
Modulation of Endoplasmic Reticulum Stress in Experimental Anti-Cancer Therapy


Abstract
1. Introduction
2. Endoplasmic Reticulum Stress and the Unfolded Protein Response
3. ERS in Cancer Cells
3.1. Ca2+ Homeostasis, SERCA, and Cancer
3.2. UPR Components and Cancer

{kind=link}
| Effector | Expression Profile | Cancer Type | Clinical Outcome | References |
|---|---|---|---|---|
| PERK | Overexpression | Prostate cancer | Poor prognosis | [56] |
| Pancreatic duct adenocarcinoma | [57] | |||
| Kidney renal papillary cell carcinoma | [58] | |||
| Brain glioma | [58] | |||
| Breast carcinoma | [58] | |||
| Thyroid carcinoma | [58] | |||
| Head and neck squamous cell carcinoma | Favorable prognosis | [58] | ||
| eIF2α | Overexpression, Phosphorylation | Melanoma, colonic adenoma and adenocarcinoma | - | [59] |
| Aggressive thyroid carcinoma and papillary carcinoma | [60] | |||
| Hodgkin’s lymphoma | [61] | |||
| Prostate cancer | Poor prognosis Lower overall and disease-free survival Metastasis Chemoresistance | [56] | ||
| Pancreatic duct adenocarcinoma | [57] | |||
| Pancreatic adenocarcinoma | [65] | |||
| Hepatocellular carcinoma | [66] | |||
| Brain meningioma, astrocytoma and oligodendroglial tumors | [67] | |||
| Overexpression, Phosphorylation | Stomach, colon, sigma-rectum carcinoma | Longer survival Better disease-free survival | [68] | |
| Non-small cell lung cancer | [69] | |||
| Triple-negative breast cancer | [70] | |||
| IRE1α | Overexpression | Prostate cancer | Higher recurrence | [56,71] |
| Lung adenocarcinoma | Favorable prognosis | [72] | ||
| XBP1/sXBP1 | Overexpression Splicing | Hepatocellular carcinoma | - | [73] |
| Breast adenocarcinoma | [74] | |||
| Melanoma | [75] | |||
| Glioblastoma | [76] | |||
| Diffuse large B-cell lymphoma | Shorter overall and disease-free survival Poor response to therapy | [77] | ||
| Acute lymphoblastic leukemia | [78] | |||
| Aggressive luminal B breast cancer | [79] | |||
| ER+ breast cancer | [80] | |||
| Prostate cancer | [81] | |||
| Acute myeloid leukemia | Better disease-free and overall survival Lower relapse rate | [82,83] | ||
| Pancreatic cancer | No correlation with survival | [85] | ||
| ATF6 | Overexpression | Hepatocellular carcinoma | Poor prognosis Chemoresistance, lower overall survival | [73] |
| Osteosarcoma | [86] | |||
| Ovarian cancer | [87] | |||
| Gastric cancer | [88] | |||
| Pancreatic cancer | [85] | |||
| Oral squamous cell carcinoma | [89] | |||
| Head and neck squamous carcinoma | [90] | |||
| Biliopancreatic carcinoma | No correlation with survival | [91] | ||
| Prostate cancer | [56] | |||
| Colon cancer | [92] | |||
| GRP78 | Overexpression | Breast adenocarcinoma | - | [74,93,94] |
| Hepatocellular carcinoma | Poor prognosis Lower overall survival Invasion Metastasis Chemoresistance | [73,95,96] | ||
| Gastric cancer | [97] | |||
| Gliomas | [98] | |||
| Prostate cancer | [99] | |||
| Pancreatic duct adenocarcinoma | [100] | |||
| Lung cancer | [101,102] | |||
| Head and neck squamous cell carcinomas | [103] | |||
| Colorectal cancer | Improved survival | [104] |
4. SERCA as a Target for Cancer Treatment
4.1. Terpenoids
4.2. Curcumin and Its Analogues
4.3. Flavonoids
4.4. Other SERCA Inhibitors
| Cell Type | Treatment | Molecular Changes | Cellular Effects | References |
|---|---|---|---|---|
| Terpenoids | ||||
| Breast cancer MCF7 and MDA-MB-231 cells | TG 6–100 nM 6–48 h | ↓ ER Ca2+, ↑ROS ↑ Cleaved PARP ↑ Caspases-8, 9, 3 | ↑ Cells in subG1 phase ↓ Proliferation ↓ Viability, ↑ Apoptosis | [118,119] |
| Breast cancer cells MDA-MB-231 and MDA-MB-436 | TG 2–10 µM 6 or 24 h | ↑ Cytoplasmic Ca2+ ↑ p-Myosin light chain 2 kinase ↑ p-Myosin phosphatase 1 | Actin contraction and rearrangement Changed morphology | [121] |
| Prostate cancer LNCaP, PC3 cells | TG 30–100 nM 6–48 h | ↓ ER Ca2+, ↑ GRP94, ↑ GRP78, ↑ ATF4, ↑ cleaved PARP, ↑ CHOP | ↓ Proliferation, ↑Death Changed morphology | [118] |
| Transfected prostate cancer LNCaP cells | TG 100 nM 30–48 h | ↑ DR5, ↑ PERK, ↑ ATF4, ↑ CHOP ↑ IRE1, ↑ XBP1, ↑ JNK ↑ Cleaved PARP, ↑ Caspases-3, 8 | ↑ Cell death | [122] |
| Patient-derived stem cell-enriched glioblastoma culture | TG 1–10 µM 24–48 h | ↑ p-PERK, ↑ ATF4, ↑ CHOP, ↑GRP78, ↑ sXBP1, ↑ ATF6, ↓ SOX2, ↑ Cleaved PARP, ↑ Caspases-3/7 | ↓ Viability ↓ Neurosphere-forming ability | [123] |
| Neuroblastoma SH-SY5Y cells | TG 300 nM 30 min or 4 h | ↓ ER Ca2+, ↑ ROS ↑ Hypodiploid nuclei, ↑ GRP78, ↑ ATF4, ↑ p-PERK, ↑ Caspase-4 | ↓ Viability | [124] |
| ACC SW-13 and NCI-H295R cells | TG 0.5–32 µM 48 h | ↑ p-JNK/JNK, ↑ PERK, ↑ ATF6, ↑ LC3B, ↑ HSAP, ↑ Bcl-2 | ↓ Viability, ↑ Apoptosis, ↓ Migration, invasion | [125] |
| Mice with SW-13 cell xenografts | TG 1 mg/kg 14 days | ↑ p-JNK/JNK, ↑ p-ERK/ERK, ↑ p-PERK/PERK, ↑ GRP78, ↑ IRE1 | ↓ Tumor growth | [125] |
| Patient-derived PTC YUMC cells resistant to PTX, SOR, and LEN | TG 10–200 µM + PTX, or SOR, or LEN 10–200 µM 40 h | ↑ CHOP, ↑p-PERK ↑ Cytochrome c ↑ Cleaved caspase-3 | ↑ Sensitivity to PTX, SOR or LEN ↓ Viability | [129,130] |
| Mice with PTC YUMC xenografts resistant to PTX, SOR, and LEN | TG 25 mg/kg PO + PTX 25 mg/kg IP, or SOR 80 mg/kg PO, or LEN 10 mg/kg PO | ↑ CHOP | ↓ Tumor weight ↑ Sensitivity to PTX, SOR or LEN | [129,130] |
| Oral cancer CAL 27 and Ca9–22 cells | TG 10 nM + Manoalide 5–10 µM 24 h | ↑ Caspase 3/7 | ↓ Viability, ↑ Autophagy ↑ Sensitivity to manoalide | [131] |
| Breast cancer MCF7 and MDA-MB-231s cells | TG 3 µM + nodakenin 40 µM 24 h | ↑ p-PERK ↑ p-eIF2α ↑ CHOP, ↑ ATF4 | ↑ Sensitivity to nodakenin ↑ Cell death | [132] |
| Hepatocellular carcinoma | G-202 40 mg on 1–3 d, or 40 mg on 1 d and 66.8 mg on 2–3 d of 28-d cycle | No complete response No progressive disease Stable disease Partial response | [135,136] | |
| Glioblastoma multiforme | G-202 IV for 3 days of 28-d cycle | No clear conclusions | [137] | |
| Lung cancer A549 and H460 cells | Lathyrol 30–120 µg/mL 24 h or 14 d | ↑ Cytosolic Ca2+, ↑ GRP78, ↑ PERK ↑ p-eIF2α, ↑ CHOP, ↑ ATF4, ↑ Bax, ↓ Bcl-2, ↑ Caspase-3, ↑ Cyt C | ↓ Viability, ↑ Apoptosis | [138] |
| Mice with H460 cell xenografts | Lathyrol 10–40 mg/kg IP 16 d | ↓ Tumor volume and weight | [138] | |
| Renal cell carcinoma 786-O cells | Lathyrol 10–375 µg/mL 24 h | ↓ Bcl-2, ↑ Bax, ↓ p-Akt, ↓ MMP2, ↓ MMP9, ↓ Ki67, ↑ Caspase-3, 9 | ↓ Viability, ↓ Invasion, ↓ Migration, ↑ Apoptosis | [139] |
| Curcumin and its analogues | ||||
| Human liposarcoma SW872 cells | Curcumin 5–20 µM 24 or 48 h | ↓ Ca2+-ATPase activity, ↑ DR5, ↑ Caspase-8, ↑ Caspase-3, ↑ Bid, ↑ PARP, ↑ CHOP, ↑ p-eIF2a, ↑ ATF4 | ↓ Cell growth ↑ Apoptosis | [126] |
| SCID mice injected with SW872 cells | Curcumin 100 mg/kg IP 40 d | ↑ Caspase-8, ↑ Caspase-3, ↑ Cleaved PARP, ↑ CHOP, | ↓ Tumor growth | [126] |
| Human glioma LN229 and U87 cells | Curcumin 8–32 μM 24–72 h | ↓ Cyclin D1, ↓ CDK46/6 ↓ Bcl-2, ↓ Bcl-XL, ↓ MMP-2, ↓ MMP-9, ↓ p-ERK1/2 | ↓ Proliferation ↓ Migration and invasion | [148] |
| Mice with subcutaneous LN229 xenografts | Curcumin 60 mg/kg/d 4 w | ↓ MMP-2 and MMP-9 ↓ CD147 | ↓ Tumor growth | [148] |
| CIS-resistant NSCLC A549 and H1299 cells | Curcumin 2.5 μg/mL + CIS 2 μg/mL 48 h | ↑ Cleaved PARP, ↑ Caspase-3 ↑ GRP78, ↑ ATF6, ↑ XBP1 ↑ Caspase-4, ↑ CHOP | ↓ Viability ↑ Apoptosis ↑ Sensitivity to CIS | [149] |
| Glioblastoma U87 and LN18 cells | Curcumin 10 µg/mL + TMZ 200 µM | Alterations in actin network | Cell cycle arrest, ↓ Viability, ↑ Apoptosis, ↑ Sensitivity to TMZ | [150] |
| Colon carcinoma SW480 cells | F36 1–10 µM 24–72 h | ↑ Cleaved PARP, ↑ Caspase-3, ↑ CHOP, ↑ ATF4, ↑ p-eIF2a | ↓ Proliferation ↑ Apoptosis | [33] |
| Advanced metastatic breast cancer | Curcumin 6 g (7 d) + DOC 100 mg/m2 every 3 w 6 cycles | No significant difference in the objective response rate and 12-month overall survival | [153] | |
| Colon carcinoma SW480 cells | RL71 0.5–10 µM 24–72 h | ↑ GRP78, ↑ ATF4, ↑ CHOP, ↑ cleaved PARP | ↓ Viability G2/M cell cycle arrest | [155] |
| Mice with SW480 xenografts | RL71 1–4 mg/kg 14 d | ↑ Cleaved PARP, ↑ CHOP, ↑ cleaved Caspase-3 | ↓ Tumor growth | [155] |
| Flavonoids | ||||
| HCC SNU-449 and Hep-3B cells | Quercetin 6.5–75 µM 24–48 h | ↓ p-PI3K, ↓ p-Akt, ↓ p-mTOR, ↓ Bcl-2, ↑ Bax, ↑ cleaved PARP, ↑ cleaved Caspase-3, ↓ P4HA2 | ↓ Viability, ↑ Apoptosis, ↓ Proliferation, ↓ Colony formation | [159] |
| AML HL-60 cells | Quercetin 25–100 µM 24–72 h | ↑ LC3II/I ↓ Bcl-2 ↑ Bax ↑ p-AMPK ↓ p-mTOR ↑ Caspase-3 | ↓ Viability, ↑ Apoptosis, ↑ Autophagy, ↓ Colony formation | [160] |
| Melanoma A375 cells | Quercetin 1–100 µM 24–72 h | ↑ p-ERK, ↑ p-Akt, ↑ GPER, ↑ c-Myc | ↓ Viability, Cell cycle arrest, Changed morphology, ↑ Apoptosis/necrosis | [161] |
| NSCLC A549 and H1299 cells | Luteolin 0.1–1000 µM 12–72 h or 50 µM 24 h | ↓ WDR72, ↓ Bcl-2, ↑ Caspase-3, ↓ p-Akt, ↓ E-cadherin, ↓ β-catenin, ↓ N-cadherin, ↓ ZEB1 | ↓ Viability, ↓ Migration and invasion, ↓ Proliferation | [163] |
| Mice with NSCLC H1299 xenografts | Luteolin 50 mg/kg IP once/2 d 21 d | ↓ WDR72 mRNA | ↓ Tumor growth | [163] |
| Bladder cancer EJ138 cells | Luteolin 20–50 µM 24–48 h | ↑ P53, ↑ ULK1, ↑ ATG12, ↓ BCL2 | ↓ Viability, ↑ Apoptosis G2/M phase arrest | [164] |
| DLBCL U2932 and OCI-LY10 cells | Luteolin 5–20 µM 24 h | ↑ Bax, ↓ Bcl-2, ↑ cleaved PARP, ↑ Caspase-3, ↓ p-JAK2, ↓ p-STAT3 | ↑ Apoptosis | [165] |
| Mice with U2932 tumors | Luteolin 12.5–50 mg/kg IP 14 d | ↑ Bax, ↓ Bcl-2, ↑ cleaved PARP, ↑ cleaved Caspase-3, ↓ p-JAK2, ↓ p-STAT3 | ↓ Tumor volume and weight | [165] |
| Other SERCA inhibitors | ||||
| Patient-derived PTX-, SOR-, or LEN- resistant PTC YUMC cells | Compounds 7, 13, 40, 42 (10–200 µM) + PTX, SOR, LEN 10–200 µM 40 h | ↑ p-PERK, ↑ CHOP, ↑ Cytochrome c, ↑ Cleaved caspase-3 | ↑ Sensitivity to PTX, SOR, or LEN ↓ Viability | [129,130] |
| Mice with xenografts of PTX-, SOR-, or LEN-sensitive and -resistant YUMC PTC cells | Compounds 7, 13, 40, 42 (25 mg/kg) + PTX 25 mg/kg, or SOR 80 mg/kg, or LEN 10 mg/kg | ↓ Tumor weight ↑ Sensitivity to PTX, SOR, or LEN | [129,130] | |
| NSCLC | Diphyllin | ↓ Ca2+ levels in the ER, ↑ ROS, ↓ MMP, Cytochrome C release | ↓ Proliferation, ↓ Migration, ↑ Apoptois, Synergy with CIS | [171] |
5. UPR Modulators
5.1. PERK Inhibitors
5.2. eIF2α Inhibitors
5.3. IRE1α/XBP1 Inhibitors
5.4. ATF6 Inhibitors
5.5. GRP78 Inhibitors
| Cell Type | Treatment | Molecular Changes | Cellular Effects | References |
|---|---|---|---|---|
| PERK inhibitors | ||||
| PDAC SW1990 cells with ↑ BZW1 | GSK2606414 10 µM 12 h | ↓ p-eIF2α, ↓ HIF1α, ↓ c-Myc, ↓ HIF1A, ↓ MYC IRES | ↓ Cell survival ↓ Proliferation | [175] |
| Mice with PDAC SW1990 BZW1 xenografts | GSK2606414 100 mg/kg IP twice/w | ↓ Ki67 staining ↓ TUNEL staining | ↓ Tumor growth ↓ Cell proliferation ↑ Apoptosis | [175] |
| Glioblastoma U87 and U251 cells | GSK2606414 1–20 µM + simvastatin + TMZ 72 h | ↑ p62, ↓ p-eIF2α, ↓ LC3B-II/I in U87 cells, ↑ LC3B-II/I in U251 cells | ↓ Viability ↑ Sensitivity to simvastatin+TMZ | [176] |
| Multidrug-resistant colorectal cancer S1-M1–80 cells | GSK2606414 1–3 µM + mitoxantrone or DOX 10 µM 24–72 h | ↑ Sensitivity to mitoxantrone and DOX | [178] | |
| Human myeloma L363, H929, U266, and KMS11 cells | GSK2606414 1–100 µM or 10 µM + BTZ 4 nM 24–48 h | ↓ PERK, ↓ ATF4, ↓ eIF2α, Changes in the expression of UPR genes | ↓ Cell survival ↑ Apoptosis ↑ Sensitivity to BTZ | [179] |
| SCLC H1688 and H446 cells | GSK2606414 10 µM + Oridonin 20 µM 24 h | ↓ p62 ↓ LC3B-II/LC3B-I | ↑ Apoptosis, ↑ Oridonin effect, ↑ Autophagy | [183] |
| Mice with SCLC H1688 cell xenografts | GSK2606414 50 mg/kg + oridonin 10 mg/kg | ↓ GRP78, ↓ p-PERK, ↓ p-eIF2α, ↓ ATF4, ↓ CHOP | ↓ Tumor size | [183] |
| Himan pancreatic adenocarcinoma BxPC3 cells | GSK2656157 1 µM + tunicamycin or TG 6 h | ↓ p-PERK, ↓ ATF4, ↓ p-eIF2α, ↓ CHOP, ↓ UPR gene expression | [185] | |
| Mice with pancreatic cancer xenografts | GSK2656157 50 or 150 mg/kg twice/d OR 14 d | ↓ p-PERK, changes in genes expression | ↓ Tumor growth ↓ Blood vessel density | [185] |
| Myeloid leukemia K562 and LAMA 84 cells | GSK2656157 0.1–10 µM + TG 100 nM + IMA 1 μM 16 h | ↓ CHOP mRNA ↓ GADD34 mRNA | [187] | |
| Mice subcutaneous K562 xenograft | GSK2656157 20 mg/kg/d + IMA 50 mg/kg twice/d 2 w | No significant decrease in the tumor mass | [187] | |
| Intact and ER-stressed NSCLC A549 lines | NCI 159456 3–100 or 50 µM + TG 500 nM 24 h | DNA damage, ↑ ATF4, ↑ DDTI3, ↑ BAX, ↓ BCL2, ↑ Caspase-3, ↑ ROS | ↓ Viability ↑ Apoptosis | [188] |
| eIF2 inhibitors | ||||
| Mice with prostate cancer xenografts | ISRIB 10 mg/kg, 6 w | ↓ Tumor growth, ↓ Metastases, ↑ Survival | [64] | |
| Mice with PDAC SW1990 BZW1 cell xenografts | ISRIB 2.5 mg/kg IP twice/w | ↓ Tumor volume ↑ Animal survival | [175] | |
| TNBC Hs576T and MDA-MB-231/ETHE1 cells | ISRIB 200 nM 24 h | ↓ p-eIF2 ↓ ATF4 | ↓ Migration ↓ Invasion | [192] |
| Mice with subcutaneous TNBC MDA-MB-231 cell xenografts | ISRIB for 15 d + DOX 6 injections | ↑ Cleaved caspase-3 | ↓ Tumor volume ↓ Tumor weight ↑ Sensitivity to DOX | [193] |
| Human ML K562 and LAMA84 cells | ISRIB 250 nM + IMA 0.5–1 µM 16 h | ↓ p-STAT, ↓ m-TOR, ↓ p-GSK3 | ↓ Proliferation | [187] |
| Mice with subcutaneous K562 xenografts | ISRIB 2 mg/kg/d + IMA 100 mg/kg/d 2 w | ↑ Sensitivity to IMA ↓ Tumor mass | [187] | |
| Inflammatory breast cancer SUM149PT and SUM190PT cells | Salubrinal 10 µM 24–48 h | ↑ p-eIF2a, ↓ PERK, ↑ CHOP, ↓ GRP78, ↑ ATF4, ↑ ROS, ↑ Bax, ↑ cleaved PARP, ↑ Caspase-3, ↓ p-Akt, ↓ p-NFkB | ↓ Proliferation | [197] |
| ACC SW-13 and NCI–H295R cells | Salubrinal 100 µM 24 h | ↑ p-eIF2α, ↑ p-PERK, ↑ ATF4, ↑ Ca2+, ↑Bcl-2 | ↓ Viability, migration ↑ Apoptosis | [198] |
| Primary pediatric GB SU-DIPG and KNS-42 lines | Salubrinal 2.5–8 µM + irradiation | ↑ p-eIF2a | ↑ Sensitivity to irradiation ↓ Cell survival | [199] |
| HNSCC SCC4 and FaDu cells, patient-derived 3D spheres | Salubrinal 10–50 µM 24–72 h | ↑ p-eIF2a, ↓ p-RB1, ↓ E2F1, ↓ Cyclin A, ↑ p21 | ↓ Viability ↓ Clonogenic ability Cell cycle arrest | [200] |
| Glioblastoma U87 and U251 cells | Salubrinal 1–20 µM or 15 µM + simvastatin + temozolomide 72 h | ↑ p-eIF2α | ↓ Viability No synergistic effect with cytotoxic drugs | [176] |
| Melanoma UACC 903 cells | Salubrinal 40 µM + 4E1RCat 10 µM 48 h | ↓ Protein synthesis, ↓ Cyclins, ↓ CDK2, ↓ Polysomes | ↓ Cell cycle progression ↓ Viability | [202] |
| Mice with subcutaneous UACC 903 melanoma | Salubrinal 1 mg/kg + 4E1RCat 2.5–15 mg/kg I P one/2 d 3–4 w | ↓Tumor volume | [202] | |
| TNBS BT549, SUM159, and MCF-10A cells | Salubrinal 5–10 µM + AgNPs 24 h | ↑ p-eIF2a, ↑ CHOP, ↑ cleaved Caspases-3/7/9 | ↓ Viability ↑ AgN-induced death | [203] |
| IRE1 inhibitors | ||||
| PDAC cells Panc3.27, Pan02, Miapaca-2 | Sunitinib 10 µM + GEM 100–250 nM 48–72 h | ↓ Lysosomal degradation, DNA fragmentation | ↓ Viability, ↑ Apoptosis, ↓ Autophagy, ↑ GEM effect | [100] |
| Mice with orthotopic PDAC Pan02 or KPCP1 xenografts | Sunitinib 25 mg/kg/d OR + GEM 25 mg/kg/w IP + PTX 10 mg/kg/w IP until mortality or for 4 w | ↓ Ki67-positive cells ↓ TUNEL-positive cells ↓ GRP78 immunosignal | ↓ Tumor growth ↑ Overall survival ↑ Chemotherapy effect | [100] |
| Myeloma patient-derived INA6 and RPMI 8226 cells | MKC-3946 10 µM + BTZ 2.5–10 nM or 17-AAG 125–1000 nM 2–24 h | ↓ XBP1s, ↑ CHOP, ↑ ATF4, ↑ p-eIF2α, ↑ Caspase-3, ↑ cleaved PARP | ↑ Growth inhibition ↑ Apoptosis ↑ BTZ, 17-AAG effect | [207] |
| Mice with subcutaneous RPMI 8226 myeloma | MKC-3946 100 mg/kg/d IP + BTZ 0.15 mg/kg IV 2/w 21 d | ↓ XBP1s ↑ CHOP mRNA | ↓ Tumor growth ↑ Overall survival | [207] |
| GBM patient-derived U87MG, A172, BAH1 TMZ-resistant cells | MKC-3496 10 μM + TMZ 50 μM 24–72 h | ↓ sXBP1 mRNA | ↓ Colony formation ↓ Viability ↑ Efficacy of TMZ | [76] |
| Mouse prostate cancer LNCaP, VCap, 22Rv1, and C4–2B cells xenograft models | MKC8866 300 mg/kg/d or once/2 d OR + enzalutamide, abiraterone acetate, cabazitaxel, PTX | ↓ sXBP1 ↑ Cleaved Caspase-3 ↓ PCNA | ↓ Tumor growth ↓ Proliferation, Additive/synergic effects with anti-cancer drugs, ↑ apoptosis | [81] |
| Mice with subcutaneous Myc-CaP prostate cancer xenografts | MKC8866 150–300 mg/kg/2 d + anti-PD-1 10 mg/kg/w IP 18–38 d | ↓ Tumor volume/weight ↑ Anti-PD-1 immunotherapy | [71] | |
| Breast cancer cells MCF7, SKBR3, MDA-MB-231 | MKC8866 5–20 μM + PTX 10 nM 72 h | ↓ XBP1s, ↓ IL-6, ↓ IL-8, ↓ CXCL1, ↓ TGFβ | ↓ Proliferation Cell cycle arrest ↓ Mammospheres | [219] |
| Mice with MDA-MB-231 xenografts | MKC8866 300 mg/kg/d OR + PTX 10 mg/kg/w IV up to 60 d | ↓ XBP1s | ↓ Tumor growth, ↑ sensitivity to PTX ↑ Survival | [219] |
| Glioblastoma U87 and U251 cells | MKC8866 10–80 µM or 30 µM + SIM and TMZ 72 h | ↓ p62, ↓ Beclin-1, ↓ XBP1s, ↑ LC3B-II/LC3B-I in U251 cells | ↓ Viability No synergistic effect with TMZ and SIM on death | [176] |
| Mice with intracerebral GL261-Luc cell glioblastoma | MKC8866 + IR 2 Gy + TMZ 25 mg/kg 5 d → TMZ 30–50 mg/kg 4 w | ↑ Active caspase-3 | ↑ Survival ↑ Apoptosis | [220] |
| HCC HepG2, Huh7 + stellate LX2 cells | 4µ8C 50–100 µM 48 h | ↓ PCNA mRNA ↓ ROS | ↓ Proliferation ↓ Migration | [223] |
| Mice with DEN-induced HCC | 4µ8C 10 mg/g 2/w until 25th week | ↓ Oncogenic proteins ↓ PCNA, ↓ HCC promoters PRDX5 and DDAH1, ↓ sXBP1/XBP1 | ↓ Tumor growth ↓ Collagen deposition ↓ Smooth muscle actin | [223] |
| HCC cells (HepG2, SNU449, Huh7) and patient-derived organoids | 4µ8C 10–1000 µM + DOX 1 µM 24 h | ↓ sXBP1 ↓ ATF4 ↓ Lipid metabolism genes expression | ↓ Viability, ↑ Death, ↓ lipid metabolism, ↑ DOX effect, ↓ oxygen consumption | [224] |
| Mice HCC tumor induced by DEN | 4µ8C 10 mg/g IP + DOX 4 mg/g IV bi-weekly 3 w | ↑ Caspase-3, ↓ ATF4, ↓ Ki67-positive cells, ↓ aSMA mRNA, ↓ CHOP, ↓ CXCL4, ↓ IL-1, ↓ ALT, | ↓ Tumors, ↑ DOX effect, ↓ CD68, ↓ Triglycerides, ↓ Inflammation, ↓ Fibrosis, ↓ Collagen | [224] |
| Patient-derived and blast AML cells | STF-083010 50 μM 24 h | ↓ sXBP1 ↑ miR-34a expression | ↑ Cytotoxicity | [83] |
| Mice with breast cancer MCF-7-TAM-resistant xenografts | STF-083010 30 mg/kg/w + TAM 100 µg/kg/d IP | ↓ sXBP1 | ↑ Effect of TAM ↓ Tumor growth ↑ Caspase-3 | [80] |
| Patient-derived AML blast cells | HNA 2–25 μM + BTZ 2.5–10 μM 48–72 h | ↓ sXBP1, ↑ CHOP, ↓ Bcl-2, ↑ Bim, ↓ Cyclin D, ↑ p21cip1, ↑ p27kip1, ↑ Cleaved PARP, ↑ Caspase-3, ↑ miR-34a | ↑ Cytotoxicity, ↑ BTZ effect, ↓ Colony formation, ↓ Viability, ↑ Apoptosis | [83] |
| Pancreatic cancer cells MiaPaCa-2, SU8686, Panc0403, and Panc0327 | HNA 10–50 μM 6–24 h | ↓ sXBP1, ↑ CHOP, ↑ p-JNK, ↑ cells in sub-G1 phase, ↑ cleaved PARP, ↓ Bcl-2, ↑ Bim | ↑ Apoptosis, ↓ Colony formation, ↓ Proliferation, ↓ MMP | [227] |
| Pancreatic cancer cells MiaPaCa-2, SU8686, Panc0403, and Panc0327 | Toyocamycin 0.5–5 μM 24 h | ↑ Cleaved PARP ↓ Bcl-2 ↑ CHOP | ↓ Proliferation ↓ Colony formation ↓ Mitochondrial membrane potential | [227] |
| Pancreatic cancer cells MiaPaCa-2, SU8686, Panc0403, and Panc0327 | 3ETH 1–10 μM 24 h | ↓ Proliferation ↓ Colony formation | [227] | |
| Mice with pancreatic BxPc3 xenografts | 3ETH 20 mg/kg 3 times/w 4 w | ↓ Tumor growth | [227] | |
| Glioblastoma U87 cells | Z4 0.5–25 μM 4–24 h | ↓ sXBP1, ↓ p-IRE1, ↓ SPARC | ↓ Viability, ↓ Migration | [228] |
| Mice with orthotopic glioblastoma | Z4 300 mg/kg/d 182 d +TMZ 10 mg/kg/d 21d | ↑ Effect of TMZ ↑ Relapse-free survival | [228] | |
| GRP78 inhibitors | ||||
| Pancreatic cancer PaCa-2, PANC, and BxPC-3 cells | YUM70 1–5–15 µM 24–48 h | ↑ GRP78, ↑ CHOP, ↑ FAM 129A, ↑ p-eIF2α, ↑ ATF4, ↓ c-MYC, ↓ eIF4A, eIF4E, ↓ eIF5A, ↑ 4E-BP1, ↓ p-4E-BP1, ↑ cleaved PARP, ↑ Caspase-3/7 | ↓ Viability, ↑ Apoptosis, ↓ Cell proliferation, ↓ Colony formation, Synergistic/additive effects with topotecan, vorinostat, or 5-FU | [232] |
| Mice bearing PaCa-2 cell xenografts | YUM70 30 mg/kg IP 5 d/w 7 w | ↓ Ki67 staining, ↑ CHOP, ↑ FAM 129A, ↑ Caspase-3 | ↓ Tumor growth ↑ Apoptosis | [233,234] |
| HNSCC cells SCC15, SCC25, and SCC351 | YUM70 1.25–30 µM + CIS 12–24 µM 48 h | ↑ GRP78, ↑ CHOP, ↑ cleaved PARP, ↑ Caspase-7 | ↓ Viability, ↑ Apoptosis, ↓ Clonogenicity, ↑ Sensitivity to CIS | [235,236] |
| TNBC MDA-MB-231 cells and HNSCC SCC15 and SCC25 cells | YUM70 10 μM 24 h | ↓ c-MYC, ↓ eIF4A, ↓ eIF4E, ↓ eIF5A, ↑ 4E-BP1, ↓ p-4E-BP1, ↑ cleaved PARP | ↓ Viability ↑ Apoptosis | [235] |
| Melanoma A375, Mel501, SKMel28, and patient-derived cells | HA15 10 μM, 48 h or 1–24 h | ↑ p-PERK/EIF2AK3, ↑ p-elF2a, ↑ ATF4, ↑ ERN1, ↑ ATF6, ↑ DDIT3, ↑ LC3, ↑ sXBP1, ↑ JUN, ↑ BCL2 | ↓ Viability ↑ ER stress ↑ Apoptosis ↑ Autophagy | [236] |
| Mice with melanoma A375 xenografts sensitive/resistant to BRAF inhibitors | HA15 0.7 mg/day 2 w | ↑ CHOP ↑ LC3B ↑ Autophagosomes | ↓ Tumor growth ↑ Apoptosis ↑ Autophagy | [236] |
| Lung cancer A549, H460, and H1975 cells | HA15 2–10 μM 48 h or 10 μM 24 h | ↑ ATF4, ↑ ATF6, ↑ XBP1, ↑ IRE1, ↑ CHOP, ↑ Atg5, ↑ Atg7, ↑ Atg12, ↑ LC3, ↑ ULK1, ↑ Bax, ↑ CHOP | ↓ Proliferation, Cell cycle arrest, ↑ Autophagosomes, ↓ Viability, ↑ Apoptosis | [239] |
| Pancreatic, lung, and colon cancer cells, KRAS mutant | HA15 10 μM 2 4–48 h | ↑ Cleaved PARP, ↑ CHOP, ↑ Caspase-7 | ↓ Viability ↑ Apoptosis | [233] |
| HNSSCC SCC25 and SCC15 cells, TNBC MDA-MB-231 cells | HA15 10 μM 24–48 h | ↓ c-MYC, ↑ 4E-BP1, ↓ p-4E-BP1, ↓ eIF4A, ↓ eIF4E, ↓ eIF5A | ↓ Viability ↑ Apoptosis | [101] |
| Breast cancer MDA-MB-231 and T-47D cells | EGCG 10 μM + ETO 20–40 μM 24-48 h | ↑ Caspase-7, ↓ GRP78/caspase-7 complex | ↑ Apoptosis ↓ Colony formation ↑ Sensitivity to ETO | [241] |
| Breast cancer cells 4T1, MCF-7, and MDA-MB-231 | EGCG 20 μM + PTX 1 μM 48 h | ↑ p-JNK ↓ GRP78 | ↑ Apoptosis ↑ Sensitivity to PTX | [242] |
| Mice with breast 4T1 xenografts | EGCG 30 mg/kg + PTX 10 mg/kg IP 24 d | ↑ p-JNK ↓ GRP78 | ↓ Tumor growth | [242] |
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cherubini, A.; Zito, E. ER stress as a trigger of UPR and ER-phagy in cancer growth and spread. Front. Oncol. 2022, 12, 997235. [Google Scholar] [CrossRef] [PubMed]
- Ghemrawi, R.; Kremesh, S.; Mousa, W.K.; Khair, M. The role of ER stress and the unfolded protein response in cancer. Cancer Genom. Proteom. 2025, 22, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Bonsignore, G.; Martinotti, S.; Ranzato, E. Endoplasmic reticulum stress and cancer: Could unfolded protein response be a druggable target for cancer therapy? Int. J. Mol. Sci. 2023, 24, 1566. [Google Scholar] [CrossRef] [PubMed]
- Lines, C.L.; McGrath, M.J.; Dorwart, T.; Conn, C.S. The integrated stress response in cancer progression: A force for plasticity and resistance. Front. Oncol. 2023, 13, 1206561. [Google Scholar] [CrossRef]
- Urra, H.; Aravena, R.; González-Johnson, L.; Hetz, C. The UPRising connection between endoplasmic reticulum stress and the tumor microenvironment. Trends Cancer 2024, 10, 1161–1173. [Google Scholar] [CrossRef]
- Mishra, T.; Dubey, N.; Basu, S. Small molecules for impairing endoplasmic reticulum in cancer. Org. Biomol. Chem. 2024, 22, 8689–8699. [Google Scholar] [CrossRef]
- Choudhary, M.K.; Pancholi, B.; Kumar, M.; Babu, R.; Garabadu, D. A review on endoplasmic reticulum-dependent anti-breast cancer activity of herbal drugs: Possible challenges and opportunities. J. Drug Target. 2025, 33, 206–231. [Google Scholar] [CrossRef]
- Kaczmarzyk, I.; Nowak-Perlak, M.; Woźniak, M. Promising approaches in plant-based therapies for thyroid cancer: An overview of In Vitro, In Vivo, and clinical trial studies. Int. J. Mol. Sci. 2024, 25, 4463. [Google Scholar] [CrossRef]
- Hetz, C.; Dillin, A. Central role of the ER proteostasis network in healthy aging. Trends Cell Biol. 2024, S0962-8924(24)00209-5. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic reticulum stress signalling—From basic mechanisms to clinical applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Akman, M.; Belisario, D.C.; Salaroglio, I.C.; Kopecka, J.; Donadelli, M.; De Smaele, E.; Riganti, C. Hypoxia, endoplasmic reticulum stress and chemoresistance: Dangerous liaisons. J. Exp. Clin. Cancer Res. 2021, 40, 28. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Alvear, D.; Harnoss, J.M.; Walter, P.; Ashkenazi, A. Homeostasis control in health and disease by the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2025, 26, 193–212. [Google Scholar] [CrossRef] [PubMed]
- Kapuy, O. Mechanism of decision making between autophagy and apoptosis induction upon endoplasmic reticulum stress. Int. J. Mol. Sci. 2024, 25, 4368. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Lv, Y.; Zhao, N.; Guan, G.; Wang, J. Protein kinase R-like ER kinase and its role in endoplasmic reticulum stress-decided cell fate. Cell Death Dis. 2015, 6, e1822. [Google Scholar] [CrossRef]
- Ong, G.; Ragetli, R.; Mnich, K.; Doble, B.W.; Kammouni, W.; Logue, S.E. IRE1 signaling increases PERK expression during chronic ER stress. Cell Death Dis. 2024, 15, 276. [Google Scholar] [CrossRef]
- Wortel, I.M.; van der Meer, L.T.; Kilberg, M.S.; van Leeuwen, F.N. Surviving stress: Modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol. Metab. 2017, 28, 794–806. [Google Scholar] [CrossRef]
- Siwecka, N.; Rozpędek-Kamińska, W.; Wawrzynkiewicz, A.; Pytel, D.; Diehl, J.A.; Majsterek, I. The structure, activation and signaling of IRE1 and its role in determining cell fate. Biomedicines 2021, 9, 156. [Google Scholar] [CrossRef]
- Park, S.M.; Kang, T.I.; So, J.S. Roles of XBP1s in transcriptional regulation of target genes. Biomedicines 2021, 9, 791. [Google Scholar] [CrossRef]
- Maurel, M.; Chevet, E.; Tavernier, J.; Gerlo, S. Getting RIDD of RNA: IRE1 in cell fate regulation. Trends Biochem. Sci. 2014, 39, 245–254. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER stress: Mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Fu, X.; Cui, J.; Meng, X.; Jiang, P.; Zheng, Q.; Zhao, W.; Chen, X. Endoplasmic reticulum stress, cell death and tumor: Association between endoplasmic reticulum stress and the apoptosis pathway in tumors (Review). Oncol. Rep. 2021, 45, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Varghese, E.; Samuel, S.M.; Sadiq, Z.; Kubatka, P.; Liskova, A.; Benacka, J.; Pazinka, P.; Kruzliak, P.; Büsselberg, D. Anti-cancer agents in proliferation and cell death: The calcium connection. Int. J. Mol. Sci. 2019, 20, 3017. [Google Scholar] [CrossRef] [PubMed]
- Papp, B.; Launay, S.; Gélébart, P.; Arbabian, A.; Enyedi, A.; Brouland, J.P.; Carosella, E.D.; Adle-Biassette, H. Endoplasmic reticulum calcium pumps and tumor cell differentiation. Int. J. Mol. Sci. 2020, 21, 3351. [Google Scholar] [CrossRef] [PubMed]
- Pontisso, I.; Ornelas-Guevara, R.; Chevet, E.; Combettes, L.; Dupont, G. Gradual ER calcium depletion induces a progressive and reversible UPR signaling. PNAS Nexus 2024, 3, 229. [Google Scholar] [CrossRef]
- Cui, C.; Merritt, R.; Fu, L.; Pan, Z. Targeting calcium signaling in cancer therapy. Acta Pharm. Sin. B 2017, 7, 3–17. [Google Scholar] [CrossRef]
- Capitani, C.; Chioccioli Altadonna, G.; Santillo, M.; Lastraioli, E. Ion channels in lung cancer: Biological and clinical relevance. Front. Pharmacol. 2023, 14, 1283623. [Google Scholar] [CrossRef]
- Su, M.; Zheng, S.; Liu, H.; Tang, T.S.; Hu, Y. Ca2+ homeostasis: A potential target for cancer therapies. Biophys. Rep. 2024, 10, 283–292. [Google Scholar] [CrossRef]
- Makio, T.; Chen, J.; Simmen, T. ER stress as a sentinel mechanism for ER Ca2+ homeostasis. Cell Calcium 2024, 124, 102961. [Google Scholar] [CrossRef]
- Song, W.; Zhang, Q.; Cao, Z.; Jing, G.; Zhan, T.; Yuan, Y.; Kang, N.; Zhang, Q. Targeting SERCA2 in anti-tumor drug discovery. Curr. Drug Targets 2025, 26, 1–16. [Google Scholar] [CrossRef]
- Christodoulou, P.; Yiallouris, A.; Michail, A.; Christodoulou, M.I.; Politis, P.K.; Patrikios, I. Altered SERCA expression in breast cancer. Medicina 2021, 57, 1074. [Google Scholar] [CrossRef]
- Zhang, G.; Shang, H.; Liu, B.; Wu, G.; Wu, D.; Wang, L.; Li, S.; Wang, Z.; Wang, S.; Yuan, J. Increased ATP2A1 predicts poor prognosis in patients with colorectal carcinoma. Front. Genet. 2022, 13, 661348. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.Y.; Lin, S.R.; Lu, C.Y.; Yeh, C.S.; Chen, F.M.; Hsieh, J.S.; Huang, T.J.; Wang, J.Y. Sarco/endoplasmic reticulum calcium-ATPase 2 expression as a tumor marker in colorectal cancer. Am. J. Surg. Pathol. 2006, 30, 969–974. [Google Scholar] [CrossRef]
- Fan, L.; Li, A.; Li, W.; Cai, P.; Yang, B.; Zhang, M.; Gu, Y.; Shu, Y.; Sun, Y.; Shen, Y.; et al. Novel role of Sarco/endoplasmic reticulum calcium ATPase 2 in development of colorectal cancer and its regulation by F36, a curcumin analog. Biomed. Pharmacother. 2014, 68, 1141–1148. [Google Scholar] [CrossRef] [PubMed]
- Youssef, H.M.; Radi, D.A.; Abd El-Azeem, M.A. Expression of TSP50, SERCA2 and IL-8 in colorectal adenoma and carcinoma: Correlation to clinicopathological factors. Pathol. Oncol. Res. 2021, 27, 1609990. [Google Scholar] [CrossRef]
- Xu, X.Y.; Gou, W.F.; Yang, X.; Wang, G.L.; Takahashi, H.; Yu, M.; Mao, X.Y.; Takano, Y.; Zheng, H.C. Aberrant SERCA3 expression is closely linked to pathogenesis, invasion, metastasis, and prognosis of gastric carcinomas. Tumour Biol. 2012, 33, 1845–1854. [Google Scholar] [CrossRef]
- Meneses-Morales, I.; Izquierdo-Torres, E.; Flores-Peredo, L.; Rodríguez, G.; Hernández-Oliveras, A.; Zarain-Herzberg, A. Epigenetic regulation of the human ATP2A3 gene promoter in gastric and colon cancer cell lines. Mol. Carcinog. 2019, 58, 887–897. [Google Scholar] [CrossRef]
- Li, J.; Li, X.; Huang, H.; Tao, L.; Zhang, C.; Xie, Y.; Jiang, Y. Role of SERCA3 in the prognosis and immune function in pan-cancer. J. Oncol. 2022, 2022, 9359879. [Google Scholar] [CrossRef]
- Gou, W.F.; Niu, Z.F.; Zhao, S.; Takano, Y.; Zheng, H.C. Aberrant SERCA3 expression during the colorectal adenoma-adenocarcinoma sequence. Oncol. Rep. 2014, 31, 232–240. [Google Scholar] [CrossRef]
- Aït-Ghezali, L.; Arbabian, A.; Jeibmann, A.; Hasselblatt, M.; Hallaert, G.G.; Broecke, C.V.D.; Gray, F.; Brouland, J.-P.; Varin-Blank, N.; Papp, B. Loss of endoplasmic reticulum calcium pump expression in choroid plexus tumours. Neuropathol. Appl. Neurobiol. 2014, 40, 726–735. [Google Scholar] [CrossRef]
- Adle-Biassette, H.; Ricci, R.; Martin, A.; Martini, M.; Ravegnini, G.; Kaci, R.; Gélébart, P.; Poirot, B.; Sándor, Z.; Lehman-Che, J.; et al. Sarco/endoplasmic reticulum calcium ATPase 3 (SERCA3) expression in gastrointestinal stromal tumours. Pathology 2024, 56, 343–356. [Google Scholar] [CrossRef]
- Papp, B.; Brouland, J.-P. Altered endoplasmic reticulum calcium pump expression during breast tumorigenesis. Breast Cancer Basic Clin. Res. 2011, 5, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Simbilyabo, L.Z.; Yang, L.; Wen, J.; Liu, Z. The unfolded protein response machinery in glioblastoma genesis, chemoresistance and as a druggable target. CNS Neurosci. Ther. 2024, 30, e14839. [Google Scholar] [CrossRef] [PubMed]
- Borrello, M.T.; Martin, M.B.; Pin, C.L. The unfolded protein response: An emerging therapeutic target for pancreatitis and pancreatic ductal adenocarcinoma. Pancreatology 2022, 22, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Botrus, G.; Miller, R.M.; Uson Junior, P.L.S.; Kannan, G.; Han, H.; Von Hoff, D.D. Increasing stress to induce apoptosis in pancreatic cancer via the unfolded protein response (UPR). Int. J. Mol. Sci. 2023, 24, 577. [Google Scholar] [CrossRef]
- Jadhav, A.; Menon, A.; Gupta, K.; Singh, N. Molecular and therapeutic insight into ER Stress signaling in NSCLC. J. Drug Target. 2025, 33, 877–886. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK induces resistance to cell death elicited by endoplasmic reticulum stress and chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, Q.; Zeng, A.; Song, L. Regulatory function of endoplasmic reticulum stress in colorectal cancer: Mechanism, facts, and perspectives. Int. Immunopharmacol. 2025, 147, 114024. [Google Scholar] [CrossRef]
- Farahani, N.; Alimohammadi, M.; Raei, M.; Nabavi, N.; Aref, A.R.; Hushmandi, K.; Daneshi, S.; Razzaghi, A.; Taheriazam, A.; Hashemi, M. Exploring the dual role of endoplasmic reticulum stress in urological cancers: Implications for tumor progression and cell death interactions. J. Cell Commun. Signal. 2024, 18, e12054. [Google Scholar] [CrossRef]
- Ali, M.U.; Ur Rahman, M.S.; Jia, Z.; Jiang, C. Eukaryotic translation initiation factors and cancer. Tumor Biol. 2017, 39, 1010428317709805. [Google Scholar] [CrossRef]
- Hao, P.; Yu, J.; Ward, R.; Liu, Y.; Hao, Q.; An, S.; Xu, T. Eukaryotic translation initiation factors as promising targets in cancer therapy. Cell Commun. Signal. 2020, 18, 175. [Google Scholar] [CrossRef]
- Féral, K.; Jaud, M.; Philippe, C.; Di Bella, D.; Pyronnet, S.; Rouault-Pierre, K.; Mazzolini, L.; Touriol, C. ER stress and unfolded protein response in leukemia: Friend, foe, or both? Biomolecules 2021, 11, 199. [Google Scholar] [CrossRef] [PubMed]
- Wiese, W.; Siwecka, N.; Wawrzynkiewicz, A.; Rozpędek-Kamińska, W.; Kucharska, E.; Majsterek, I. IRE1α inhibitors as a promising therapeutic strategy in blood malignancies. Cancers 2022, 14, 2526. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.; Sarkar, T.; Niemeyer, C.Y.; Harnoss, J.C.; Schneider, M.; Strowitzki, M.J.; Harnoss, J.M. IRE1 is a promising therapeutic target in pancreatic cancer. Am. J. Physiol. Cell Physiol. 2025, 328, C806–C824. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.; Mo, Y.; Li, C.M.; Liu, Y.Z.; Feng, X.P. GRP78 as a potential therapeutic target in cancer treatment: An updated review of its role in chemoradiotherapy resistance of cancer cells. Med. Oncol. 2025, 42, 49. [Google Scholar] [CrossRef]
- Song, W.; Rahimian, N.; Bashkandi, A.H. GRP78: A new promising candidate in colorectal cancer pathogenesis and therapy. Eur. J. Pharmacol. 2025, 995, 177308. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, M.; Li, J.; Wang, D.; He, Y.; He, J.; Gao, F.; Mai, L.; Li, Y.; Liang, Y.; et al. Activation of UPR signaling pathway is associated with the malignant progression and poor prognosis in prostate cancer. Prostate 2017, 77, 274–281. [Google Scholar] [CrossRef]
- Wang, E.M.; Akasaka, H.; Zhao, J.; Varadhachary, G.R.; Lee, J.E.; Maitra, A.; Fleming, J.B.; Hung, M.C.; Wang, H.; Katz, M.H. Expression and clinical significance of Protein Kinase RNA-like endoplasmic reticulum kinase and phosphorylated Eukaryotic Initiation Factor 2α in pancreatic ductal adenocarcinoma. Pancreas 2019, 48, 323–328. [Google Scholar] [CrossRef]
- Wang, P.; Han, L.; Yu, M.; Cao, Z.; Li, X.; Shao, Y.; Zhu, G. The prognostic value of PERK in cancer and its relationship with immune cell infiltration. Front. Mol. Biosci. 2021, 8, 648752. [Google Scholar] [CrossRef]
- Rosenwald, I.B.; Wang, S.; Savas, L.; Woda, B.; Pullman, J. Expression of translation initiation factor eIF-2alpha is increased in benign and malignant melanocytic and colonic epithelial neoplasms. Cancer 2003, 98, 1080–1088. [Google Scholar] [CrossRef]
- Wang, S.; Lloyd, R.V.; Hutzler, M.J.; Rosenwald, I.B.; Safran, M.S.; Patwardhan, N.A.; Khan, A. Expression of eukaryotic translation initiation factors 4E and 2alpha correlates with the progression of thyroid carcinoma. Thyroid 2001, 11, 1101–1107. [Google Scholar] [CrossRef]
- Rosenwald, I.B.; Koifman, L.; Savas, L.; Chen, J.J.; Woda, B.A.; Kadin, M.E. Expression of the translation initiation factors eIF-4E and eIF-2* is frequently increased in neoplastic cells of Hodgkin lymphoma. Hum. Pathol. 2008, 39, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sun, H.; Li, Y.; Bai, X.; Dong, X.; Zhao, N.; Meng, J.; Sun, B.; Zhang, D. High expression of eIF4E is associated with tumor macrophage infiltration and leads to poor prognosis in breast cancer. BMC Cancer 2021, 21, 1305. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.H.; Chu, H.; Kim, H.; Huh, S.; Kim, E.K.; Kang, U.B.; Shin, H.C. Comparative profiling by data-independent acquisition mass spectrometry reveals featured plasma proteins in breast cancer: A pilot study. Ann. Surg. Treat. Res. 2024, 106, 195–202. [Google Scholar] [CrossRef]
- Nguyen, H.G.; Conn, C.S.; Kye, Y.; Xue, L.; Forester, C.M.; Cowan, J.E.; Hsieh, A.C.; Cunningham, J.T.; Truillet, C.; Tameire, F.; et al. Development of a stress response therapy targeting aggressive prostate cancer. Sci. Transl. Med. 2018, 10, eaar2036. [Google Scholar] [CrossRef]
- Cao, Z.; Jing, Y.; Cheng, C.; Wang, F.; Guan, M.; Zhang, K.; Jiao, J.; Ruan, L.; Chen, Z. EIF2Ss, a novel c-Myc-correlated gene family, is associated with poor prognosis and immune infiltration in pancreatic adenocarcinoma. Front. Biosci. 2024, 29, 119. [Google Scholar] [CrossRef]
- Ji, P.; Wang, H.; Cheng, Y.; Liang, S. Prognostic prediction and gene regulation network of EIF2S2 in hepatocellular carcinoma based on data mining. J. Gastrointest. Oncol. 2021, 12, 3061–3078. [Google Scholar] [CrossRef]
- Tejada, S.; Lobo, M.V.; García-Villanueva, M.; Sacristán, S.; Pérez-Morgado, M.I.; Salinas, M.; Martín, M.E. Eukaryotic initiation factors (eIF) 2alpha and 4E expression, localization, and phosphorylation in brain tumors. J. Histochem. Cytochem. 2009, 57, 503–512. [Google Scholar] [CrossRef]
- Lobo, M.V.; Martín, M.E.; Pérez, M.I.; Alonso, F.J.; Redondo, C.; Alvarez, M.I.; Salinas, M. Levels, phosphorylation status and cellular localization of translational factor eIF2 in gastrointestinal carcinomas. Histochem. J. 2000, 32, 139–150. [Google Scholar] [CrossRef]
- He, Y.; Correa, A.M.; Raso, M.G.; Hofstetter, W.L.; Fang, B.; Behrens, C.; Roth, J.A.; Zhou, Y.; Yu, L.; Wistuba, I.I.; et al. The role of PKR/eIF2alpha signaling pathway in prognosis of non-small cell lung cancer. PLoS ONE 2011, 6, e24855. [Google Scholar] [CrossRef]
- Guo, L.; Chi, Y.; Xue, J.; Ma, L.; Shao, Z.; Wu, J. Phosphorylated EIF2 predicts disease-free survival in triple-negative breast cancer patients. Sci. Rep. 2017, 7, 44674. [Google Scholar] [CrossRef]
- Unal, B.; Kuzu, O.F.; Jin, Y.; Osorio, D.; Kildal, W.; Pradhan, M.; Kung, S.H.; Oo, H.Z.; Daugaard, M.; Vendelbo, M.; et al. Targeting IRE1α reprograms the tumor microenvironment and enhances anti-tumor immunity in prostate cancer. Nat. Commun. 2024, 15, 8895. [Google Scholar] [CrossRef] [PubMed]
- Sakatani, T.; Maemura, K.; Hiyama, N.; Amano, Y.; Watanabe, K.; Kage, H.; Fukayama, M.; Nakajima, J.; Yatomi, Y.; Nagase, T.; et al. High expression of IRE1 in lung adenocarcinoma is associated with a lower rate of recurrence. Jpn. J. Clin. Oncol. 2017, 47, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: A possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Scriven, P.; Coulson, S.; Haines, R.; Balasubramanian, S.; Cross, S.; Wyld, L. Activation and clinical significance of the unfolded protein response in breast cancer. Br. J. Cancer 2009, 101, 1692–1698. [Google Scholar] [CrossRef]
- Chen, C.; Zhang, X. IRE1α-XBP1 pathway promotes melanoma progression by regulating IL-6/STAT3 signaling. J. Transl. Med. 2017, 15, 42. [Google Scholar] [CrossRef]
- Dowdell, A.; Marsland, M.; Faulkner, S.; Gedye, C.; Lynam, J.; Griffin, C.P.; Marsland, J.; Jiang, C.C.; Hondermarck, H. Targeting XBP1 mRNA splicing sensitizes glioblastoma to chemotherapy. FASEB Bioadv. 2023, 5, 211–220. [Google Scholar] [CrossRef]
- Balague, O.; Mozos, A.; Martinez, D.; Hernandez, L.; Colomo, L.; Mate, J.L.; Teruya-Feldstein, J.; Lin, O.; Campo, E.; Lopez-Guillermo, A.; et al. Activation of the Endoplasmic Reticulum Stress-Associated Transcription Factor X Box-Binding Protein-1 Occurs in a Subset of Normal Germinal-Center B Cells and in Aggressive B-Cell Lymphomas with Prognostic Implications. Am. J. Pathol. 2009, 174, 2337–2346. [Google Scholar] [CrossRef]
- Masouleh, B.K.; Geng, H.; Hurtz, C.; Chan, L.N.; Logan, A.C.; Chang, M.S.; Huang, C.; Swaminathan, S.; Sun, H.; Paietta, E.; et al. Mechanistic rationale for targeting the unfolded protein response in pre-B acute lymphoblastic leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E2219–E2228. [Google Scholar] [CrossRef]
- Zhang, K.; Liu, H.; Song, Z.; Jiang, Y.; Kim, H.; Samavati, L.; Nguyen, H.M.; Yang, Z.Q. The UPR transducer IRE1 promotes breast cancer malignancy by degrading tumor suppressor microRNAs. iScience 2020, 23, 101503. [Google Scholar] [CrossRef]
- Ming, J.; Ruan, S.; Wang, M.; Ye, D.; Fan, N.; Meng, Q.; Tian, B.; Huang, T. A novel chemical, STF-083010, reverses Tamoxifen-related drug resistance in breast cancer by inhibiting IRE1/XBP1. Oncotarget 2015, 6, 40692–40703. [Google Scholar] [CrossRef]
- Sheng, X.; Nenseth, H.Z.; Qu, S.; Kuzu, O.F.; Frahnow, T.; Simon, L.; Greene, S.; Zeng, Q.; Fazli, L.; Rennie, P.S.; et al. IRE1α-XBP1s pathway promotes prostate cancer by activating c-MYC signaling. Nat. Commun. 2019, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- Schardt, J.A.; Weber, D.; Eyholzer, M.; Mueller, B.U.; Pabst, T. Activation of the Unfolded protein response is associated with favorable prognosis in acute myeloid leukemia. Clin. Cancer Res. 2009, 15, 3834–3841. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Lin, D.C.; Guo, X.; Kharabi Masouleh, B.; Gery, S.; Cao, Q.; Alkan, S.; Ikezoe, T.; Akiba, C.; Paquette, R.; et al. Inhibition of IRE1α-driven pro-survival pathways is a promising therapeutic application in acute myeloid leukemia. Oncotarget 2016, 7, 18736–18749. [Google Scholar] [CrossRef] [PubMed]
- Philippe, C.; Jaud, M.; Féral, K.; Gay, A.; Van Den Berghe, L.; Farce, M.; Bousquet, M.; Pyronnet, S.; Mazzolini, L.; Rouault-Pierre, K.; et al. Pivotal role of the endoplasmic reticulum stress-related XBP1s/miR-22/SIRT1 axis in acute myeloid leukemia apoptosis and response to chemotherapy. Leukemia 2024, 38, 1764–1776. [Google Scholar] [CrossRef]
- Xiao, W.; Cao, R.C.; Yang, W.J.; Tan, J.H.; Liu, R.Q.; Kan, H.P.; Zhou, L.; Zhang, N.; Chen, Z.Y.; Chen, X.M.; et al. Roles and clinical significances of ATF6, EMC6, and APAF1 in prognosis of pancreatic cancer. Front. Genet. 2022, 12, 730847. [Google Scholar] [CrossRef]
- Yarapureddy, S.; Abril, J.; Foote, J.; Kumar, S.; Asad, O.; Sharath, V.; Faraj, J.; Daniel, D.; Dickman, P.; White-Collins, A.; et al. ATF6α activation enhances survival against chemotherapy and serves as a prognostic indicator in osteosarcoma. Neoplasia 2019, 21, 516–532. [Google Scholar] [CrossRef]
- Meng, J.; Liu, K.; Shao, Y.; Feng, X.; Ji, Z.; Chang, B.; Wang, Y.; Xu, L.; Yang, G. ID1 confers cancer cell chemoresistance through STAT3/ATF6-mediated induction of autophagy. Cell Death Dis. 2020, 11, 137. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhu, X.D.; Sun, Q.N.; Wang, D. The UPR signaling molecule ATF6 is a poor prognostic marker in gastric cancer. Asian J. Surg. 2022, 45, 2836–2837. [Google Scholar] [CrossRef]
- Wu, Y.; Xie, Q.; Wu, L.; Li, Z.; Li, X.; Zhang, L.; Zhang, B. Identification of activating transcription factor 6 (ATF6) as a novel prognostic biomarker and potential target in oral squamous cell carcinoma. Gene 2024, 915, 148436. [Google Scholar] [CrossRef]
- Miao, Y.; Chen, Q.; Liu, X.; Bu, J.; Zhang, Z.; Liu, T.; Yue, Z.; Huang, L.; Sun, S.; Li, H.; et al. Comprehensive analysis of endoplasmic reticulum stress related signature in head and neck squamous carcinoma. Sci. Rep. 2024, 14, 16972. [Google Scholar] [CrossRef]
- Martinez-Useros, J.; Georgiev-Hristov, T.; Borrero-Palacios, A.; Fernandez-Aceñero, M.J.; Rodríguez-Remírez, M.; Del Puerto-Nevado, L.; Cebrian, A.; Gomez Del Pulgar, M.T.; Cazorla, A.; Vega-Bravo, R.; et al. Identification of poor-outcome biliopancreatic carcinoma patients with two-marker signature based on ATF6α and p-p38 “STARD compliant”. Medicine 2015, 94, e1972. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Hsu, C.C.; Huang, T.T.; Lee, C.H.; Chen, J.L.; Yang, S.H.; Jiang, J.K.; Chen, W.S.; Lee, K.D.; Teng, H.W. ER stress-related ATF6 upregulates CIP2A and contributes to poor prognosis of colon cancer. Mol. Oncol. 2018, 12, 1706–1717. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.; Tabbara, S.; Jacobs, L.; Manning, F.; Tsangaris, T.; Schwartz, A.; Kennedy, K.; Patierno, S. Overexpression of the glucoseregulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res. Treat. 2000, 59, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Bartkowiak, K.; Effenberger, K.E.; Harder, S.; Andreas, A.; Buck, F.; Peter-Katalinic, J.; Pantel, K.; Brandt, B.H. Discovery of a novel unfolded protein response phenotype of cancer stem/progenitor cells from the bone marrow of breast cancer patients. J. Proteome Res. 2010, 9, 3158–3168. [Google Scholar] [CrossRef]
- Al-Rawashdeh, F.Y.; Scriven, P.; Cameron, I.C.; Vergani, P.V.; Wyld, L. Unfolded protein response activation contributes to chemoresistance in hepatocellular carcinoma. Eur. J. Gastroenterol. Hepatol. 2010, 22, 1099–1105. [Google Scholar] [CrossRef]
- Su, R.; Li, Z.; Li, H.; Song, H.; Bao, C.; Wei, J.; Cheng, L. Grp78 promotes the invasion of hepatocellular carcinoma. BMC Cancer 2010, 10, 20. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, Y.; Jia, Z.; Li, Q.; Gong, W.; Wang, L.; Wei, D.; Yao, J.; Fang, S.; Xie, K. Association of elevated GRP78 expression with increased lymph node metastasis and poor prognosis in patients with gastric cancer. Clin. Exp. Metastasis 2006, 23, 401–410. [Google Scholar] [CrossRef]
- Pyrko, P.; Schönthal, A.H.; Hofman, F.M.; Chen, T.C.; Lee, A.S. The unfolded protein response regulator GRP78/BiP as a novel target for increasing chemosensitivity in malignant gliomas. Cancer Res. 2007, 67, 9809–9816. [Google Scholar] [CrossRef]
- Daneshmand, S.; Quek, M.L.; Lin, E.; Lee, C.; Cote, R.J.; Hawes, D.; Cai, J.; Groshen, S.; Lieskovsky, G.; Skinner, D.G.; et al. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum. Pathol. 2007, 38, 1547–1552. [Google Scholar] [CrossRef]
- Thakur, P.C.; Miller-Ocuin, J.L.; Nguyen, K.; Matsuda, R.; Singhi, A.D.; Zeh, H.J.; Bahary, N. Inhibition of endoplasmic-reticulum-stress-mediated autophagy enhances the effectiveness of chemotherapeutics on pancreatic cancer. J. Transl. Med. 2018, 16, 190. [Google Scholar] [CrossRef]
- Wu, J.; Wu, Y.; Lian, X. Targeted inhibition of GRP78 by HA15 promotes apoptosis of lung cancer cells accompanied by ER stress and autophagy. Biol. Open 2020, 9, bio053298. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.; Duan, W.; Liu, W.; Zhang, X.; Wang, Q. GRP78 in lung cancer. J. Transl. Med. 2021, 19, 118. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Winkler, K.; Kell, R.; Pfaffl, M.W.; Atkinson, M.J.; Moertl, S. The Chaperone protein GRP78 promotes survival and migration of head and neck cancer after direct radiation exposure and extracellular vesicle-transfer. Front. Oncol. 2022, 12, 842418. [Google Scholar] [CrossRef] [PubMed]
- Thornton, M.; Aslam, M.A.; Tweedle, E.M.; Ang, C.; Campbell, F.; Jackson, R.; Costello, E.; Rooney, P.S.; Vlatković, N.; Boyd, M.T. The unfolded protein response regulator GRP78 is a novel predictive biomarker in colorectal cancer. Int. J. Cancer 2013, 133, 1408–1418. [Google Scholar] [CrossRef]
- Chen, Q.; Li, C.; Wei, W.; Li, J.; Liu, F.; Fu, Y.; Tang, L.; Han, F. Endoplasmic reticulum stress response pathway-mediated cell death in ovarian cancer. Front. Oncol. 2024, 14, 1446552. [Google Scholar] [CrossRef]
- Xu, D.; Liu, Z.; Liang, M.X.; Fei, Y.J.; Zhang, W.; Wu, Y.; Tang, J.H. Endoplasmic reticulum stress targeted therapy for breast cancer. Cell Commun. Signal. 2022, 20, 174. [Google Scholar] [CrossRef]
- Markouli, M.; Strepkos, D.; Papavassiliou, A.G.; Piperi, C. Targeting of endoplasmic reticulum (ER) stress in gliomas. Pharmacol. Res. 2020, 157, 104823. [Google Scholar] [CrossRef]
- Zhai, K.; Mazurakova, A.; Koklesova, L.; Kubatka, P.; Büsselberg, D. Flavonoids synergistically enhance the anti-glioblastoma effects of chemotherapeutic drugs. Biomolecules 2021, 11, 1841. [Google Scholar] [CrossRef]
- Yu, M.; Lun, J.; Zhang, H.; Wang, L.; Zhang, G.; Zhang, H.; Fang, J. Targeting UPR branches, a potential strategy for enhancing efficacy of cancer chemotherapy. Acta Biochim. Biophys. Sin. 2021, 53, 1417–1427. [Google Scholar] [CrossRef]
- Mu, W.; Zhi, Y.; Zhou, J.; Wang, C.; Chai, K.; Fan, Z.; Lv, G. Endoplasmic reticulum stress and quality control in relation to Cisplatin resistance in tumor cells. Front. Pharmacol. 2024, 15, 1419468. [Google Scholar] [CrossRef]
- Michelangeli, F.; East, J.M. A diversity of SERCA Ca2+ pump inhibitors. Biochem. Soc. Trans. 2011, 39, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Peterková, L.; Kmoníčková, E.; Ruml, T.; Rimpelová, S. Sarco/endoplasmic reticulum calcium ATPase inhibitors: Beyond anticancer perspective. J. Med. Chem. 2020, 63, 1937–1963. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Ma, H.; Inesi, G.; Al-Shawi, M.K.; Toyoshima, C. Specific structural requirements for the inhibitory effect of Thapsigargin on the Ca2+ ATPase SERCA. J. Biol. Chem. 2004, 279, 17973–17979. [Google Scholar] [CrossRef]
- Suresh, A.; Bagchi, D.; Kaliappan, K.P. Thapsigargin: A promising natural product with diverse medicinal potential—A review of synthetic approaches and total syntheses. Org. Biomol. Chem. 2024, 22, 8551–8569. [Google Scholar] [CrossRef]
- Jaskulska, A.; Janecka, A.E.; Gach-Janczak, K. Thapsigargin-from traditional medicine to anticancer drug. Int. J. Mol. Sci. 2020, 22, 4. [Google Scholar] [CrossRef]
- Khurram, I.; Khan, M.U.; Ibrahim, S.; Ghani, M.U.; Amin, I.; Falzone, L.; Herrera-Bravo, J.; Setzer, W.N.; Sharifi-Rad, J.; Calina, D. Thapsigargin and its prodrug derivatives: Exploring novel approaches for targeted cancer therapy through calcium signaling disruption. Med. Oncol. 2024, 42, 7. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Jakobsen, C.M.; Janssen, S.; Khan, S.R.; Garrett, E.S.; Lilja, H.; Christensen, S.B.; Isaacs, J.T. Prostate-specific antigen-activated Thapsigargin prodrug as targeted therapy for prostate cancer. J. Natl. Cancer Inst. 2003, 95, 990–1000. [Google Scholar] [CrossRef]
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Møller, J.V. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by Thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J. Biol. Chem. 2017, 292, 19656–19673. [Google Scholar] [CrossRef]
- Yu, T.J.; Shiau, J.P.; Tang, J.Y.; Farooqi, A.A.; Cheng, Y.B.; Hou, M.F.; Yen, C.H.; Chang, H.W. Physapruin A exerts endoplasmic reticulum stress to trigger breast cancer cell apoptosis via oxidative stress. Int. J. Mol. Sci. 2023, 24, 8853. [Google Scholar] [CrossRef]
- Kotnova, A.P.; Lyanova, B.M.; Dukhanina, E.A.; Portseva, T.N.; Ilyin, Y.V.; Georgieva, S.G.; Stepchenko, A.G.; Pankratova, E.V. Thapsigargin, inhibitor of sarco-endoplasmic Ca2+-ATPase, effectively suppresses the expression of S100A4 protein in human breast cancer cell line. Dokl. Biochem. Biophys. 2019, 486, 181–183. [Google Scholar] [CrossRef]
- Chang, K.T.; Thompson, K.N.; Pratt, S.J.P.; Ju, J.A.; Lee, R.M.; Mathias, T.J.; Mull, M.L.; Annis, D.A.; Ory, E.C.; Stemberger, M.B.; et al. Elevation of cytoplasmic calcium suppresses microtentacle formation and function in breast tumor cells. Cancers 2023, 15, 884. [Google Scholar] [CrossRef] [PubMed]
- Lindner, P.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Cell death induced by the ER stressor Thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun. Signal. 2020, 18, 12. [Google Scholar] [CrossRef]
- Peñaranda-Fajardo, N.M.; Meijer, C.; Liang, Y.; Dijkstra, B.M.; Aguirre-Gamboa, R.; den Dunnen, W.F.; Kruyt, F.A. ER stress and UPR activation in glioblastoma: Identification of a noncanonical PERK mechanism regulating GBM stem cells through SOX2 modulation. Cell Death Dis. 2019, 10, 690. [Google Scholar] [CrossRef] [PubMed]
- Pecoraro, M.; Serra, A.; Pascale, M.; Franceschelli, S. The ER stress induced in human neuroblastoma cells can be reverted by lumacaftor, a CFTR corrector. Curr. Issues Mol. Biol. 2024, 46, 9342–9358. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Huang, X.; Kuang, Y.; Xing, Z.; Deng, X.; Luo, Z. Thapsigargin induces apoptosis in adrenocortical carcinoma by activating endoplasmic reticulum stress and the JNK signaling pathway: An in vitro and in vivo study. Drug Des. Devel. Ther. 2019, 13, 2787–2798. [Google Scholar] [CrossRef]
- Wang, L.; Wang, L.; Song, R.; Shen, Y.; Sun, Y.; Gu, Y.; Shu, Y.; Xu, Q. Targeting sarcoplasmic/endoplasmic reticulum Ca2+-ATPase 2 by curcumin induces ER stress-associated apoptosis for treating human liposarcoma. Mol. Cancer Ther. 2011, 10, 461–471. [Google Scholar] [CrossRef]
- Al-Taweel, N.; Varghese, E.; Florea, A.M.; Büsselberg, D. Cisplatin (CDDP) triggers cell death of MCF-7 cells following disruption of intracellular calcium ([Ca2+]i) homeostasis. J. Toxicol. Sci. 2014, 39, 765–774. [Google Scholar] [CrossRef]
- Balakrishnan, P.; Arasu, A.; Velusamy, T. Targeting altered calcium homeostasis and uncoupling protein-2 promotes sensitivity in drug-resistant breast cancer cells. J. Biochem. Mol. Toxicol. 2024, 38, e23575. [Google Scholar] [CrossRef]
- Kim, S.M.; Park, K.; Lim, J.H.; Yun, H.J.; Kim, S.Y.; Choi, K.H.; Kim, C.W.; Lee, J.H.; Weicker, R.; Pan, C.H.; et al. Potential therapeutic agents against Paclitaxel- and Sorafenib-resistant papillary thyroid carcinoma. Int. J. Mol. Sci. 2022, 23, 10378. [Google Scholar] [CrossRef]
- Kim, J.; Chang, H.S.; Yun, H.J.; Chang, H.J.; Park, K.C. New small-molecule SERCA inhibitors enhance treatment efficacy in Lenvatinib-resistant papillary thyroid cancer. Int. J. Mol. Sci. 2024, 25, 10646. [Google Scholar] [CrossRef]
- Peng, S.Y.; Tang, J.Y.; Lan, T.H.; Shiau, J.P.; Chen, K.L.; Jeng, J.H.; Yen, C.Y.; Chang, H.W. Oxidative-stress-mediated ER stress is involved in regulating Manoalide-induced antiproliferation in oral cancer cells. Int. J. Mol. Sci. 2023, 24, 3987. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W. Nodakenin induces ROS-dependent apoptotic cell death and ER stress in radioresistant breast cancer. Antioxidants 2023, 12, 492. [Google Scholar] [CrossRef] [PubMed]
- Doan, N.T.; Christensen, S.B. Thapsigargin, origin, chemistry, structure-activity relationships and prodrug development. Curr. Pharm. Des. 2015, 21, 5501–5517. [Google Scholar] [CrossRef]
- Isaacs, J.T.; Brennen, W.N.; Christensen, S.B.; Denmeade, S.R. Mipsagargin: The beginning—Not the end—Of Thapsigargin prodrug-based cancer therapeutics. Molecules 2021, 26, 7469. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulas, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel Thapsigargin-based PSMA-activated prodrug: Results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br. J. Cancer 2016, 114, 986–994. [Google Scholar] [CrossRef]
- Mahalingam, D.; Mahalingam, D.; Arora, S.P.; Sarantopoulos, J.; Peguero, J.; Campos, L.; Cen, P.; Rowe, J.; Allgood, V.; Tubb, B.; et al. A phase II, multicenter, single-arm study of Mipsagargin (G-202) as a second-line therapy following sorafenib for adult patients with progressive advanced hepatocellular carcinoma. Cancers 2019, 11, 833. [Google Scholar] [CrossRef]
- NCT02067156; Efficacy, Safety and CNS Exposure of G-202 (Mipsagargin) in Patients with Recurrent or Progressive Glioblastoma. GenSpera, Inc.: San Antonio, TX, USA, 2024.
- Chen, P.; Li, Y.; Zhou, Z.; Pan, C.; Zeng, L. Lathyrol promotes ER stress-induced apoptosis and proliferation inhibition in lung cancer cells by targeting SERCA2. Biomed. Pharmacother. 2023, 158, 114123. [Google Scholar] [CrossRef]
- Song, S.; Tai, L.; Zhou, L.; Jiang, J.; Zhao, J. Lathyrol affects the expression of AR and PSA and inhibits the malignant behavior of RCC cells. Open Med. 2025, 20, 20241136. [Google Scholar] [CrossRef]
- Rani, K.; Chand Sahu, R.; Chaudhuri, A.; Kumar, D.N.; Arora, S.; Kumar, D.; Agrawal, A.K. Exploring combinations of dihydroartemisinin for cancer therapy: A comprehensive review. Biochem. Biophys. Res. Commun. 2025, 765, 151854. [Google Scholar] [CrossRef]
- Jang, E.; Lee, J.H. Promising anticancer activities of Alismatis rhizome and its triterpenes via p38 and PI3K/Akt/mTOR signaling pathways. Nutrients 2021, 13, 2455. [Google Scholar] [CrossRef]
- Bilmen, J.G.; Khan, S.Z.; Javed, M.H.; Michelangeli, F. Inhibition of the SERCA Ca2+ pumps by curcumin. Curcumin putatively stabilizes the interaction between the nucleotide-binding and phosphorylation domains in the absence of ATP. Eur. J. Biochem. 2001, 268, 6318–6327. [Google Scholar] [CrossRef] [PubMed]
- Paula, S.; Floruta, S.; Pajazetovic, K.; Sobota, S.; Almahmodi, D. The molecular determinants of calcium ATPase inhibition by curcuminoids. Biochim. Biophys. Acta Biomembr. 2024, 1866, 184367. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Tommonaro, G. Curcumin and cancer. Nutrients 2019, 11, 2376. [Google Scholar] [CrossRef]
- Mayo, B.; Penroz, S.; Torres, K.; Simón, L. Curcumin administration routes in breast cancer treatment. Int. J. Mol. Sci. 2024, 25, 11492. [Google Scholar] [CrossRef]
- Shi, W.; Wang, Q.; Jiang, S.; Wu, Y.; Li, C.; Xie, Y.; Chen, Q.; Luo, X. Evaluating the efficacy of Curcumin in the management of oral potentially malignant disorders: A systematic review and meta-analysis. PeerJ 2024, 12, e18492. [Google Scholar] [CrossRef]
- Seo, J.A.; Kim, B.; Dhanasekaran, D.N.; Tsang, B.K.; Song, Y.S. Curcumin induces apoptosis by inhibiting sarco/endoplasmic reticulum Ca2+ ATPase activity in ovarian cancer cells. Cancer Lett. 2016, 371, 30–37. [Google Scholar] [CrossRef]
- Wang, P.; Hao, X.; Li, X.; Yan, Y.; Tian, W.; Xiao, L.; Wang, Z.; Dong, J. Curcumin inhibits adverse psychological stress-induced proliferation and invasion of glioma cells via down-regulating the ERK/MAPK pathway. J. Cell Mol. Med. 2021, 25, 7190–7203. [Google Scholar] [CrossRef]
- Wang, L.; Hu, R.; Dai, A. Curcumin increased the sensitivity of non-small-cell lung cancer to Cisplatin through the endoplasmic reticulum stress pathway. Evid. Based Complement. Alternat. Med. 2022, 2022, 6886366. [Google Scholar] [CrossRef]
- Serafino, A.; Krasnowska, E.K.; Romanò, S.; De Gregorio, A.; Colone, M.; Dupuis, M.L.; Bonucci, M.; Ravagnan, G.; Stringaro, A.; Fuggetta, M.P. The synergistic combination of curcumin and polydatin improves temozolomide efficacy on glioblastoma cells. Int. J. Mol. Sci. 2024, 25, 10572. [Google Scholar] [CrossRef]
- Luís, Â.; Amaral, L.; Domingues, F.; Pereira, L.; Cascalheira, J.F. Action of Curcumin on glioblastoma growth: A systematic review with meta-analysis of animal model studies. Biomedicines 2024, 12, 268. [Google Scholar] [CrossRef]
- Gutsche, L.C.; Dörfler, J.; Hübner, J. Curcumin as a complementary treatment in oncological therapy: A systematic review. Eur. J. Clin. Pharmacol. 2024, 81, 1–33. [Google Scholar] [CrossRef]
- Judith, P.J.; Maureen, B.; Mélanie, P.; Fabrice, K.; Isabelle, V.D.; Pascale, D.L.; Catherine, A.; Jean-Marc, N.; Hervé, C.; Valérie, D.; et al. Curcumin’s effect in advanced and metastatic breast cancer patients treated with first or second-line Docetaxel: A randomized trial. Health Sci. Rep. 2024, 7, e70052. [Google Scholar] [CrossRef] [PubMed]
- Kuzminska, J.; Szyk, P.; Mlynarczyk, D.T.; Bakun, P.; Muszalska-Kolos, I.; Dettlaff, K.; Sobczak, A.; Goslinski, T.; Jelinska, A. Curcumin derivatives in medicinal chemistry: Potential applications in cancer treatment. Molecules 2024, 29, 5321. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhang, M.; Gao, J.; Li, J.; Fan, L.; Xiang, G.; Wang, X.; Wang, X.; Wu, X.; Sun, Y.; et al. Small molecule RL71 targets SERCA2 at a novel site in the treatment of human colorectal cancer. Oncotarget 2015, 6, 37613–37625. [Google Scholar] [CrossRef] [PubMed]
- Rezbarikova, P.; Viskupicova, J.; Majekova, M.; Horakova, L. Interaction of quercetin and its derivatives with Ca2+-ATPase from sarcoplasmic reticulum: Kinetic and molecular modeling studies. Gen. Physiol. Biophys. 2023, 42, 457–468. [Google Scholar] [CrossRef]
- Silva-Pinto, P.A.; de Pontes, J.T.C.; Aguilar-Morón, B.; Canales, C.S.C.; Pavan, F.R.; Roque-Borda, C.A. Phytochemical insights into flavonoids in cancer: Mechanisms, therapeutic potential, and the case of quercetin. Heliyon 2025, 11, e42682. [Google Scholar] [CrossRef]
- Hoinoiu, T.; Dumitrascu, V.; Pit, D.; Schipor, D.A.; Jabri-Tabrizi, M.; Hoinoiu, B.; Petreuș, D.E.; Seiman, C. Quercetin as a potential therapeutic agent for malignant melanoma-a review of current evidence and future directions. Medicina 2025, 61, 656. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, J.; Qian, Y.; Yu, L.; Ma, J.; Gu, B.; Tang, W.; Li, Y.; Li, H.; Wu, W. Quercetin induces apoptosis through downregulating P4HA2 and inhibiting the PI3K/Akt/mTOR axis in hepatocellular carcinoma cells: An In Vitro study. Cancer Rep. 2025, 8, e70220. [Google Scholar] [CrossRef]
- Xiao, J.; Zhang, B.; Yin, S.; Xie, S.; Huang, K.; Wang, J.; Yang, W.; Liu, H.; Zhang, G.; Liu, X.; et al. Quercetin induces autophagy-associated death in HL-60 cells through CaMKKbeta/AMPK/mTOR signal pathway. Acta Biochim. Biophys. Sin. 2022, 54, 1244–1256. [Google Scholar] [CrossRef]
- Hussein, S.A.; Ababneh, N.A.; Tarawneh, N.; Ismail, M.A.; Awidi, A.; Abdalla, S. Antitumor effects of quercetin and luteolin in A375 cutaneous melanoma cell line are mediated by upregulation of P-ERK, c-Myc, and the upstream GPER. Life 2025, 15, 417. [Google Scholar] [CrossRef]
- Zhang, C.; Zeng, Y.; Wu, B.; Wang, L.; Wu, P.; Shen, A.; Wang, L. Anti-cancer properties and mechanistic insights of Dihydroquercetin. Curr. Pharm. Biotechnol, 2025; Online ahead of print. [Google Scholar] [CrossRef]
- Shi, G.; Wei, J.; Rahemu, S.; Zhou, J.; Li, X. Study on the regulatory mechanism of luteolin inhibiting WDR72 on the proliferation and metastasis of non small cell lung cancer. Sci. Rep. 2025, 15, 12398. [Google Scholar] [CrossRef] [PubMed]
- Vatankhah, M.A.; Ziyabakhsh, A.; Vakili Ojarood, M. Regulation of apoptosis, autophagy, and metastasis by luteolin in human bladder cancer EJ138 cells: An experimental study. Iran. J. Pharm. Res. 2024, 23, e153408. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.Z.; Bo, Y.W.; Zhang, Y.; Zhang, H.D.; Shang, Z.H.; Yu, H.; Chen, X.L.; Kong, X.T.; Zhao, W.Z.; Teimonen, T.; et al. Luteolin inhibits diffuse large B-cell lymphoma cell growth through the JAK2/STAT3 signaling pathway. Front. Pharmacol. 2025, 16, 1545779. [Google Scholar] [CrossRef]
- Pawar, C.S.; Balamurugan, K.; Baskar, S.; Prasad, N.R.; Khan, H.A. Enhancing chemosensitivity in drug-resistant breast cancer cells using beta-cyclodextrin-loaded Quercetin and Doxorubicin inclusion complex via modulating SRC/PI3K/Akt Pathway. Appl. Biochem. Biotechnol. 2025, 197, 4068–4095. [Google Scholar] [CrossRef]
- Tiburzi, S.; Lezcano, V.; Principe, G.; Montiel Schneider, M.G.; Miravalles, A.B.; Lassalle, V.; Bruzzone, A.; González-Pardo, V. Quercetin-loaded magnetic nanoparticles: A promising tool for antitumor treatment in human breast cancer cells. J. Drug Target. 2025, 13, 1–16. [Google Scholar] [CrossRef]
- Sun, C.; Xie, F.; Zhang, H.; Feng, L.; Wang, Y.; Huang, C.; Cui, Z.; Luo, C.; Zhang, L.; Wang, Q. Paclitaxel/Luteolin coloaded dual-functional liposomes for esophageal cancer therapy. Adv. Sci. 2025, 29, e2411930. [Google Scholar] [CrossRef]
- Mukherjee, A.; Ghosh, S.; Ganguli, S.; Basu, J.; Basu, B. Antiproliferative and apoptotic efficacy of nano-PLGA encapsulated Quercetin molecules by downregulation of Akt in K-ras mutated NSCLC cell lines, A549 and H460. J. Biochem. Mol. Toxicol. 2025, 39, e70240. [Google Scholar] [CrossRef]
- Kim, S.M.; Park, K.; Yun, H.J.; Kim, J.M.; Choi, K.H.; Park, K.C. Identification of new small molecules for selective inhibition of SERCA1 in patient-derived metastatic papillary thyroid cancer. Br. J. Pharmacol. 2025, 182, 2392–2408. [Google Scholar] [CrossRef]
- Xu, Z.; Shi, Y.; Zhu, L.; Luo, J.; Hu, Q.; Jiang, S.; Xiao, M.; Jiang, X.; Wang, H.; Xu, Y.; et al. Novel SERCA2 inhibitor Diphyllin displays anti-tumor effect in non-small cell lung cancer by promoting endoplasmic reticulum stress and mitochondrial dysfunction. Cancer Lett. 2024, 598, 217075. [Google Scholar] [CrossRef]
- Li, Y.; Lu, Q.; Xiao, R.; Ma, J.; Tang, Y.; Chen, W.; Zhang, R.; Jiang, L.; Chen, H.; Shen, B.; et al. Synthesis and anti-tumor activity of nitrogen-containing derivatives of the natural product Diphyllin. Eur. J. Med. Chem. 2022, 243, 114708. [Google Scholar] [CrossRef]
- Tang, Y.; Pan, X.; Shang, F.F.; Li, Y.; Zhang, C.; Ma, H.; Zhang, A.; Wang, X.; Ding, C.; Chen, W. Synthesis and pharmacological evaluation of natural product Diphyllin derivatives against head and neck squamous cell carcinoma. Eur. J. Med. Chem. 2025, 285, 117215. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Medina, J.R.; Feng, Y.; Shu, A.; Romeril, S.P.; Grant, S.W.; Li, W.H.; Heerding, D.A.; Minthorn, E.; Mencken, T.; et al. Discovery of 7-methyl-5-(1-{[3-(trifluoromethyl)phenyl]acetyl}-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK). J. Med. Chem. 2012, 55, 7193–7207. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Ge, Y.; Dong, J.; Wang, H.; Zhao, T.; Wang, X.; Liu, J.; Gao, S.; Shi, L.; Yang, S.; et al. BZW1 Facilitates glycolysis and promotes tumor growth in pancreatic ductal adenocarcinoma through potentiating eIF2alpha phosphorylation. Gastroenterology 2022, 162, 1256–1271. [Google Scholar] [CrossRef]
- Dastghaib, S.; Shojaei, S.; Mostafavi-Pour, Z.; Sharma, P.; Patterson, J.B.; Samali, A.; Mokarram, P.; Ghavami, S. Simvastatin induces unfolded protein response and enhances Temozolomide-induced cell death in glioblastoma cells. Cells 2020, 9, 2339. [Google Scholar] [CrossRef]
- Ketkar, M.; Desai, S.; Rana, P.; Thorat, R.; Epari, S.; Dutt, A.; Dutt, S. Inhibition of PERK-mediated unfolded protein response acts as a switch for reversal of residual senescence and as senolytic therapy in glioblastoma. Neuro Oncol. 2024, 26, 2027–2043. [Google Scholar] [CrossRef]
- Yu, Z.Z.; Xu, B.Q.; Wang, Y.Y.; Zhang, P.W.; Shu, Y.B.; Shi, Z. GSK2606414 Sensitizes ABCG2-overexpressing multidrug-resistant colorectal cancer cells to chemotherapeutic drugs. Biomedicines 2023, 11, 3103. [Google Scholar] [CrossRef]
- Bagratuni, T.; Patseas, D.; Mavrianou-Koutsoukou, N.; Liacos, C.I.; Sklirou, A.D.; Rousakis, P.; Gavriatopoulou, M.; Terpos, E.; Tsitsilonis, O.E.; Trougakos, I.P.; et al. Characterization of a PERK kinase inhibitor with anti-myeloma activity. Cancers 2020, 12, 2864. [Google Scholar] [CrossRef]
- McLaughlin, M.; Pedersen, M.; Roulstone, V.; Bergerhoff, K.F.; Smith, H.G.; Whittock, H.; Kyula, J.N.; Dillon, M.T.; Pandha, H.S.; Vile, R.; et al. The PERK inhibitor GSK2606414 enhances reovirus infection in head and neck squamous cell carcinoma via an ATF4-dependent mechanism. Mol. Ther. Oncolytics 2020, 16, 238–249. [Google Scholar] [CrossRef]
- Cai, W.; Sun, X.; Jin, F.; Xiao, D.; Li, H.; Sun, H.; Wang, Y.; Lu, Y.; Liu, J.; Huang, C.; et al. PERK-eIF2α-ERK1/2 axis drives mesenchymal-endothelial transition of cancer-associated fibroblasts in pancreatic cancer. Cancer Lett. 2021, 515, 86–95. [Google Scholar] [CrossRef]
- Zhang, X.H.; Wang, X.Y.; Zhou, Z.W.; Bai, H.; Shi, L.; Yang, Y.X.; Zhou, S.F.; Zhang, X.C. The combination of Digoxin and GSK2606414 exerts synergistic anticancer activity against leukemia In Vitro and In Vivo. Biofactors 2017, 43, 812–820. [Google Scholar] [CrossRef]
- Xu, L.; Jiang, Y.; Bi, Y.; Zheng, S.; Wu, Y.; Wu, Y.; Xu, Y.; Chen, J. Suppression of PERK/eIF2α/CHOP pathway enhances Oridonin-induced apoptosis by inhibiting autophagy in small-cell lung cancer cells. Biomed. Pharmacother. 2024, 175, 116684. [Google Scholar] [CrossRef] [PubMed]
- Axten, J.M.; Romeril, S.P.; Shu, A.; Ralph, J.; Medina, J.R.; Feng, Y.; Li, W.H.; Grant, S.W.; Heerding, D.A.; Minthorn, E.; et al. Discovery of GSK2656157: An optimized PERK inhibitor selected for preclinical development. ACS Med. Chem. Lett. 2013, 4, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Atkins, C.; Liu, Q.; Minthorn, E.; Zhang, S.Y.; Figueroa, D.J.; Moss, K.; Stanley, T.B.; Sanders, B.; Goetz, A.; Gaul, N.; et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res. 2013, 73, 1993–2002. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Q.; Wang, X.; Zheng, K.; Liu, K.S.; Wang, S.X.; Xie, C.H. Simultaneous targeting PI3K and PERK pathways promotes cell death and improves the clinical prognosis in esophageal squamous carcinoma. Biochem. Biophys. Res. Commun. 2017, 493, 534–541. [Google Scholar] [CrossRef]
- Dudka, W.; Hoser, G.; Mondal, S.S.; Turos-Korgul, L.; Swatler, J.; Kusio-Kobialka, M.; Wołczyk, M.; Klejman, A.; Brewinska-Olchowik, M.; Kominek, A.; et al. Targeting integrated stress response with ISRIB combined with Imatinib treatment attenuates RAS/RAF/MAPK and STAT5 signaling and eradicates chronic myeloid leukemia cells. BMC Cancer 2022, 22, 1254. [Google Scholar] [CrossRef]
- Rozpędek-Kamińska, W.; Galita, G.; Siwecka, N.; Granek, Z.; Barczuk, J.; Saramowicz, K.; Majsterek, I. NCI 159456 PERK inhibitor as a targeted therapy for lung cancer: An In Vitro study. Biomedicines 2024, 12, 889. [Google Scholar] [CrossRef]
- McCarthy, N.; Dolgikh, N.; Logue, S.; Patterson, J.B.; Zeng, Q.; Gorman, A.M.; Samali, A.; Fulda, S. The IRE1 and PERK arms of the unfolded protein response promote survival of rhabdomyosarcoma cells. Cancer Lett. 2020, 490, 76–88. [Google Scholar] [CrossRef]
- Anand, A.A.; Walter, P. Structural insights into ISRIB, a memory-enhancing inhibitor of the integrated stress response. FEBS J. 2020, 287, 239–245. [Google Scholar] [CrossRef]
- Ghaddar, N.; Wang, S.; Woodvine, B.; Krishnamoorthy, J.; van Hoef, V.; Darini, C.; Kazimierczak, U.; Ah-Son, N.; Popper, H.; Johnson, M.; et al. The integrated stress response is tumorigenic and constitutes a therapeutic liability in KRAS-riven lung cancer. Nat. Commun. 2021, 12, 4651. [Google Scholar] [CrossRef]
- Yang, S.Y.; Liao, L.; Hu, S.Y.; Deng, L.; Andriani, L.; Zhang, T.M.; Zhang, Y.L.; Ma, X.Y.; Zhang, F.L.; Liu, Y.Y.; et al. ETHE1 accelerates triple-negative breast cancer metastasis by activating GCN2/eIF2alpha/ATF4 signaling. Int. J. Mol. Sci. 2023, 24, 14566. [Google Scholar] [CrossRef]
- Wang, X.; Chen, T.; Li, C.; Li, W.; Zhou, X.; Li, Y.; Luo, D.; Zhang, N.; Chen, B.; Wang, L.; et al. CircRNA-CREIT inhibits stress granule assembly and overcomes doxorubicin resistance in TNBC by destabilizing PKR. J. Hematol. Oncol. 2022, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Boretti, A.; Banik, B. Maximizing ISRIB Potential requires addressing specificity, long-term safety, and disease-specific considerations. Curr. Med. Chem. 2024. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, M.; Komoike, Y. Experimental evidence shows Salubrinal, an eIF2α dephosphorylation inhibitor, reduces xenotoxicant-induced cellular damage. Int. J. Mol. Sci. 2015, 16, 16275–16287. [Google Scholar] [CrossRef] [PubMed]
- da Silva, D.C.; Valentao, P.; Andrade, P.B.; Pereira, D.M. Endoplasmic reticulum stress signaling in cancer and neurodegenerative disorders: Tools and strategies to understand its complexity. Pharmacol. Res. 2020, 155, 104702. [Google Scholar] [CrossRef]
- Alsterda, A.; Asha, K.; Powrozek, O.; Repak, M.; Goswami, S.; Dunn, A.M.; Memmel, H.C.; Sharma-Walia, N. Salubrinal exposes anticancer properties in inflammatory breast cancer cells by manipulating the endoplasmic reticulum stress pathway. Front. Oncol. 2021, 11, 654940. [Google Scholar] [CrossRef]
- Wu, L.; Liang, C.; Huang, X.; Deng, X.; Jiang, J.; Luo, Z. Salubrinal regulates the apoptosis of adrenocortical carcinoma cells via the PERK/eIF2α/ATF4 signaling pathway. Int. J. Endocrinol. 2021, 2021, 5038130. [Google Scholar] [CrossRef]
- Eytan, K.; Versano, Z.; Oren, R.; Jacob-Hirsch, J.; Leitner, M.; Harmelin, A.; Rechavi, G.; Toren, A.; Paglin, S.; Yalon, M. Pediatric glioblastoma cells are sensitive to drugs that inhibit eIF2a dephosphorylation and its phosphomimetic S51D variant. Front. Oncol. 2022, 12, 959133. [Google Scholar] [CrossRef]
- Cyran, A.M.; Kleinegger, F.; Nass, N.; Naumann, M.; Haybaeck, J.; Arens, C. Inhibition of EIF2α dephosphorylation decreases cell viability and synergizes with standard-of-care chemotherapeutics in head and neck squamous cell carcinoma. Cancers 2023, 15, 5350. [Google Scholar] [CrossRef]
- Chen, M.C.; Hsu, L.L.; Wang, S.F.; Pan, Y.L.; Lo, J.F.; Yeh, T.S.; Tseng, L.M.; Lee, H.C. Salubrinal enhances cancer cell death during glucose deprivation through the upregulation of xCT and mitochondrial oxidative stress. Biomedicines 2021, 9, 1101. [Google Scholar] [CrossRef]
- Kardos, G.R.; Gowda, R.; Dinavahi, S.S.; Kimball, S.; Robertson, G.P. Salubrinal in combination with 4E1RCat synergistically impairs melanoma development by disrupting the protein synthetic machinery. Front. Oncol. 2020, 10, 834. [Google Scholar] [CrossRef]
- Snyder, C.M.; Mateo, B.; Patel, K.; Fahrenholtz, C.D.; Rohde, M.M.; Carpenter, R.; Singh, R.N. Enhancement of triple-negative breast cancer-specific induction of cell death by silver nanoparticles by combined treatment with proteotoxic stress response inhibitors. Nanomaterials 2024, 14, 1564. [Google Scholar] [CrossRef] [PubMed]
- Shao, A.; Xu, Q.; Spalek, W.T.; Cain, C.F.; Kang, C.W.; Tang, C.A.; Del Valle, J.R.; Hu, C.A. Development of tumor-targeting IRE-1 inhibitors for B-cell cancer therapy. Mol. Cancer Ther. 2020, 19, 2432–2444. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Perera, B.G.; Hari, S.B.; Bhhatarai, B.; Backes, B.J.; Seeliger, M.A.; Schurer, S.C.; Oakes, S.A.; Papa, F.R.; Maly, D.J. Divergent allosteric control of the IRE1a endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 2012, 8, 982–989. [Google Scholar] [CrossRef]
- Carlesso, A.; Chintha, C.; Gorman, A.M.; Samali, A.; Eriksson, L.A. Effect of kinase inhibiting RNase attenuator (KIRA) compounds on the formation of face-to-face dimers of inositol-requiring enzyme 1: Insights from computational modeling. Int. J. Mol. Sci. 2019, 20, 5538. [Google Scholar] [CrossRef]
- Mimura, N.; Fulciniti, M.; Gorgun, G.; Tai, Y.T.; Cirstea, D.; Santo, L.; Hu, Y.; Fabre, C.; Minami, J.; Ohguchi, H.; et al. Blockade of XBP1 splicing by inhibition of IRE1α is a promising therapeutic option in multiple myeloma. Blood 2012, 119, 5772–5781. [Google Scholar] [CrossRef]
- Cross, B.C.; Bond, P.J.; Sadowski, P.G.; Jha, B.K.; Zak, J.; Goodman, J.M.; Silverman, R.H.; Neubert, T.A.; Baxendale, I.R.; Ron, D.; et al. The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc. Natl. Acad. Sci. USA 2012, 109, E869–E878. [Google Scholar] [CrossRef]
- Volkmann, K.; Lucas, J.L.; Vuga, D.; Wang, X.; Brumm, D.; Stiles, C.; Kriebel, D.; Der-Sarkissian, A.; Krishnan, K.; Schweitzer, C.; et al. Potent and selective inhibitors of the inositol-requiring enzyme 1 endoribonuclease. J. Biol. Chem. 2011, 286, 12743–12755. [Google Scholar] [CrossRef]
- Shao, A.; Xu, Q.; Kang, C.W.; Cain, C.F.; Lee, A.C.; Tang, C.A.; Del Valle, J.R.; Hu, C.A. IRE-1-targeting caged prodrug with endoplasmic reticulum stress-inducing and XBP-1S-inhibiting activities for cancer therapy. Mol. Pharm. 2022, 19, 1059–1067. [Google Scholar] [CrossRef]
- Aldin, A.; Besiroglu, B.; Adams, A.; Monsef, I.; Piechotta, V.; Tomlinson, E.; Hornbach, C.; Dressen, N.; Goldkuhle, M.; Maisch, P.; et al. First-line therapy for adults with advanced renal cell carcinoma: A systematic review and network meta-analysis. Cochrane Database Syst. Rev. 2023, 5, CD013798. [Google Scholar] [CrossRef]
- Wen, W.; Han, E.S.; Dellinger, T.H.; Lu, L.X.; Wu, J.; Jove, R.; Yim, J.H. Synergistic anti-tumor activity by targeting multiple signaling pathways in ovarian cancer. Cancers 2020, 12, 2586. [Google Scholar] [CrossRef]
- Fullana, B.; Morales, S.; Petit, A.; Alay, A.; Verdaguer, H.; Climent, F.; Navarro-Perez, V.; Cejuela, M.; Galvan, P.; Gumà, A.; et al. Neoadjuvant Sunitinib plus Exemestane in post-menopausal women with hormone receptor-positive/HER2-negative early-stage breast cancer (SUT_EXE-08): A phase I/II trial. Sci. Rep. 2024, 14, 23626. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Porta, C.; Eto, M.; Hutson, T.E.; Rha, S.Y.; Merchan, J.R.; Winquist, E.; Gurney, H.; Grünwald, V.; George, S.; et al. Biomarker analyses from the phase III randomized CLEAR trial: Lenvatinib plus Pembrolizumab versus Sunitinib in advanced renal cell carcinoma. Ann. Oncol. 2025, 36, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, T.; Bick, F.; Peters, K.; Mohta, V.; Tirosh, B.; Patterson, J.B.; Kharabi-Masouleh, B.; Huber, M. Infliction of proteotoxic stresses by impairment of the unfolded protein response or proteasomal inhibition as a therapeutic strategy for mast cell leukemia. Oncotarget 2017, 9, 2984–3000. [Google Scholar] [CrossRef] [PubMed]
- Harnoss, J.M.; Le Thomas, A.; Shemorry, A.; Marsters, S.A.; Lawrence, D.A.; Lu, M.; Chen, Y.A.; Qing, J.; Totpal, K.; Kan, D.; et al. Disruption of IRE1α through its kinase domain attenuates multiple myeloma. Proc. Natl. Acad. Sci. USA 2019, 116, 16420–16429. [Google Scholar] [CrossRef]
- Kern, J.; Schilling, D.; Schneeweis, C.; Schmid, R.M.; Schneiderm, G.; Combs, S.E.; Dobiasch, S. Identification of the unfolded protein response pathway as target for radiosensitization in pancreatic cancer. Radiother. Oncol. 2024, 191, 110059. [Google Scholar] [CrossRef]
- Li, X.; Bo, Y.; Zeng, Q.; Diao, L.; Greene, S.; Patterson, J.; Liu, L.; Yang, F. Population pharmacokinetic model for oral ORIN1001 in Chinese patients with advanced solid tumors. Front. Pharmacol. 2024, 15, 1322557. [Google Scholar] [CrossRef]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef]
- Le Reste, P.J.; Pineau, R.; Voutetakis, K.; Samal, J.; Jégou, G.; Lhomond, S.; Gorman, A.M.; Samali, A.; Patterson, J.B.; Zeng, Q.; et al. Local intracerebral inhibition of IRE1 by MKC8866 sensitizes glioblastoma to irradiation/chemotherapy In Vivo. Cancer Lett. 2020, 494, 73–83. [Google Scholar] [CrossRef]
- Vieri, M.; Preisinger, C.; Schemionek, M.; Salimi, A.; Patterson, J.B.; Samali, A.; Brümmendorf, T.H.; Appelmann, I.; Kharabi Masouleh, B. Targeting of BCR-ABL1 and IRE1α induces synthetic lethality in Philadelphia-positive acute lymphoblastic leukemia. Carcinogenesis 2021, 42, 272–284. [Google Scholar] [CrossRef]
- Xiao, R.; You, L.; Zhang, L.; Guo, X.; Guo, E.; Zhao, F.; Yang, B.; Li, X.; Fu, Y.; Lu, F.; et al. Inhibiting the IRE1α axis of the unfolded protein response enhances the antitumor effect of AZD1775 in TP53 mutant ovarian cancer. Adv. Sci. 2022, 9, e2105469. [Google Scholar] [CrossRef]
- Pavlović, N.; Calitz, C.; Thanapirom, K.; Mazza, G.; Rombouts, K.; Gerwins, P.; Heindryckx, F. Inhibiting IRE1α-endonuclease activity decreases tumor burden in a mouse model for hepatocellular carcinoma. Elife 2020, 9, e55865. [Google Scholar] [CrossRef] [PubMed]
- Kopsida, M.; Clavero, A.L.; Khaled, J.; Balgoma, D.; Luna-Marco, C.; Chowdhury, A.; Nyman, S.S.; Rorsman, F.; Ebeling Barbier, C.; Bergsten, P.; et al. Inhibiting the endoplasmic reticulum stress response enhances the effect of D221oxorubicin by altering the lipid metabolism of liver cancer cells. Mol. Metab. 2024, 79, 101846. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Zhang, H.S.; Xu, Y.M.; Zhang, R.J.; Chen, Y.; Fan, L.; Qin, Y.Q.; Liu, Y.; Li, M.; Fang, J. Knockdown of IRE1alpha inhibits colonic tumorigenesis through decreasing beta-catenin and IRE1alpha targeting suppresses colon cancer cells. Oncogene 2017, 36, 6738–6746. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Li, X.X.; Xu, Y.M.; Zhang, J.Z.; Rong, S.D.; Qin, Y.Q.; Fang, J. IRE1alpha-targeting downregulates ABC transporters and overcomes drug resistance of colon cancer cells. Cancer Lett. 2020, 476, 67–74. [Google Scholar] [CrossRef]
- Chien, W.; Ding, L.W.; Sun, Q.Y.; Torres-Fernandez, L.A.; Tan, S.Z.; Xiao, J.; Lim, S.L.; Garg, M.; Lee, K.L.; Kitajima, S.; et al. Selective inhibition of unfolded protein response induces apoptosis in pancreatic cancer cells. Oncotarget 2014, 5, 4881–4894. [Google Scholar] [CrossRef]
- Pelizzari-Raymundo, D.; Doultsinos, D.; Pineau, R.; Sauzay, C.; Koutsandreas, T.; Langlais, T.; Carlesso, A.; Gkotsi, E.; Negroni, L.; Avril, T.; et al. A novel IRE1 kinase inhibitor for adjuvant glioblastoma treatment. iScience 2023, 26, 106687. [Google Scholar] [CrossRef]
- Okada, T.; Haze, K.; Nadanaka, S.; Yoshida, H.; Seidah, N.G.; Hirano, Y.; Sato, R.; Negishi, M.; Mori, K. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane bound transcription factor ATF6. J. Biol. Chem. 2003, 278, 31024–31032. [Google Scholar] [CrossRef]
- Gallagher, C.M.; Garri, C.; Cain, E.L.; Ang, K.K.; Wilson, C.G.; Chen, S.; Hearn, B.R.; Jaishankar, P.; Aranda-Diaz, A.; Arkin, M.R.; et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch. Elife 2016, 5, e11878. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, T.; Chen, L.; Cui, F.; Xu, C.; Peng, J.; Ma, W.; Huang, J.; Sheng, X.; Liu, M.; et al. The functional implication of ATF6α in castration-resistant prostate cancer cells. FASEB J. 2023, 37, e22758. [Google Scholar] [CrossRef]
- Samanta, S.; Yang, S.; Debnath, B.; Xue, D.; Kuang, Y.; Ramkumar, K.; Lee, A.S.; Ljungman, M.; Neamati, N. The hydroxyquinoline analogue YUM70 inhibits GRP78 to induce ER stress-mediated apoptosis in pancreatic cancer. Cancer Res. 2021, 81, 1883–1895. [Google Scholar] [CrossRef]
- Ha, D.P.; Huang, B.; Wang, H.; Rangel, D.F.; Van Krieken, R.; Liu, Z.; Samanta, S.; Neamati, N.; Lee, A.S. Targeting GRP78 suppresses oncogenic KRAS protein expression and reduces viability of cancer cells bearing various KRAS mutations. Neoplasia 2022, 33, 100837. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, V.; Wang, B.; Lee, A.S. Suppression of head and neck cancer cell survival and Cisplatin resistance by GRP78 small molecule inhibitor YUM70. Front. Oncol. 2023, 12, 104469. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, V.; Ha, D.P.; Liu, Z.; Huang, M.; Samanta, S.; Neamati, N.; Lee, A.S. GRP78 inhibitor YUM70 upregulates 4E-BP1 and suppresses c-MYC expression and viability of oncogenic c-MYC tumors. Neoplasia 2024, 55, 101020. [Google Scholar] [CrossRef]
- Cerezo, M.; Lehraiki, A.; Millet, A.; Rouaud, F.; Plaisant, M.; Jaune, E.; Botton, T.; Ronco, C.; Abbe, P.; Amdouni, H.; et al. Compounds triggering ER stress exert anti-melanoma effects and overcome BRAF inhibitor resistance. Cancer Cell 2016, 29, 805–819. [Google Scholar] [CrossRef]
- Ruggiero, C.; Doghman-Bouguerra, M.; Ronco, C.; Benhida, R.; Rocchi, S.; Lalli, E. The GRP78/BiP inhibitor HA15 synergizes with mitotane action against adrenocortical carcinoma cells through convergent activation of ER stress pathways. Mol. Cell. Endocrinol. 2018, 474, 57–64. [Google Scholar] [CrossRef]
- Cheng, C.C.; Yang, B.L.; Chen, W.C.; Ho, A.S.; Sie, Z.L.; Lin, H.C.; Chang, C.C. STAT3 mediated miR-30a-5p Inhibition enhances proliferation and inhibits apoptosis in colorectal cancer cells. Int. J. Mol. Sci. 2020, 21, 7315. [Google Scholar] [CrossRef]
- Park, J.; Purushothaman, B.; Hong, S.; Choi, M.; Jegal, K.H.; Park, M.; Song, J.M.; Kang, K.W. GRP78 blockade overcomes acquired resistance to EGFR-tyrosine kinase inhibitors in non-small cell lung cancer. Life Sci. 2024, 348, 122681. [Google Scholar] [CrossRef]
- Ermakova, S.P.; Kang, B.S.; Choi, B.Y.; Choi, H.S.; Schuster, T.F.; Ma, W.Y.; Bode, A.M.; Dong, Z. (−)-Epigallocatechin gallate overcomes resistance to Etoposide-induced cell death by targeting the molecular chaperone glucose-regulated protein 78. Cancer Res. 2006, 66, 9260–9269. [Google Scholar] [CrossRef]
- Le, C.T.; Leenders, W.P.; Molenaar, R.J.; van Noorden, C.J. Effects of the green tea polyphenol Epigallocatechin-3-Gallate on glioma: A critical evaluation of the literature. Nutr. Cancer 2018, 70, 317–333. [Google Scholar] [CrossRef]
- Luo, T.; Wang, J.; Yin, Y.; Hua, H.; Jing, J.; Sun, X.; Li, M.; Zhang, Y.; Jiang, Y. (−)-Epigallocatechin gallate sensitizes breast cancer cells to Paclitaxel in a murine model of breast carcinoma. Breast Cancer Res. 2010, 12, R8. [Google Scholar] [CrossRef]
- Wu, P.P.; Kuo, S.C.; Huang, W.W.; Yang, J.S.; Lai, K.C.; Chen, H.J.; Lin, K.L.; Chiu, Y.J.; Huang, L.J.; Chung, J.G. (−)-Epigallocatechin gallate induced apoptosis in human adrenal cancer NCI-H295 cells through caspase-dependent and caspase-independent pathway. Anticancer Res. 2009, 29, 1435–1442. [Google Scholar] [PubMed]
- La, X.; Zhang, L.; Li, Z.; Li, H.; Yang, Y. (−)-Epigallocatechin gallate (EGCG) enhances the sensitivity of colorectal cancer cells to 5-FU by inhibiting GRP78/NF-κB/miR-155-5p/MDR1 pathway. J. Agric. Food Chem. 2019, 67, 2510–2518. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.C.; Wang, W.; Golden, E.B.; Thomas, S.; Sivakumar, W.; Hofman, F.M.; Louie, S.G.; Schönthal, A.H. Green tea Epigallocatechin Gallate enhances therapeutic efficacy of temozolomide in orthotopic mouse glioblastoma models. Cancer Lett. 2011, 302, 100–108. [Google Scholar] [CrossRef] [PubMed]
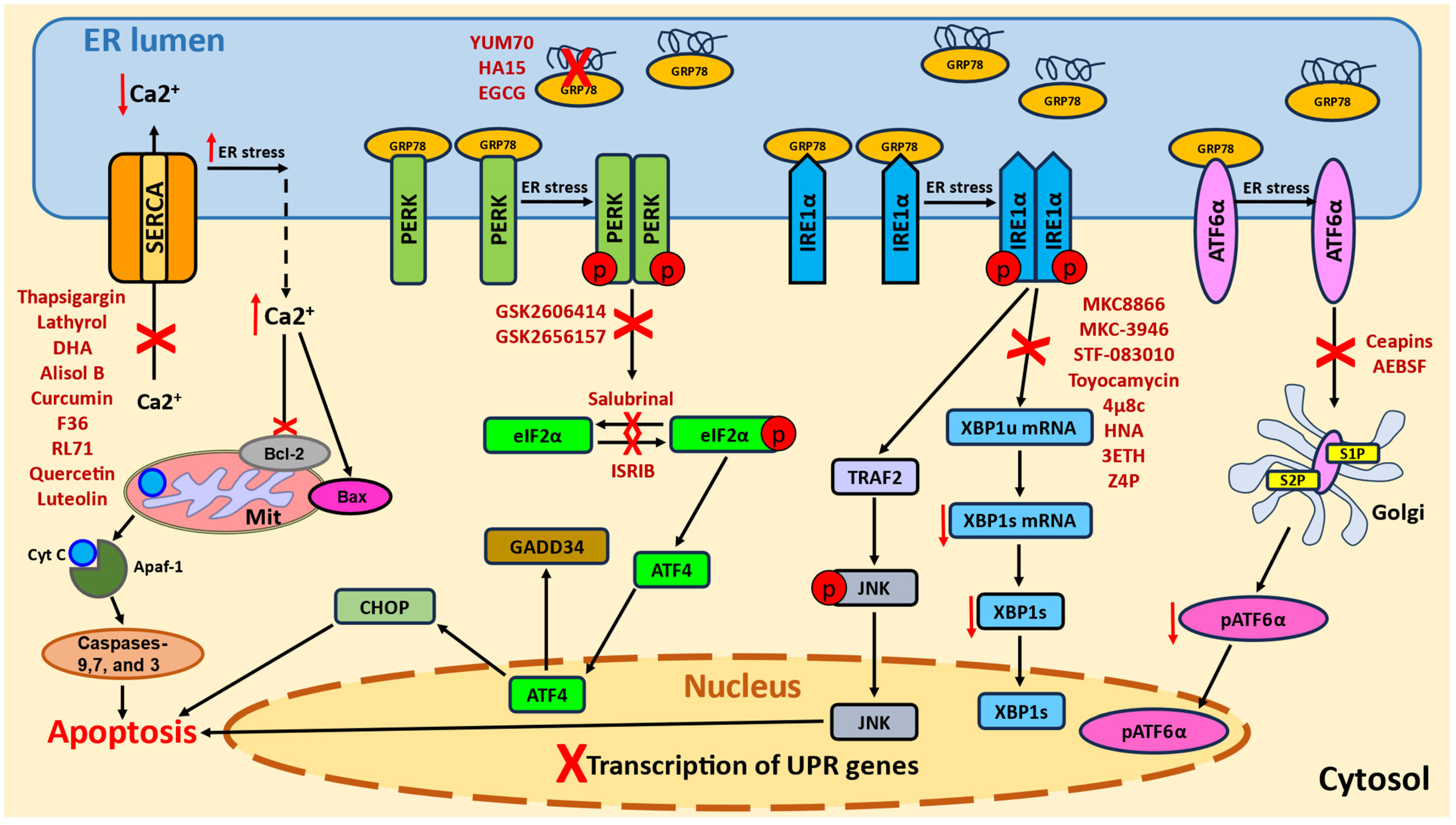
| Scheme | Expression Profile | Cancer Type | Clinical Outcome | References |
|---|---|---|---|---|
| SERCA1 | Overexpression | Breast cancer | Poor prognosis Reduced survival | [30] |
| Colorectal carcinoma | [31] | |||
| SERCA2 | Overexpression | Colon and rectal adenomas and carcinomas | Lower survival Invasion Metastasis | [32,33,34] |
| SERCA3 | Downregulation | Gastric carcinoma | Metastasis Poor prognosis | [35,36] |
| Glioma/glioblastoma | [37] | |||
| Colorectal carcinoma | No correlation with survival | [38] | ||
| Choroid plexus papillomas and carcinomas | - | [39] | ||
| Breast carcinomas | [41] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agalakova, N.I. Modulation of Endoplasmic Reticulum Stress in Experimental Anti-Cancer Therapy. Int. J. Mol. Sci. 2025, 26, 6407. https://doi.org/10.3390/ijms26136407
Agalakova NI. Modulation of Endoplasmic Reticulum Stress in Experimental Anti-Cancer Therapy. International Journal of Molecular Sciences. 2025; 26(13):6407. https://doi.org/10.3390/ijms26136407
Chicago/Turabian StyleAgalakova, Natalia Ivanovna. 2025. "Modulation of Endoplasmic Reticulum Stress in Experimental Anti-Cancer Therapy" International Journal of Molecular Sciences 26, no. 13: 6407. https://doi.org/10.3390/ijms26136407
APA StyleAgalakova, N. I. (2025). Modulation of Endoplasmic Reticulum Stress in Experimental Anti-Cancer Therapy. International Journal of Molecular Sciences, 26(13), 6407. https://doi.org/10.3390/ijms26136407