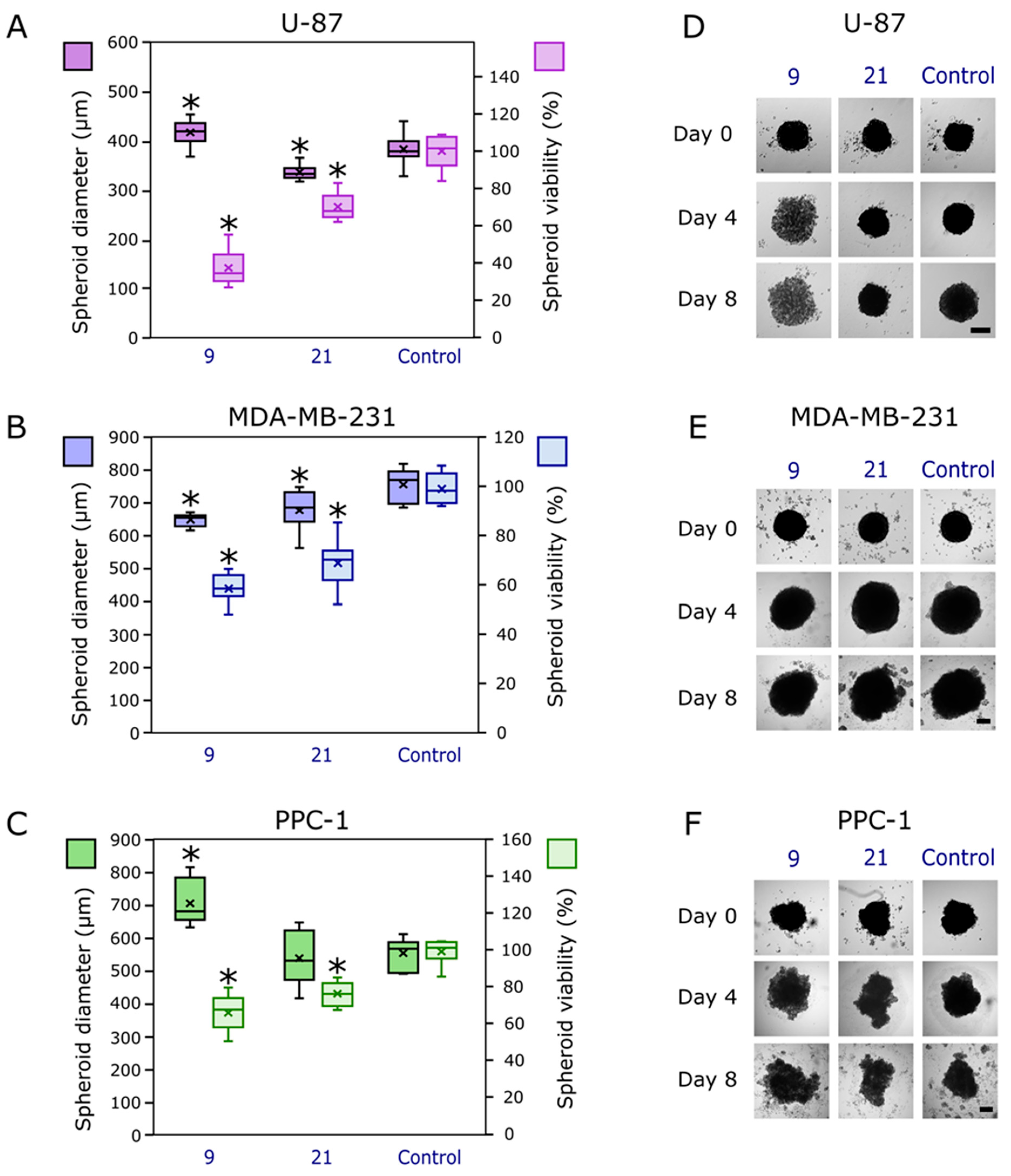
2.4. Biology
3. Materials and Methods
3.1. Chemistry
3.2. Affinity Measurements
3.3. Crystallization and Structure Solution
3.4. Biological Experiments in Cancer Cells In Vitro
3.5. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rutkauskas, K.; Zubrienė, A.; Tumosienė, I.; Kantminienė, K.; Mickevičius, V.; Matulis, D. Benzenesulfonamides Bearing Pyrrolidinone Moiety as Inhibitors of Carbonic Anhydrase IX: Synthesis and Binding Studies. Med. Chem. Res. 2017, 26, 235–246. [Google Scholar] [CrossRef]
- Balandis, B.; Ivanauskaitė, G.; Smirnovienė, J.; Kantminienė, K.; Matulis, D.; Mickevičius, V.; Zubrienė, A. Synthesis and Structure–Affinity Relationship of Chlorinated Pyrrolidinone-Bearing Benzenesulfonamides as Human Carbonic Anhydrase Inhibitors. Bioorg. Chem. 2020, 97, 103658. [Google Scholar] [CrossRef]
- Vaškevičienė, I.; Paketurytė, V.; Pajanok, N.; Žukauskas, Š.; Sapijanskaitė, B.; Kantminienė, K.; Mickevičius, V.; Zubrienė, A.; Matulis, D. Pyrrolidinone-Bearing Methylated and Halogenated Benzenesulfonamides as Inhibitors of Carbonic Anhydrases. Bioorg. Med. Chem. 2019, 27, 322–337. [Google Scholar] [CrossRef]
- Afifi, N.; Barrero, C.A. Understanding Breast Cancer Aggressiveness and Its Implications in Diagnosis and Treatment. JCM 2023, 12, 1375. [Google Scholar] [CrossRef] [PubMed]
- Roda, D.; Veiga, P.; Melo, J.B.; Carreira, I.M.; Ribeiro, I.P. Principles in the Management of Glioblastoma. Genes 2024, 15, 501. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Ward, C.; Meehan, J.; Mullen, P.; Supuran, C.; Dixon, J.M.; Thomas, J.S.; Winum, J.-Y.; Lambin, P.; Dubois, L.; Pavathaneni, N.-K.; et al. Evaluation of Carbonic Anhydrase IX as a Therapeutic Target for Inhibition of Breast Cancer Invasion and Metastasis Using a Series of in Vitro Breast Cancer Models. Oncotarget 2015, 6, 24856–24870. [Google Scholar] [CrossRef]
- Haapasalo, J.; Nordfors, K.; Haapasalo, H.; Parkkila, S. The Expression of Carbonic Anhydrases II, IX and XII in Brain Tumors. Cancers 2020, 12, 1723. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Schäfer, A.; Zhang, Z.; Elsässer, K.; Culmsee, C.; Zhong, L.; Pagenstecher, A.; Nimsky, C.; Bartsch, J.W. Inhibition of Carbonic Anhydrase 2 Overcomes Temozolomide Resistance in Glioblastoma Cells. Int. J. Mol. Sci. 2021, 23, 157. [Google Scholar] [CrossRef]
- Smyth, L.G.; O’Hurley, G.; O’Grady, A.; Fitzpatrick, J.M.; Kay, E.; Watson, R.W.G. Carbonic Anhydrase IX Expression in Prostate Cancer. Prostate Cancer Prostatic Dis. 2010, 13, 178–181. [Google Scholar] [CrossRef]
- Ivanov, S.; Liao, S.-Y.; Ivanova, A.; Danilkovitch-Miagkova, A.; Tarasova, N.; Weirich, G.; Merrill, M.J.; Proescholdt, M.A.; Oldfield, E.H.; Lee, J.; et al. Expression of Hypoxia-Inducible Cell-Surface Transmembrane Carbonic Anhydrases in Human Cancer. Am. J. Pathol. 2001, 158, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Chiche, J.; Ilc, K.; Laferrie`re, J.; Trottier, E.; Dayan, F.; Mazure, N.M.; Brahimi-Horn, M.C.; Pouysségur, J. Hypoxia-Inducible Carbonic Anhydrase IX and XII Promote Tumor Cell Growth by Counteracting Acidosis through the Regulation of the Intracellular pH. Cancer Res. 2009, 69, 358–368. [Google Scholar] [CrossRef]
- 13 Pastorekova, S.; Gillies, R.J. The role of carbonic anhydrase IX in cancer development: Links to hypoxia, acidosis, and beyond. Cancer Metastasis Rev. 2019, 38, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Mboge, M.Y.; McKenna, R.; Frost, S.C. Advances in Anti-Cancer Drug Development Targeting Carbonic Anhydrase IX and XII. Top. Anticancer Res. 2015, 5, 3–42. [Google Scholar] [PubMed] [PubMed Central]
- Kirsh, S.M.; Pascetta, S.A.; Uniacke, J. Spheroids as a 3D Model of the Hypoxic Tumor Microenvironment. In The Tumor Microenvironment; Ursini-Siegel, J., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2023; Volume 2614, pp. 273–285. ISBN 978-1-07-162913-0. [Google Scholar]
- Riffle, S.; Hegde, R.S. Modeling Tumor Cell Adaptations to Hypoxia in Multicellular Tumor Spheroids. J. Exp. Clin. Cancer Res. 2017, 36, 102. [Google Scholar] [CrossRef]
- Sulfanilamide|Drug|Britannica. Available online: https://www.britannica.com/science/sulfanilamide (accessed on 10 May 2025).
- Gaffer, H.E.; Mahmoud, S.A.; El-Sedik, M.S.; Aysha, T.; Abdel-Rhman, M.H.; Abdel-latif, E. Synthesis, Molecular Modelling, and Antibacterial Evaluation of New Sulfonamide-Dyes Based Pyrrole Compounds. Sci. Rep. 2024, 14, 10973. [Google Scholar] [CrossRef]
- Abd El Salam, H.A.; Abdel-Aziz, M.S.; El-Sawy, E.R.; Shaban, E. Synthesis and Antibacterial Activity of Azo-Sulfa-Based Disperse Dyes and Their Application in Polyester Printing. Fibers Polym. 2023, 24, 2751–2760. [Google Scholar] [CrossRef]
- Bringmann, G.; Dreyer, M.; Faber, J.H.; Dalsgaard, P.W.; Stærk, D.; Jaroszewski, J.W.; Ndangalasi, H.; Mbago, F.; Brun, R.; Christensen, S.B. Ancistrotanzanine C and Related 5,1′- and 7,3′-Coupled Naphthylisoquinoline Alkaloids from Ancistrocladus tanzaniensis. J. Nat. Prod. 2004, 67, 743–748. [Google Scholar] [CrossRef]
- Souza, A.O.D.; Galetti, F.C.S.; Silva, C.L.; Bicalho, B.; Parma, M.M.; Fonseca, S.F.; Marsaioli, A.J.; Trindade, A.C.L.B.; Gil, R.P.F.; Bezerra, F.S.; et al. Antimycobacterial and Cytotoxicity Activity of Synthetic and Natural Compounds. Quím. Nova 2007, 30, 1563–1566. [Google Scholar] [CrossRef]
- Raczuk, E.; Dmochowska, B.; Samaszko-Fiertek, J.; Madaj, J. Different Schiff Bases—Structure, Importance and Classification. Molecules 2022, 27, 787. [Google Scholar] [CrossRef]
- Ceramella, J.; Iacopetta, D.; Catalano, A.; Cirillo, F.; Lappano, R.; Sinicropi, M.S. A Review on the Antimicrobial Activity of Schiff Bases: Data Collection and Recent Studies. Antibiotics 2022, 11, 191. [Google Scholar] [CrossRef]
- Rehman, W.; Baloch, M.K.; Muhammad, B.; Badshah, A.; Khan, K.M. Characteristic Spectral Studies Andin Vitro Antifungal Activity of Some Schiff Bases and Their Organotin (IV) Complexes. Chin. Sci. Bull. 2004, 49, 119–122. [Google Scholar] [CrossRef]
- Karthikeyan, M.S.; Prasad, D.J.; Poojary, B.; Subrahmanya Bhat, K.; Holla, B.S.; Kumari, N.S. Synthesis and Biological Activity of Schiff and Mannich Bases Bearing 2,4-Dichloro-5-Fluorophenyl Moiety. Bioorg. Med. Chem. 2006, 14, 7482–7489. [Google Scholar] [CrossRef] [PubMed]
- Kayser, O.; Kiderlen, A.F.; Croft, S.L. Natural Products as Antiparasitic Drugs. Parasitol. Res. 2003, 90 (Suppl. S2), S55–S62. [Google Scholar] [CrossRef] [PubMed]
- Przybylski, P.; Huczynski, A.; Pyta, K.; Brzezinski, B.; Bartl, F. Biological Properties of Schiff Bases and Azo Derivatives of Phenols. Curr. Org. Chem. 2009, 13, 124–148. [Google Scholar] [CrossRef]
- Murtaza, S.; Akhtar, M.S.; Kanwal, F.; Abbas, A.; Ashiq, S.; Shamim, S. Synthesis and Biological Evaluation of Schiff Bases of 4-Aminophenazone as an Anti-Inflammatory, Analgesic and Antipyretic Agent. J. Saud. Chem. Soc. 2017, 21, S359–S372. [Google Scholar] [CrossRef]
- Gujjarappa, R.; Kabi, A.K.; Sravani, S.; Garg, A.; Vodnala, N.; Tyagi, U.; Kaldhi, D.; Velayutham, R.; Singh, V.; Gupta, S.; et al. Overview on Biological Activities of Imidazole Derivatives. In Nanostructured Biomaterials; Swain, B.P., Ed.; Materials Horizons: From Nature to Nanomaterials; Springer: Singapore, 2022; pp. 135–227. ISBN 9789811683985. [Google Scholar]
- Luca, L. Naturally Occurring and Synthetic Imidazoles: Their Chemistry and Their Biological Activities. Curr. Med. Chem. 2006, 13, 1–23. [Google Scholar] [CrossRef]
- Rashid, M.; Maqbool, A.; Shafiq, N.; Bin Jardan, Y.A.; Parveen, S.; Bourhia, M.; Nafidi, H.-A.; Khan, R.A. The Combination of Multi-Approach Studies to Explore the Potential Therapeutic Mechanisms of Imidazole Derivatives as an MCF-7 Inhibitor in Therapeutic Strategies. Front. Chem. 2023, 11, 1197665. [Google Scholar] [CrossRef]
- Balandis, B.; Mickevičius, V.; Petrikaitė, V. Exploration of Benzenesulfonamide-Bearing Imidazole Derivatives Activity in Triple-Negative Breast Cancer and Melanoma 2D and 3D Cell Cultures. Pharmaceuticals 2021, 14, 1158. [Google Scholar] [CrossRef]
- Holzer, W.; Hahn, K. Synthesis of Substituted 3-phenyl-6 h -pyrazolo[4,3-d]Isoxazoles from Corresponding 4-benzoyl-5-hydroxypyrazoles. J. Heterocycl. Chem. 2003, 40, 303–308. [Google Scholar] [CrossRef]
- Chourasiya, S.S.; Kathuria, D.; Wani, A.A.; Bharatam, P.V. Azines: Synthesis, Structure, Electronic Structure and Their Applications. Org. Biomol. Chem. 2019, 17, 8486–8521. [Google Scholar] [CrossRef]
- DeLeeuw, L.W.; Monsen, R.C.; Petrauskas, V.; Gray, R.D.; Baranauskiene, L.; Matulis, D.; Trent, J.O.; Chaires, J.B. POT1 Stability and Binding Measured by Fluorescence Thermal Shift Assays. PLoS ONE 2021, 16, e0245675. [Google Scholar] [CrossRef]
- Gedgaudas, M.; Baronas, D.; Kazlauskas, E.; Petrauskas, V.; Matulis, D. Thermott: A Comprehensive Online Tool for Protein–Ligand Binding Constant Determination. Drug Discov. Today 2022, 27, 2076–2079. [Google Scholar] [CrossRef]
- Matulienė, J.; Žvinys, G.; Petrauskas, V.; Kvietkauskaitė, A.; Zakšauskas, A.; Shubin, K.; Zubrienė, A.; Baranauskienė, L.; Kačenauskaitė, L.; Kopanchuk, S.; et al. Picomolar Fluorescent Probes for Compound Affinity Determination to Carbonic Anhydrase IX Expressed in Live Cancer Cells. Sci. Rep. 2022, 12, 17644. [Google Scholar] [CrossRef] [PubMed]
- Cherinka, B.; Andrews, B.H.; Sánchez-Gallego, J.; Brownstein, J.; Argudo-Fernández, M.; Blanton, M.; Bundy, K.; Jones, A.; Masters, K.; Law, D.R.; et al. Marvin: A Tool Kit for Streamlined Access and Visualization of the SDSS-IV MaNGA Data Set. Astron. J. 2019, 158, 74. [Google Scholar] [CrossRef]
- Alterio, V.; Di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lu, Y. Optimizing a 3D Culture System to Study the Interaction between Epithelial Breast Cancer and Its Surrounding Fibroblasts. J. Cancer 2011, 2, 458–466. [Google Scholar] [CrossRef]
- Roque Marques, K.M.; Do Desterro, M.R.; De Arruda, S.M.; De Araújo Neto, L.N.; Do Carmo Alves De Lima, M.; De Almeida, S.M.V.; Da Silva, E.C.D.; De Aquino, T.M.; Da Silva-Júnior, E.F.; De Araújo-Júnior, J.X.; et al. 5-Nitro-Thiophene-Thiosemicarbazone Derivatives Present Antitumor Activity Mediated by Apoptosis and DNA Intercalation. Curr. Top. Med. Chem. 2019, 19, 1075–1091. [Google Scholar] [CrossRef]
- Daunys, S.; Petrikaitė, V. The Roles of Carbonic Anhydrases IX and XII in Cancer Cell Adhesion, Migration, Invasion and Metastasis. Biol. Cell 2020, 112, 383–397. [Google Scholar] [CrossRef]
- Bonardi, A.; Supuran, C.T. Polypharmacology of Carbonic Anhydrase Inhibitors and Activators. Expert. Opin. Pharmacother. 2025, 26, 567–580. [Google Scholar] [CrossRef]
- Ovung, A.; Bhattacharyya, J. Sulfonamide Drugs: Structure, Antibacterial Property, Toxicity, and Biophysical Interactions. Biophys. Rev. 2021, 13, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Freake, H.C.; Sankavaram, K. Zinc: Physiology, Dietary Sources, and Requirements. In Encyclopedia of Human Nutrition; Elsevier: Amsterdam, The Netherlands, 2013; pp. 437–443. ISBN 978-0-12-384885-7. [Google Scholar]
- Oh, S.; Moon, H.; Son, I.; Jung, J. Synthesis of Sulfonamides and Evaluation of Their Histone Deacetylase (HDAC) Activity. Molecules 2007, 12, 1125–1135. [Google Scholar] [CrossRef]
- Luchinat, E.; Barbieri, L.; Cremonini, M.; Nocentini, A.; Supuran, C.T.; Banci, L. Intracellular Binding/Unbinding Kinetics of Approved Drugs to Carbonic Anhydrase II Observed by in-Cell NMR. ACS Chem. Biol. 2020, 15, 2792–2800. [Google Scholar] [CrossRef] [PubMed]
- Dudutienė, V.; Matulienė, J.; Smirnov, A.; Timm, D.D.; Zubrienė, A.; Baranauskienė, L.; Morkunaite, V.; Smirnovienė, J.; Michailovienė, V.; Juozapaitienė, V.; et al. Discovery and Characterization of Novel Selective Inhibitors of Carbonic Anhydrase IX. J. Med. Chem. 2014, 57, 9435–9446. [Google Scholar] [CrossRef] [PubMed]
- Mickevičiūtė, A.; Juozapaitienė, V.; Michailovienė, V.; Jachno, J.; Matulienė, J.; Matulis, D. Recombinant Production of 12 Catalytically Active Human CA Isoforms. In Carbonic Anhydrase as Drug Target; Matulis, D., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 15–37. ISBN 978-3-030-12778-7. [Google Scholar]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef]
- Agirre, J.; Atanasova, M.; Bagdonas, H.; Ballard, C.B.; Baslé, A.; Beilsten-Edmands, J.; Borges, R.J.; Brown, D.G.; Burgos-Mármol, J.J.; Berrisford, J.M.; et al. The CCP 4 Suite: Integrative Software for Macromolecular Crystallography. Acta Crystallogr. D Struct. Biol. 2023, 79, 449–461. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. Molecular Replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 22–25. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC 5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Skaraitė, I.; Maccioni, E.; Petrikaitė, V. Anticancer Activity of Sunitinib Analogues in Human Pancreatic Cancer Cell Cultures under Normoxia and Hypoxia. Int. J. Mol. Sci. 2023, 24, 5422. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CAI | CAII | CAIII | CAIV | CAVA | CAVB | CAVI | CAVII | CAIX | CAXII | CAXIII | CAXIV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 130 | 17 | ≥200 | 20 | 130 | nd | 110 | 100 | 15 | 40 | 140 | 22 |
| 2 | ≥200 | 20 | ≥200 | 28 | ≥200 | nd | 89 | 120 | 16 | 33 | 130 | 26 |
| 3 | 90 | 18 | ≥200 | 29 | 140 | nd | 79 | 100 | 16 | 58 | 160 | 22 |
| 4 | 120 | 11 | ≥200 | 14 | 140 | nd | 39 | 83 | 13 | 28 | 150 | 22 |
| 5 | 150 | 23 | ≥200 | 12 | ≥200 | nd | 120 | 100 | 17 | 43 | 130 | 16 |
| 6 | 47 | 13 | ≥200 | 37 | 67 | nd | 93 | 112 | 8.4 | 54 | 98 | 20 |
| 7 | 59 | 16 | ≥200 | 5.5 | 170 | nd | 130 | 120 | 18 | 41 | ≥200 | 30 |
| 8 | 120 | 16 | ≥200 | 9.9 | nd | nd | 79 | 92 | 16 | 41 | 120 | 21 |
| 9 | 76 | 17 | ≥200 | 9.1 | 13 | nd | 80 | 31 | 9 | 65 | 100 | 0.68 |
| 10 | 3.1 | 0.14 | ≥200 | 13 | ≥200 | nd | 4.5 | 0.49 | 2.3 | 17 | 6.9 | 0.36 |
| 11 | 6 | 0.15 | ≥200 | 3.7 | 29 | nd | 12 | 0.87 | 2.1 | 19 | 12 | 0.67 |
| 12 | 0.0062 | 0.097 | ≥200 | 17 | 8 | 0.88 | 20 | 0.24 | 0.98 | 27 | 5.5 | 0.34 |
| 13 | 7.7 | 3.6 | ≥200 | 7.3 | 85 | nd | 82 | 22 | 4.3 | 37 | 26 | 5.2 |
| 14 | 0.052 | 0.16 | ≥200 | 11 | 22 | nd | 18 | 0.39 | 0.51 | 20 | 4 | 0.78 |
| 15 | 60 | 8 | ≥200 | 1.9 | ≥200 | nd | 2.4 | 17 | 1.9 | 11 | 27 | 1.6 |
| 16 | 30 | 7.3 | ≥200 | 3 | ≥200 | nd | 5.1 | 7.5 | 1.5 | 15 | 28 | 1.7 |
| 17 | 180 | 14 | ≥200 | 23 | 160 | 93 | 4.7 | 27 | 4.8 | 56 | 25 | 2.9 |
| 18 | 0.0077 | 0.11 | ≥200 | 6.6 | 14 | nd | 13 | 0.25 | 0.53 | 6.2 | 3.2 | 0.27 |
| 19 | 0.52 | 3 | ≥200 | 34 | 160 | nd | 17 | 32 | 5.3 | 25 | 15 | 3.3 |
| 20 | 21 | 10 | ≥200 | 17 | 180 | nd | 12 | 140 | 16 | 18 | 28 | 1.6 |
| 21 | ≥200 | ≥200 | ≥200 | ≥200 | nd | nd | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 |
| 22 | 28 | 9.8 | ≥200 | 7.1 | ≥200 | nd | 32 | 100 | 0.98 | 41 | 14 | 5.9 |
| 23 | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 | nd | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 |
| 24 | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 | 22 | 33 | ≥200 | ≥200 |
| 25 | 1.6 | 5.4 | ≥200 | 12 | ≥200 | nd | 120 | 150 | 4.2 | 74 | 39 | 9 |
| 26 | ≥200 | 24 | ≥200 | 7.6 | 35 | nd | ≥200 | 180 | 14 | 74 | 65 | 21 |
| 27 | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 | nd | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 |
| 28 | ≥200 | 47 | ≥200 | ≥200 | ≥200 | ≥200 | ≥200 | 100 | 14 | 57 | 90 | 46 |
| 29 | ≥200 | 58 | ≥200 | 54 | ≥200 | 96 | 140 | ≥200 | 7.1 | 75 | 47 | 34 |
| 30 | ≥200 | 22 | ≥200 | 100 | 29 | 3 | 48 | 43 | 14 | 48 | 16 | 26 |
| 31 | ≥200 | 16 | ≥200 | 24 | 120 | 1.1 | 98 | 25 | 9.7 | 59 | 19 | 14 |
| 32 | ≥200 | 11 | ≥200 | 9.2 | 150 | 4 | 150 | 18 | 8.2 | 48 | 21 | 12 |
| 33 | 160 | 16 | ≥200 | 24 | 87 | 3.7 | 120 | 28 | 8.2 | 72 | 22 | 16 |
| 34 | 110 | 10 | ≥200 | 16 | 66 | 4.9 | 96 | 15 | 7.1 | 50 | 17 | 11 |
| 35 | ≥200 | 8.1 | ≥200 | 4 | ≥200 | nd | 38 | 62 | 0.39 | 12 | 18 | 0.48 |
| 36 | ≥200 | 43 | ≥200 | 52 | ≥200 | nd | 96 | 92 | 2.3 | 6.1 | 42 | 5 |
| 37 | ≥200 | 15 | ≥200 | 2.2 | 130 | nd | 49 | 12 | 1.2 | 8.9 | 25 | 0.94 |
| 38 | ≥200 | 8.6 | ≥200 | 2.1 | 58 | nd | 2.9 | 49 | 0.83 | 9.1 | 15 | 0.92 |
| AZM * | 2.4 | 0.046 | 40 | 0.087 | 0.84 | 0.14 | 0.22 | 0.013 | 0.021 | 0.13 | 0.12 | 0.063 |
| U-104 * | 1.1 | 0.33 | ≥200 | 3.1 | 6 | 0.43 | 10 | 0.25 | 0.1 | 5.5 | 2.2 | 0.27 |
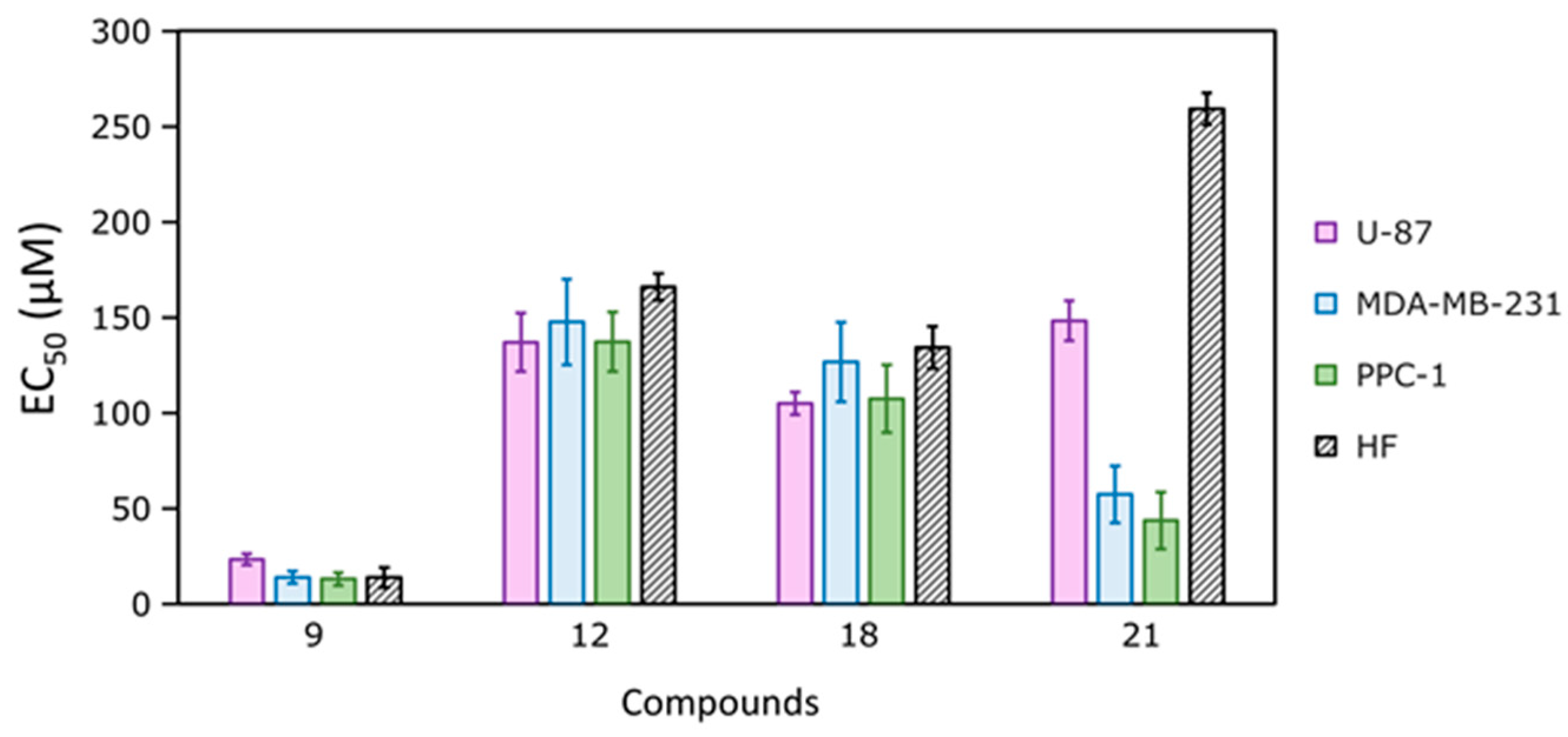
| Compound | EC50 (μM) | |||
|---|---|---|---|---|
| U-87 | MDA-MB-231 | PPC-1 | HF | |
| 9 | 23.3 ± 2.9 | 14.0 ± 3.2 | 13.0 ± 3.5 | 13.8 ± 5.2 |
| 12 | 137.0 ± 15.4 | 147.7 ± 22.5 | 137.3 ± 15.5 | 166.0 ± 7.0 |
| 18 | 105.0 ± 6.0 | 126.7 ± 20.8 | 107.5 ± 17.7 | 134.3 ± 11.1 |
| 21 | 148.3 ± 10.4 | 57.3 ± 15.0 | 43.7 ± 15.0 | 259.3 ± 8.4 |
| U-104 | ND | 239.8 ± 18.0 | ND | ND |
| AZM | >500 | >500 | >500 | >500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vainauskas, V.; Norvaišaitė, R.; Grybaitė, B.; Vaickelionienė, R.; Smirnov, A.; Kojis, T.; Baranauskiene, L.; Manakova, E.; Gražulis, S.; Zubrienė, A.; et al. Novel Derivatives of 3-Amino-4-hydroxy-benzenesulfonamide: Synthesis, Binding to Carbonic Anhydrases, and Activity in Cancer Cell 2D and 3D Cultures. Int. J. Mol. Sci. 2025, 26, 6466. https://doi.org/10.3390/ijms26136466
Vainauskas V, Norvaišaitė R, Grybaitė B, Vaickelionienė R, Smirnov A, Kojis T, Baranauskiene L, Manakova E, Gražulis S, Zubrienė A, et al. Novel Derivatives of 3-Amino-4-hydroxy-benzenesulfonamide: Synthesis, Binding to Carbonic Anhydrases, and Activity in Cancer Cell 2D and 3D Cultures. International Journal of Molecular Sciences. 2025; 26(13):6466. https://doi.org/10.3390/ijms26136466
Chicago/Turabian StyleVainauskas, Valdas, Rugilė Norvaišaitė, Birutė Grybaitė, Rita Vaickelionienė, Alexey Smirnov, Tautvydas Kojis, Lina Baranauskiene, Elena Manakova, Saulius Gražulis, Asta Zubrienė, and et al. 2025. "Novel Derivatives of 3-Amino-4-hydroxy-benzenesulfonamide: Synthesis, Binding to Carbonic Anhydrases, and Activity in Cancer Cell 2D and 3D Cultures" International Journal of Molecular Sciences 26, no. 13: 6466. https://doi.org/10.3390/ijms26136466
APA StyleVainauskas, V., Norvaišaitė, R., Grybaitė, B., Vaickelionienė, R., Smirnov, A., Kojis, T., Baranauskiene, L., Manakova, E., Gražulis, S., Zubrienė, A., Matulis, D., Mickevičius, V., & Petrikaitė, V. (2025). Novel Derivatives of 3-Amino-4-hydroxy-benzenesulfonamide: Synthesis, Binding to Carbonic Anhydrases, and Activity in Cancer Cell 2D and 3D Cultures. International Journal of Molecular Sciences, 26(13), 6466. https://doi.org/10.3390/ijms26136466