
EpInflammAge: Epigenetic-Inflammatory Clock for Disease-Associated Biological Aging Based on Deep Learning

 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
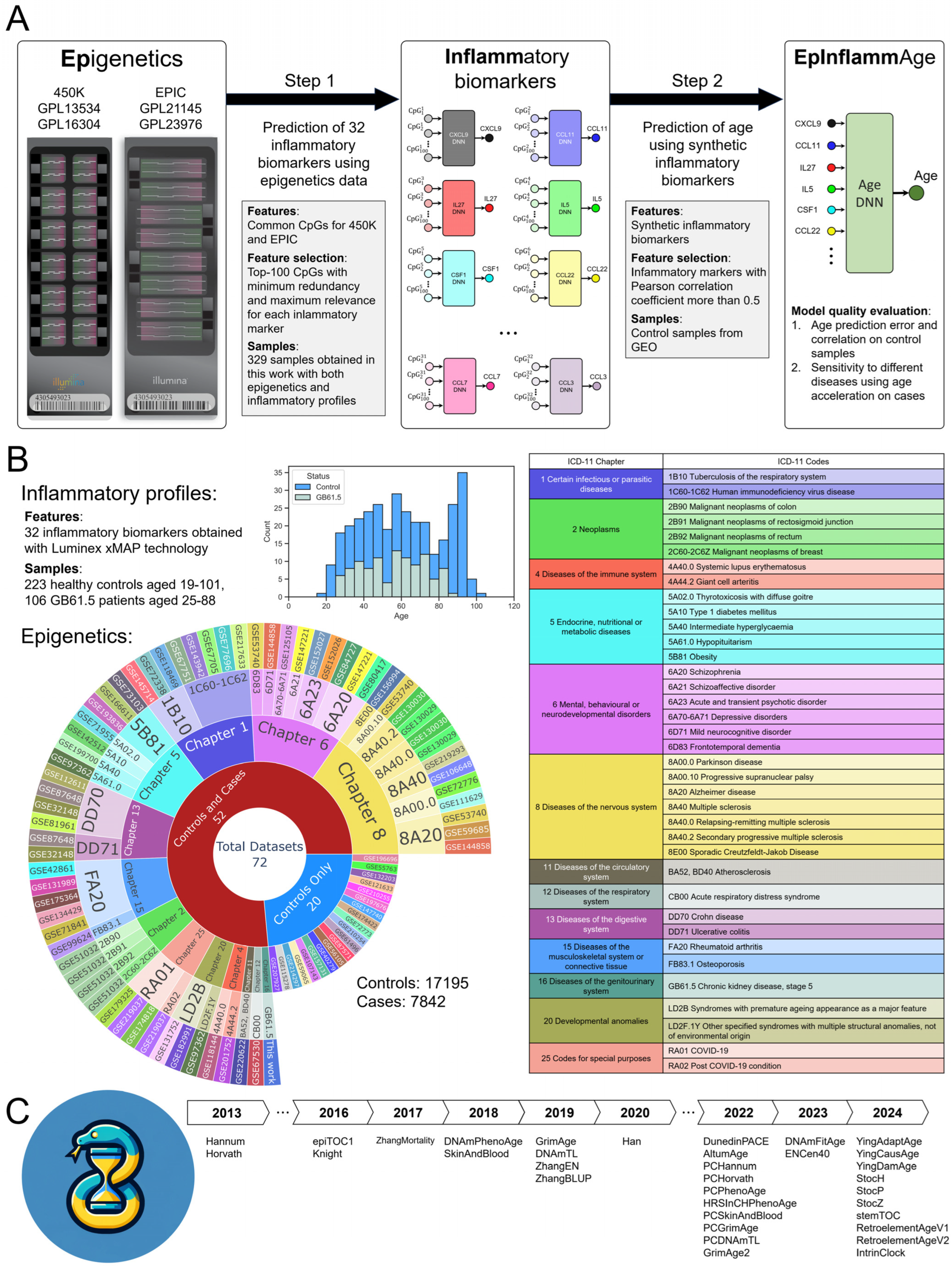
2.1. Study Design and Model Development Pipeline
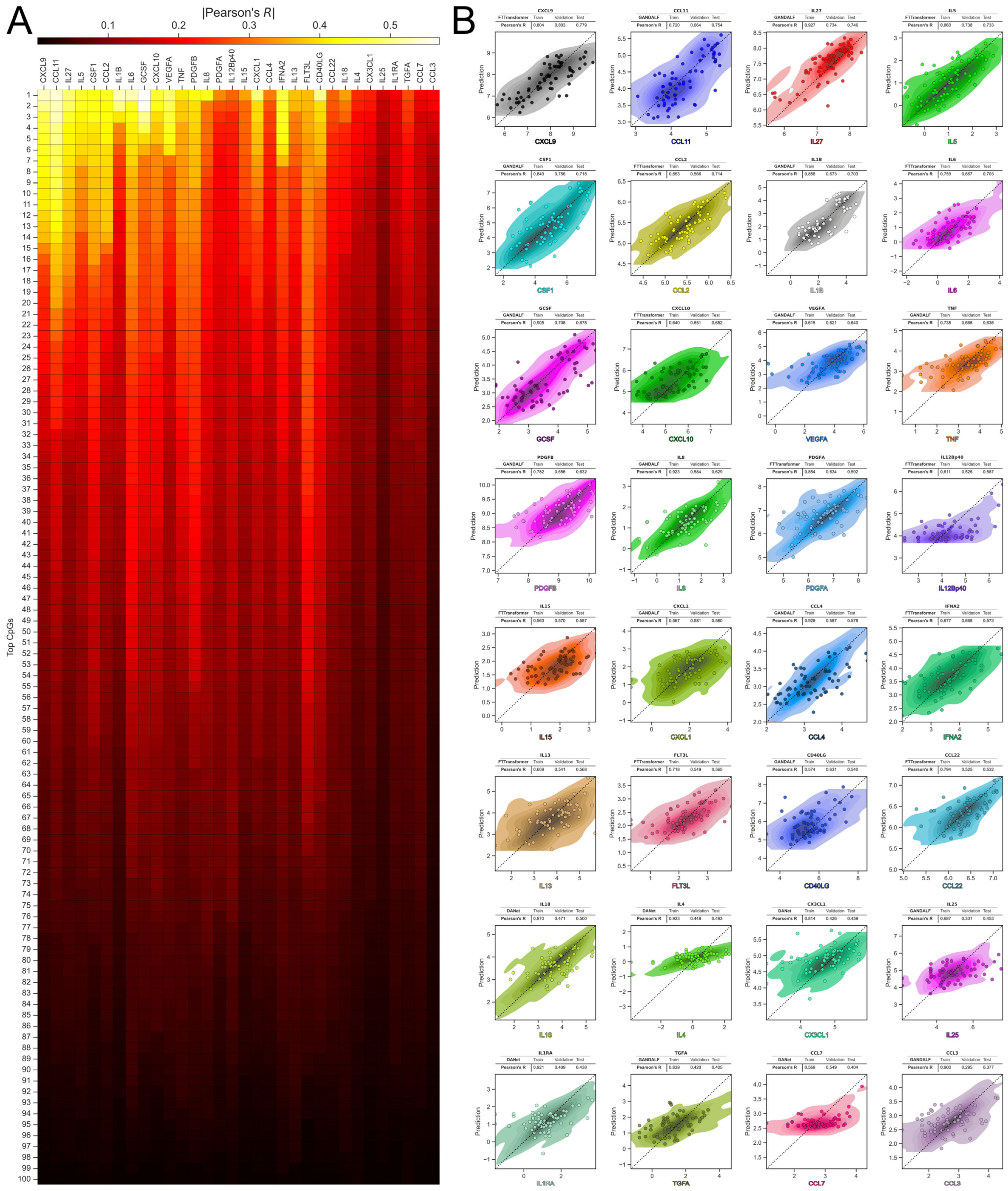
2.2. Predicting Inflammatory Biomarkers from Epigenetic Data
2.3. EpInflammAge: Epigenetic-Inflammatory Clock Model
2.4. Sensitivity of EpInflammAge and Other Epigenetic Clocks to Different Diseases
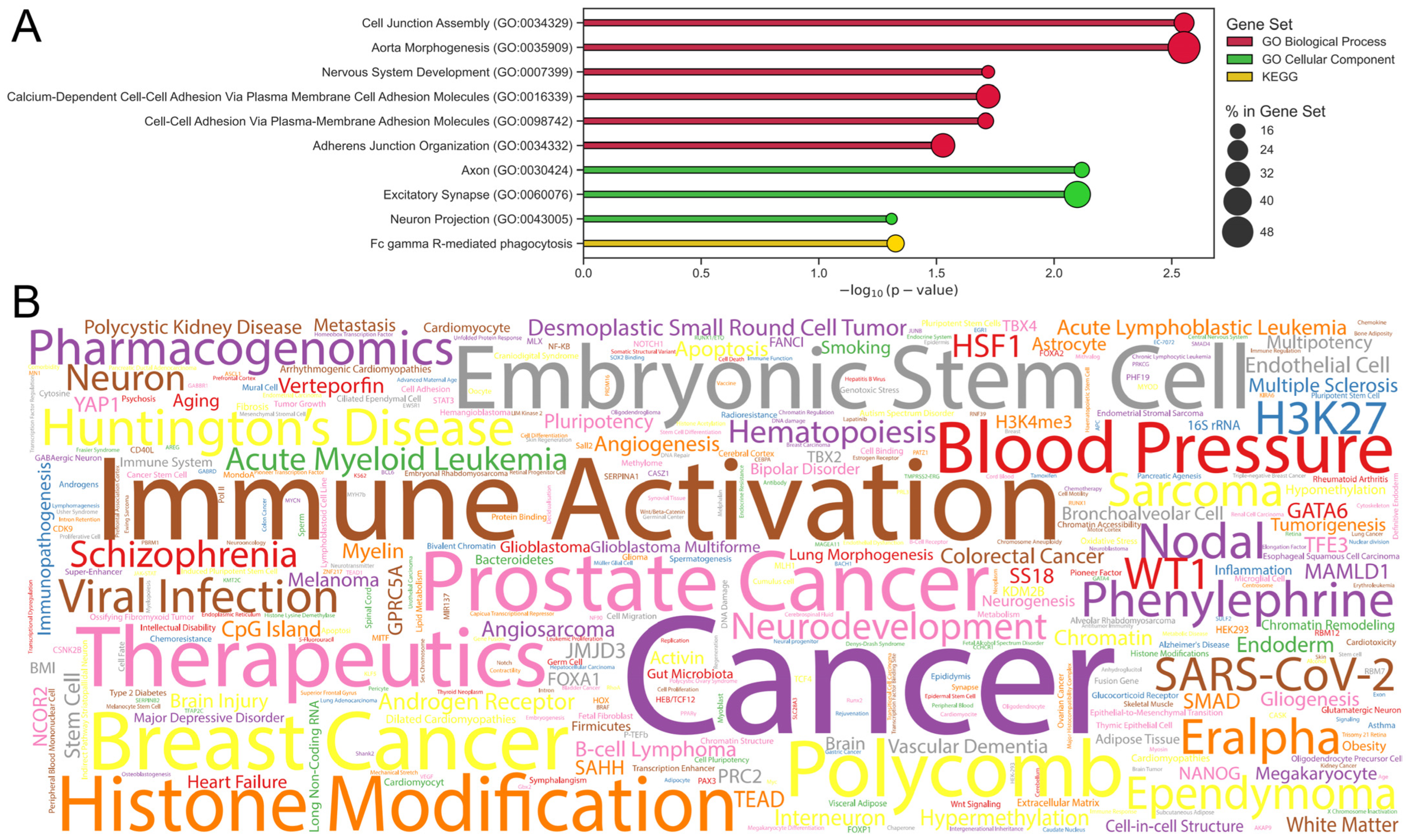
2.5. Applying XAI to the EpInflammAge Model
2.6. EpInflammAge Web Interface
3. Discussion
Limitations
4. Materials and Methods
4.1. Data Collection and Processing
4.1.1. Concurrent Data from Inflammatory and Epigenetic Profiles
4.1.2. GEO
4.2. Model Development and Architecture
4.2.1. Feature Selection
4.2.2. Models Training
4.2.3. XAI
4.3. Statistical Analysis and Validation
4.3.1. Statistical Tests
4.3.2. Epigenetic Clock Models
4.3.3. GSEA
4.4. Web Interface
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AI | Artificial Intelligence |
| COPD | Chronic Obstructive Pulmonary Disease |
| COVID-19 | Coronavirus Disease 2019 |
| DANet | Deep Abstract Network |
| DNA | Deoxyribonucleic Acid |
| FT-Transformer | Feature Tokenizer and Transformer |
| GANDALF | Gated Adaptive Network for Deep Automated Learning of Features |
| GEO | Gene Expression Omnibus |
| GFLU | Gated Feature Learning Unit |
| GO | Gene Ontology |
| GSEA | Gene-Set Enrichment Analysis |
| ICD | International Classification of Diseases |
| HIV | Human Immunodeficiency Virus |
| KDE | Kernel Density Estimation |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| kNN | k Nearest Neighbors |
| MAE | Mean Absolute Error |
| mRMR | Minimum Redundancy, Maximum Relevance |
| RNA | Ribonucleic Acid |
| SHAP | Shapley Additive Explanations |
| XAI | Explainable Artificial Intelligence |
References
- Pereira, B.; Correia, F.P.; Alves, I.A.; Costa, M.; Gameiro, M.; Martins, A.P.; Saraiva, J.A. Epigenetic Reprogramming as a Key to Reverse Ageing and Increase Longevity. Ageing Res. Rev. 2024, 95, 102204. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and Aging-Related Diseases: From Molecular Mechanisms to Interventions and Treatments. Signal Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, H.; Hu, Q.; Wang, L.; Liu, J.; Zheng, Z.; Zhang, W.; Ren, J.; Zhu, F.; Liu, G.-H. Epigenetic Regulation of Aging: Implications for Interventions of Aging and Diseases. Signal Transduct. Target. Ther. 2022, 7, 374. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Pal, S.; Tyler, J.K. Epigenetics and Aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Aging: A Common Driver of Chronic Diseases and a Target for Novel Interventions. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef]
- Wu, Z.; Qu, J.; Zhang, W.; Liu, G.-H. Stress, Epigenetics, and Aging: Unraveling the Intricate Crosstalk. Mol. Cell 2024, 84, 34–54. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Epigenetic Changes during Aging and Their Reprogramming Potential. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 63–81. [Google Scholar] [CrossRef]
- Moskalev, A.; Stambler, I.; Caruso, C. Innate and Adaptive Immunity in Aging and Longevity: The Foundation of Resilience. Aging Dis. 2020, 11, 1363–1373. [Google Scholar] [CrossRef]
- Solana, R.; Tarazona, R.; Gayoso, I.; Lesur, O.; Dupuis, G.; Fulop, T. Innate Immunosenescence: Effect of Aging on Cells and Receptors of the Innate Immune System in Humans. Semin. Immunol. 2012, 24, 331–341. [Google Scholar] [CrossRef]
- Nikolich-Žugich, J. The Twilight of Immunity: Emerging Concepts in Aging of the Immune System. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Pawelec, G.; Khalil, A.; Cohen, A.A.; Hirokawa, K.; Witkowski, J.M.; Franceschi, C. Immunology of Aging: The Birth of Inflammaging. Clin. Rev. Allergy Immunol. 2023, 64, 109. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-Aging. An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A New Immune–Metabolic Viewpoint for Age-Related Diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Santoro, A.; Bientinesi, E.; Monti, D. Immunosenescence and Inflammaging in the Aging Process: Age-Related Diseases or Longevity? Ageing Res. Rev. 2021, 71, 101422. [Google Scholar] [CrossRef] [PubMed]
- Bacos, K.; Gillberg, L.; Volkov, P.; Olsson, A.H.; Hansen, T.; Pedersen, O.; Gjesing, A.P.; Eiberg, H.; Tuomi, T.; Almgren, P.; et al. Blood-Based Biomarkers of Age-Associated Epigenetic Changes in Human Islets Associate with Insulin Secretion and Diabetes. Nat. Commun. 2016, 7, 11089. [Google Scholar] [CrossRef]
- Rönn, T.; Volkov, P.; Gillberg, L.; Kokosar, M.; Perfilyev, A.; Jacobsen, A.L.; Jørgensen, S.W.; Brøns, C.; Jansson, P.-A.; Eriksson, K.-F.; et al. Impact of Age, BMI and HbA1c Levels on the Genome-Wide DNA Methylation and mRNA Expression Patterns in Human Adipose Tissue and Identification of Epigenetic Biomarkers in Blood. Hum. Mol. Genet. 2015, 24, 3792–3813. [Google Scholar] [CrossRef]
- Bacalini, M.G.; Deelen, J.; Pirazzini, C.; De Cecco, M.; Giuliani, C.; Lanzarini, C.; Ravaioli, F.; Marasco, E.; van Heemst, D.; Suchiman, H.E.D.; et al. Systemic Age-Associated DNA Hypermethylation of ELOVL2 Gene: In Vivo and In Vitro Evidences of a Cell Replication Process. J. Gerontol. Ser. A 2017, 72, 1015–1023. [Google Scholar] [CrossRef]
- Slieker, R.C.; Relton, C.L.; Gaunt, T.R.; Slagboom, P.E.; Heijmans, B.T. Age-Related DNA Methylation Changes Are Tissue-Specific with ELOVL2 Promoter Methylation as Exception. Epigenet. Chromatin 2018, 11, 25. [Google Scholar] [CrossRef]
- Nardini, C.; Moreau, J.-F.; Gensous, N.; Ravaioli, F.; Garagnani, P.; Bacalini, M.G. The Epigenetics of Inflammaging: The Contribution of Age-Related Heterochromatin Loss and Locus-Specific Remodelling and the Modulation by Environmental Stimuli. Semin. Immunol. 2018, 40, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Tay, J.; Yang, G.-E.; Agrawal, S.; Gupta, S. Age-Associated Epigenetic Modifications in Human DNA Increase Its Immunogenicity. Aging 2010, 2, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Jylhävä, J.; Pedersen, N.L.; Hägg, S. Biological Age Predictors. eBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef]
- Horvath, S. DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-Wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An Epigenetic Biomarker of Aging for Lifespan and Healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA Methylation GrimAge Strongly Predicts Lifespan and Healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Belsky, D.W.; Caspi, A.; Arseneault, L.; Baccarelli, A.; Corcoran, D.L.; Gao, X.; Hannon, E.; Harrington, H.L.; Rasmussen, L.J.; Houts, R.; et al. Quantification of the Pace of Biological Aging in Humans through a Blood Test, the DunedinPoAm DNA Methylation Algorithm. eLife 2020, 9, e54870. [Google Scholar] [CrossRef]
- Belsky, D.W.; Caspi, A.; Corcoran, D.L.; Sugden, K.; Poulton, R.; Arseneault, L.; Baccarelli, A.; Chamarti, K.; Gao, X.; Hannon, E.; et al. DunedinPACE, a DNA Methylation Biomarker of the Pace of Aging. eLife 2022, 11, e73420. [Google Scholar] [CrossRef]
- Ying, K.; Liu, H.; Tarkhov, A.E.; Sadler, M.C.; Lu, A.T.; Moqri, M.; Horvath, S.; Kutalik, Z.; Shen, X.; Gladyshev, V.N. Causality-Enriched Epigenetic Age Uncouples Damage and Adaptation. Nat. Aging 2024, 4, 231–246. [Google Scholar] [CrossRef]
- Oblak, L.; van der Zaag, J.; Higgins-Chen, A.T.; Levine, M.E.; Boks, M.P. A Systematic Review of Biological, Social and Environmental Factors Associated with Epigenetic Clock Acceleration. Ageing Res. Rev. 2021, 69, 101348. [Google Scholar] [CrossRef] [PubMed]
- Fransquet, P.D.; Wrigglesworth, J.; Woods, R.L.; Ernst, M.E.; Ryan, J. The Epigenetic Clock as a Predictor of Disease and Mortality Risk: A Systematic Review and Meta-Analysis. Clin. Epigenet. 2019, 11, 62. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Li, W.; Wang, T.; Ran, D.; Davalos, V.; Planas-Serra, L.; Pujol, A.; Esteller, M.; Wang, X.; Yu, H. Accelerated Biological Aging in COVID-19 Patients. Nat. Commun. 2022, 13, 2135. [Google Scholar] [CrossRef]
- Sayed, N.; Huang, Y.; Nguyen, K.; Krejciova-Rajaniemi, Z.; Grawe, A.P.; Gao, T.; Tibshirani, R.; Hastie, T.; Alpert, A.; Cui, L.; et al. An Inflammatory Aging Clock (iAge) Based on Deep Learning Tracks Multimorbidity, Immunosenescence, Frailty and Cardiovascular Aging. Nat. Aging 2021, 1, 598–615. [Google Scholar] [CrossRef]
- Kalyakulina, A.; Yusipov, I.; Kondakova, E.; Bacalini, M.G.; Franceschi, C.; Vedunova, M.; Ivanchenko, M. Small Immunological Clocks Identified by Deep Learning and Gradient Boosting. Front. Immunol. 2023, 14, 1177611. [Google Scholar] [CrossRef] [PubMed]
- Murabito, J.M.; Zhao, Q.; Larson, M.G.; Rong, J.; Lin, H.; Benjamin, E.J.; Levy, D.; Lunetta, K.L. Measures of Biologic Age in a Community Sample Predict Mortality and Age-Related Disease: The Framingham Offspring Study. J. Gerontol. Ser. A 2018, 73, 757–762. [Google Scholar] [CrossRef]
- Alpert, A.; Pickman, Y.; Leipold, M.; Rosenberg-Hasson, Y.; Ji, X.; Gaujoux, R.; Rabani, H.; Starosvetsky, E.; Kveler, K.; Schaffert, S.; et al. A Clinically Meaningful Metric of Immune Age Derived from High-Dimensional Longitudinal Monitoring. Nat. Med. 2019, 25, 487–495. [Google Scholar] [CrossRef]
- Yusipov, I.; Kondakova, E.; Kalyakulina, A.; Krivonosov, M.; Lobanova, N.; Bacalini, M.G.; Franceschi, C.; Vedunova, M.; Ivanchenko, M. Accelerated Epigenetic Aging and Inflammatory/Immunological Profile (ipAGE) in Patients with Chronic Kidney Disease. GeroScience 2022, 44, 817–834. [Google Scholar] [CrossRef]
- Markov, N.T.; Lindbergh, C.A.; Staffaroni, A.M.; Perez, K.; Stevens, M.; Nguyen, K.; Murad, N.F.; Fonseca, C.; Campisi, J.; Kramer, J.; et al. Age-Related Brain Atrophy Is Not a Homogenous Process: Different Functional Brain Networks Associate Differentially with Aging and Blood Factors. Proc. Natl. Acad. Sci. USA 2022, 119, e2207181119. [Google Scholar] [CrossRef]
- Crimmins, E.M.; Klopack, E.T.; Kim, J.K. Generations of Epigenetic Clocks and Their Links to Socioeconomic Status in the Health and Retirement Study. Epigenomics 2024, 16, 1031–1042. [Google Scholar] [CrossRef]
- Yusipov, I.; Kalyakulina, A.; Trukhanov, A.; Franceschi, C.; Ivanchenko, M. Map of Epigenetic Age Acceleration: A Worldwide Analysis. Ageing Res. Rev. 2024, 100, 102418. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA Methylation-Based Biomarkers and the Epigenetic Clock Theory of Ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Grodstein, F.; Lemos, B.; Yu, L.; Klein, H.-U.; Iatrou, A.; Buchman, A.S.; Shireby, G.L.; Mill, J.; Schneider, J.A.; De Jager, P.L.; et al. The Association of Epigenetic Clocks in Brain Tissue with Brain Pathologies and Common Aging Phenotypes. Neurobiol. Dis. 2021, 157, 105428. [Google Scholar] [CrossRef]
- Chervova, O.; Panteleeva, K.; Chernysheva, E.; Widayati, T.A.; Baronik, Ž.F.; Hrbková, N.; Schneider, J.L.; Bobak, M.; Beck, S.; Voloshin, V. Breaking New Ground on Human Health and Well-Being with Epigenetic Clocks: A Systematic Review and Meta-Analysis of Epigenetic Age Acceleration Associations. Ageing Res. Rev. 2024, 102, 102552. [Google Scholar] [CrossRef]
- Moguilner, S.; Baez, S.; Hernandez, H.; Migeot, J.; Legaz, A.; Gonzalez-Gomez, R.; Farina, F.R.; Prado, P.; Cuadros, J.; Tagliazucchi, E.; et al. Brain Clocks Capture Diversity and Disparities in Aging and Dementia across Geographically Diverse Populations. Nat. Med. 2024, 30, 3646–3657. [Google Scholar] [CrossRef] [PubMed]
- Melendez, J.; Sung, Y.J.; Orr, M.; Yoo, A.; Schindler, S.; Cruchaga, C.; Bateman, R. An Interpretable Machine Learning-Based Cerebrospinal Fluid Proteomics Clock for Predicting Age Reveals Novel Insights into Brain Aging. Aging Cell 2024, 23, e14230. [Google Scholar] [CrossRef] [PubMed]
- Conole, E.L.S.; Robertson, J.A.; Smith, H.M.; Cox, S.R.; Marioni, R.E. Epigenetic Clocks and DNA Methylation Biomarkers of Brain Health and Disease. Nat. Rev. Neurol. 2025, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Myte, R.; Sundkvist, A.; Van Guelpen, B.; Harlid, S. Circulating Levels of Inflammatory Markers and DNA Methylation, an Analysis of Repeated Samples from a Population Based Cohort. Epigenetics 2019, 14, 649–659. [Google Scholar] [CrossRef]
- Stevenson, A.J.; Gadd, D.A.; Hillary, R.F.; McCartney, D.L.; Campbell, A.; Walker, R.M.; Evans, K.L.; Harris, S.E.; Spires-Jones, T.L.; McRae, A.F.; et al. Creating and Validating a DNA Methylation-Based Proxy for Interleukin-6. J. Gerontol. Ser. A 2021, 76, 2284–2292. [Google Scholar] [CrossRef]
- Gadd, D.A.; Hillary, R.F.; McCartney, D.L.; Zaghlool, S.B.; Stevenson, A.J.; Cheng, Y.; Fawns-Ritchie, C.; Nangle, C.; Campbell, A.; Flaig, R.; et al. Epigenetic Scores for the Circulating Proteome as Tools for Disease Prediction. eLife 2022, 11, e71802. [Google Scholar] [CrossRef]
- Bernabeu, E.; McCartney, D.L.; Gadd, D.A.; Hillary, R.F.; Lu, A.T.; Murphy, L.; Wrobel, N.; Campbell, A.; Harris, S.E.; Liewald, D.; et al. Refining Epigenetic Prediction of Chronological and Biological Age. Genome Med. 2023, 15, 12. [Google Scholar] [CrossRef] [PubMed]
- Schmunk, L.J.; Call, T.P.; McCartney, D.L.; Javaid, H.; Hastings, W.J.; Jovicevic, V.; Kojadinović, D.; Tomkinson, N.; Zlamalova, E.; McGee, K.C.; et al. A Novel Framework to Build Saliva-Based DNA Methylation Biomarkers: Quantifying Systemic Chronic Inflammation as a Case Study. Aging Cell 2025, 24, e14444. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef] [PubMed]
- Perera, P.-Y.; Lichy, J.H.; Waldmann, T.A.; Perera, L.P. The Role of Interleukin-15 in Inflammation and Immune Responses to Infection: Implications for Its Therapeutic Use. Microbes Infect. Inst. Pasteur 2012, 14, 247–261. [Google Scholar] [CrossRef]
- Lopez-Castejon, G.; Brough, D. Understanding the Mechanism of IL-1β Secretion. Cytokine Growth Factor Rev. 2011, 22, 189–195. [Google Scholar] [CrossRef]
- Kalyakulina, A.; Yusipov, I. Web-Interface for Calculating EpInflammAge. Available online: https://huggingface.co/spaces/UNNAILab/EpInflammAge (accessed on 30 March 2025).
- Kalyakulina, A.; Yusipov, I.; Kondakova, E.; Bacalini, M.G.; Giuliani, C.; Sivtseva, T.; Semenov, S.; Ksenofontov, A.; Nikolaeva, M.; Khusnutdinova, E.; et al. Epigenetics of the Far Northern Yakutian Population. Clin. Epigenet. 2023, 15, 189. [Google Scholar] [CrossRef]
- Ding, C.; Peng, H. Minimum Redundancy Feature Selection from Microarray Gene Expression Data. J. Bioinform. Comput. Biol. 2005, 3, 185–205. [Google Scholar] [CrossRef]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for Functional Genomics Data Sets—Update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Xie, Z.; Bailey, A.; Kuleshov, M.V.; Clarke, D.J.B.; Evangelista, J.E.; Jenkins, S.L.; Lachmann, A.; Wojciechowicz, M.L.; Kropiwnicki, E.; Jagodnik, K.M.; et al. Gene Set Knowledge Discovery with Enrichr. Curr. Protoc. 2021, 1, e90. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology Consortium The Gene Ontology Resource: Enriching a GOld Mine. Nucleic Acids Res. 2021, 49, D325–D334. [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, M.J.; Smyth, J.W. Gap Junctions and Ageing. Subcell. Biochem. 2023, 102, 113–137. [Google Scholar] [CrossRef]
- Dong, D.; Xie, W.; Liu, M. Alteration of Cell Junctions during Viral Infection. Thorac. Cancer 2020, 11, 519–525. [Google Scholar] [CrossRef]
- Ashcroft, G.S.; Horan, M.A.; Ferguson, M.W. Aging Alters the Inflammatory and Endothelial Cell Adhesion Molecule Profiles during Human Cutaneous Wound Healing. Lab. Investig. J. Tech. Methods Pathol. 1998, 78, 47–58. [Google Scholar]
- Arnesen, S.M.; Lawson, M.A. Age-Related Changes in Focal Adhesions Lead to Altered Cell Behavior in Tendon Fibroblasts. Mech. Ageing Dev. 2006, 127, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front. Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef]
- Salvadores, N.; Sanhueza, M.; Manque, P.; Court, F.A. Axonal Degeneration during Aging and Its Functional Role in Neurodegenerative Disorders. Front. Neurosci. 2017, 11, 451. [Google Scholar] [CrossRef]
- Azpurua, J.; Eaton, B.A. Neuronal Epigenetics and the Aging Synapse. Front. Cell. Neurosci. 2015, 9, 208. [Google Scholar] [CrossRef]
- Galloway, D.A.; Phillips, A.E.M.; Owen, D.R.J.; Moore, C.S. Phagocytosis in the Brain: Homeostasis and Disease. Front. Immunol. 2019, 10, 790. [Google Scholar] [CrossRef] [PubMed]
- Li, W. Phagocyte Dysfunction, Tissue Aging and Degeneration. Ageing Res. Rev. 2013, 12, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Frobel, J.; Hemeda, H.; Lenz, M.; Abagnale, G.; Joussen, S.; Denecke, B.; Šarić, T.; Zenke, M.; Wagner, W. Epigenetic Rejuvenation of Mesenchymal Stromal Cells Derived from Induced Pluripotent Stem Cells. Stem Cell Rep. 2014, 3, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Lodde, V.; Floris, M.; Munk, R.; Martindale, J.L.; Piredda, D.; Napodano, C.M.P.; Cucca, F.; Uzzau, S.; Abdelmohsen, K.; Gorospe, M.; et al. Systematic Identification of NF90 Target RNAs by iCLIP Analysis. Sci. Rep. 2022, 12, 364. [Google Scholar] [CrossRef]
- Păun, O.; Tan, Y.X.; Patel, H.; Strohbuecker, S.; Ghanate, A.; Cobolli-Gigli, C.; Llorian Sopena, M.; Gerontogianni, L.; Goldstone, R.; Ang, S.-L.; et al. Pioneer Factor ASCL1 Cooperates with the mSWI/SNF Complex at Distal Regulatory Elements to Regulate Human Neural Differentiation. Genes Dev. 2023, 37, 218–242. [Google Scholar] [CrossRef]
- Marttila, S.; Chatsirisupachai, K.; Palmer, D.; de Magalhães, J.P. Ageing-Associated Changes in the Expression of lncRNAs in Human Tissues Reflect a Transcriptional Modulation in Ageing Pathways. Mech. Ageing Dev. 2020, 185, 111177. [Google Scholar] [CrossRef]
- Fu, X.; Zhao, Y.; Cui, X.; Huang, S.; Lv, Y.; Li, C.; Gong, F.; Yang, Z.; Yang, X.; Xiao, R. Cxcl9 Modulates Aging Associated Microvascular Metabolic and Angiogenic Dysfunctions in Subcutaneous Adipose Tissue. Angiogenesis 2025, 28, 17. [Google Scholar] [CrossRef]
- Brito-de-Sousa, J.P.; Lima-Silva, M.L.; Costa-Rocha, I.A.; Campi-Azevedo, A.C.; Mambrini, J.V.d.M.; Faria, A.M.C.; Lima-Costa, M.F.; Peixoto, S.V.; Teixeira-Carvalho, A.; Torres, K.C.L.; et al. Rhythms and Shifts of Chemokines and Cytokines Interplay in a Decade Lifespan: The Longitudinal Community-Based Bambuí Health and Aging Study. Exp. Gerontol. 2025, 202, 112700. [Google Scholar] [CrossRef]
- Seo, D.H.; Corr, M.; Patel, S.; Lui, L.-Y.; Cauley, J.A.; Evans, D.; Mau, T.; Lane, N.E. Chemokine CXCL9, a Marker of Inflammaging, Is Associated with Changes of Muscle Strength and Mortality in Older Men. Osteoporos. Int. 2024, 35, 1789–1796. [Google Scholar] [CrossRef]
- Chaudhary, J.K.; Danga, A.K.; Kumari, A.; Bhardwaj, A.; Rath, P.C. Role of Chemokines in Aging and Age-Related Diseases. Mech. Ageing Dev. 2025, 223, 112009. [Google Scholar] [CrossRef]
- Maggio, M.; Guralnik, J.M.; Longo, D.L.; Ferrucci, L. Interleukin-6 in Aging and Chronic Disease: A Magnificent Pathway. J. Gerontol. A. Biol. Sci. Med. Sci. 2006, 61, 575–584. [Google Scholar] [CrossRef]
- Omoigui, S. The Interleukin-6 Inflammation Pathway from Cholesterol to Aging—Role of Statins, Bisphosphonates and Plant Polyphenols in Aging and Age-Related Diseases. Immun. Ageing 2007, 4, 1. [Google Scholar] [CrossRef]
- Didion, S.P. Cellular and Oxidative Mechanisms Associated with Interleukin-6 Signaling in the Vasculature. Int. J. Mol. Sci. 2017, 18, 2563. [Google Scholar] [CrossRef] [PubMed]
- Grebenciucova, E.; VanHaerents, S. Interleukin 6: At the Interface of Human Health and Disease. Front. Immunol. 2023, 14, 1255533. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kuan, W.P.; Tam, L.-S.; Wong, C.-K.; Ko, F.W.S.; Li, T.; Zhu, T.; Li, E.K. CXCL 9 and CXCL 10 as Sensitive Markers of Disease Activity in Patients with Rheumatoid Arthritis. J. Rheumatol. 2010, 37, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Giamarellos-Bourboulis, E.J.; Antonelli, M.; Bloos, F.; Kotsamidi, I.; Psarrakis, C.; Dakou, K.; Thomas-Rüddel, D.; Montini, L.; Briegel, J.; Damoraki, G.; et al. Interferon-Gamma Driven Elevation of CXCL9: A New Sepsis Endotype Independently Associated with Mortality. eBioMedicine 2024, 109, 105414. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Huang, Y.; Cai, W.; Chen, X.; Men, X.; Lu, T.; Wu, A.; Lu, Z. Age-Related Cerebral Small Vessel Disease and Inflammaging. Cell Death Dis. 2020, 11, 932. [Google Scholar] [CrossRef]
- Srirangan, S.; Choy, E.H. The Role of Interleukin 6 in the Pathophysiology of Rheumatoid Arthritis. Ther. Adv. Musculoskelet. Dis. 2010, 2, 247–256. [Google Scholar] [CrossRef]
- Choy, E.H.; De Benedetti, F.; Takeuchi, T.; Hashizume, M.; John, M.R.; Kishimoto, T. Translating IL-6 Biology into Effective Treatments. Nat. Rev. Rheumatol. 2020, 16, 335–345. [Google Scholar] [CrossRef]
- Hirano, T. IL-6 in Inflammation, Autoimmunity and Cancer. Int. Immunol. 2020, 33, 127–148. [Google Scholar] [CrossRef] [PubMed]
- Håkansson, L.; Dunér, P.; Broströmer, E.; Gustavsson, B.; Wettergren, Y.; Ghafouri, B.; Håkansson, A.; Clinchy, B. A New IL-6-Inducing Mechanism in Cancer with New Therapeutic Possibilities. Cancers 2024, 16, 3588. [Google Scholar] [CrossRef] [PubMed]
- Mehta, N.N.; deGoma, E.; Shapiro, M.D. IL-6 and Cardiovascular Risk: A Narrative Review. Curr. Atheroscler. Rep. 2024, 27, 12. [Google Scholar] [CrossRef] [PubMed]
- Katkenov, N.; Mukhatayev, Z.; Kozhakhmetov, S.; Sailybayeva, A.; Bekbossynova, M.; Kushugulova, A. Systematic Review on the Role of IL-6 and IL-1β in Cardiovascular Diseases. J. Cardiovasc. Dev. Dis. 2024, 11, 206. [Google Scholar] [CrossRef]
- Cau, R.; Saba, L. Interlinking Pathways: A Narrative Review on the Role of IL-6 in Cancer and Atherosclerosis. Cardiovasc. Diagn. Ther. 2024, 14, 1186–1201. [Google Scholar] [CrossRef]
- Lundin, J.I.; Peters, U.; Hu, Y.; Ammous, F.; Benjamin, E.J.; Bis, J.C.; Brody, J.A.; Cushman, M.; Fuller, H.; Gignoux, C.; et al. Epigenetic Mechanisms Underlying Variation of IL-6, a Well-Established Inflammation Biomarker and Risk Factor for Cardiovascular Disease. Atherosclerosis 2025, 407, 120219. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Calarota, S.A.; Vidali, F.; MacDonald, T.T.; Corazza, G.R. Role of IL-15 in Immune-Mediated and Infectious Diseases. Cytokine Growth Factor Rev. 2011, 22, 19–33. [Google Scholar] [CrossRef]
- Loughland, J.R.; Woodberry, T.; Oyong, D.; Piera, K.A.; Amante, F.H.; Barber, B.E.; Grigg, M.J.; William, T.; Engwerda, C.R.; Anstey, N.M.; et al. Reduced Circulating Dendritic Cells in Acute Plasmodium Knowlesi and Plasmodium Falciparum Malaria Despite Elevated Plasma Flt3 Ligand Levels. Malar. J. 2021, 20, 97. [Google Scholar] [CrossRef]
- Mahittikorn, A.; Kwankaew, P.; Rattaprasert, P.; Kotepui, K.U.; Masangkay, F.R.; Kotepui, M. Elevation of Serum Interleukin-1β Levels as a Potential Indicator for Malarial Infection and Severe Malaria: A Meta-Analysis. Malar. J. 2022, 21, 308. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in Infectious Diseases Pathogenesis and Potential Therapeutic Implications. Cytokine Growth Factor Rev. 2011, 22, 121. [Google Scholar] [CrossRef]
- Quartuccio, L.; Fabris, M.; Sonaglia, A.; Peghin, M.; Domenis, R.; Cifù, A.; Curcio, F.; Tascini, C. Interleukin 6, Soluble Interleukin 2 Receptor Alpha (CD25), Monocyte Colony-Stimulating Factor, and Hepatocyte Growth Factor Linked with Systemic Hyperinflammation, Innate Immunity Hyperactivation, and Organ Damage in COVID-19 Pneumonia. Cytokine 2021, 140, 155438. [Google Scholar] [CrossRef]
- Korobova, Z.R.; Arsentieva, N.A.; Liubimova, N.E.; Dedkov, V.G.; Gladkikh, A.S.; Sharova, A.A.; Chernykh, E.I.; Kashchenko, V.A.; Ratnikov, V.A.; Gorelov, V.P.; et al. A Comparative Study of the Plasma Chemokine Profile in COVID-19 Patients Infected with Different SARS-CoV-2 Variants. Int. J. Mol. Sci. 2022, 23, 9058. [Google Scholar] [CrossRef] [PubMed]
- D’Rozario, R.; Raychaudhuri, D.; Bandopadhyay, P.; Sarif, J.; Mehta, P.; Liu, C.S.C.; Sinha, B.P.; Roy, J.; Bhaduri, R.; Das, M.; et al. Circulating Interleukin-8 Dynamics Parallels Disease Course and Is Linked to Clinical Outcomes in Severe COVID-19. Viruses 2023, 15, 549. [Google Scholar] [CrossRef] [PubMed]
- Mandel, M.; Harari, G.; Gurevich, M.; Achiron, A. Cytokine Prediction of Mortality in COVID19 Patients. Cytokine 2020, 134, 155190. [Google Scholar] [CrossRef]
- Pius-Sadowska, E.; Niedźwiedź, A.; Kulig, P.; Baumert, B.; Sobuś, A.; Rogińska, D.; Łuczkowska, K.; Ulańczyk, Z.; Wnęk, S.; Karolak, I.; et al. CXCL8, CCL2, and CMV Seropositivity as New Prognostic Factors for a Severe COVID-19 Course. Int. J. Mol. Sci. 2022, 23, 11338. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Chemokines and Chemokine Receptors during COVID-19 Infection. Comput. Struct. Biotechnol. J. 2021, 19, 976–988. [Google Scholar] [CrossRef]
- Bekbossynova, M.; Tauekelova, A.; Sailybayeva, A.; Kozhakhmetov, S.; Mussabay, K.; Chulenbayeva, L.; Kossumov, A.; Khassenbekova, Z.; Vinogradova, E.; Kushugulova, A. Unraveling Acute and Post-COVID Cytokine Patterns to Anticipate Future Challenges. J. Clin. Med. 2023, 12, 5224. [Google Scholar] [CrossRef]
- Pan, T.; Gallo, M.E.; Donald, K.A.; Webb, K.; Bath, K.G. Elevated Risk for Psychiatric Outcomes in Pediatric Patients with Multisystem Inflammatory Syndrome (MIS-C): A Review of Neuroinflammatory and Psychosocial Stressors. Brain Behav. Immun. Health 2024, 38, 100760. [Google Scholar] [CrossRef]
- Kameda, M.; Otsuka, M.; Chiba, H.; Kuronuma, K.; Hasegawa, T.; Takahashi, H.; Takahashi, H. CXCL9, CXCL10, and CXCL11; Biomarkers of Pulmonary Inflammation Associated with Autoimmunity in Patients with Collagen Vascular Diseases–Associated Interstitial Lung Disease and Interstitial Pneumonia with Autoimmune Features. PLoS ONE 2020, 15, e0241719. [Google Scholar] [CrossRef]
- Xu, J.; Zhong, S.; Liu, J.; Li, L.; Li, Y.; Wu, X.; Li, Z.; Deng, P.; Zhang, J.; Zhong, N.; et al. Detection of Severe Acute Respiratory Syndrome Coronavirus in the Brain: Potential Role of the Chemokine Mig in Pathogenesis. Clin. Infect. Dis. 2005, 41, 1089–1096. [Google Scholar] [CrossRef]
- Stefano, A.D.; Coccini, T.; Roda, E.; Signorini, C.; Balbi, B.; Brunetti, G.; Ceriana, P. Blood MCP-1 Levels Are Increased in Chronic Obstructive Pulmonary Disease Patients with Prevalent Emphysema. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 1691. [Google Scholar] [CrossRef]
- Reynolds, D.; Vazquez Guillamet, C.; Day, A.; Borcherding, N.; Vazquez Guillamet, R.; Choreño-Parra, J.A.; House, S.L.; O’Halloran, J.A.; Zúñiga, J.; Ellebedy, A.H.; et al. Comprehensive Immunologic Evaluation of Bronchoalveolar Lavage Samples from Human Patients with Moderate and Severe Seasonal Influenza and Severe COVID-19. J. Immunol. 2021, 207, 1229–1238. [Google Scholar] [CrossRef] [PubMed]
- Rincon, M.; Irvin, C.G. Role of IL-6 in Asthma and Other Inflammatory Pulmonary Diseases. Int. J. Biol. Sci. 2012, 8, 1281. [Google Scholar] [CrossRef] [PubMed]
- Moledina, D.G.; Obeid, W.; Smith, R.N.; Rosales, I.; Sise, M.E.; Moeckel, G.; Kashgarian, M.; Kuperman, M.; Campbell, K.N.; Lefferts, S.; et al. Identification and Validation of Urinary CXCL9 as a Biomarker for Diagnosis of Acute Interstitial Nephritis. J. Clin. Investig. 2023, 133, e168950. [Google Scholar] [CrossRef]
- Araújo, L.S.; Torquato, B.G.S.; da Silva, C.A.; dos Reis Monteiro, M.L.G.; dos Santos Martins, A.L.M.; da Silva, M.V.; dos Reis, M.A.; Machado, J.R. Renal Expression of Cytokines and Chemokines in Diabetic Nephropathy. BMC Nephrol. 2020, 21, 308. [Google Scholar] [CrossRef]
- Roy, M.S.; Janal, M.N.; Crosby, J.; Donnelly, R. Markers of Endothelial Dysfunction and Inflammation Predict Progression of Diabetic Nephropathy in African Americans with Type 1 Diabetes. Kidney Int. 2015, 87, 427–433. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Junior, W.V.; Silva, A.P.F.; de Figueiredo, R.C.; Gomes, K.B.; Simões e Silva, A.C.; Dusse, L.M.S.; Rios, D.R.A. Association between Dyslipidemia and CCL2 in Patients Undergoing Hemodialysis. Cytokine 2020, 125, 154858. [Google Scholar] [CrossRef]
- Badeński, A.; Badeńska, M.; Świętochowska, E.; Janek, A.; Gliwińska, A.; Morawiec-Knysak, A.; Szczepańska, M. Assessment of Interleukin-15 (IL-15) Concentration in Children with Idiopathic Nephrotic Syndrome. Int. J. Mol. Sci. 2023, 24, 6993. [Google Scholar] [CrossRef]
- Lebherz-Eichinger, D.; Klaus, D.A.; Reiter, T.; Hörl, W.H.; Haas, M.; Ankersmit, H.J.; Krenn, C.G.; Roth, G.A. Increased Chemokine Excretion in Patients Suffering from Chronic Kidney Disease. Transl. Res. 2014, 164, 433–443.e2. [Google Scholar] [CrossRef]
- Fallahi, P.; Ferrari, S.M.; Ragusa, F.; Ruffilli, I.; Elia, G.; Paparo, S.R.; Antonelli, A. Th1 Chemokines in Autoimmune Endocrine Disorders. J. Clin. Endocrinol. Metab. 2020, 105, 1046–1060. [Google Scholar] [CrossRef]
- Ullah, A.; Ud Din, A.; Ding, W.; Shi, Z.; Pervaz, S.; Shen, B. A Narrative Review: CXC Chemokines Influence Immune Surveillance in Obesity and Obesity-Related Diseases: Type 2 Diabetes and Nonalcoholic Fatty Liver Disease. Rev. Endocr. Metab. Disord. 2023, 24, 611–631. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.-P.; Jardel, C.; Bruckert, E.; Blondy, P.; Capeau, J.; Laville, M.; Vidal, H.; Hainque, B. Elevated Levels of Interleukin 6 Are Reduced in Serum and Subcutaneous Adipose Tissue of Obese Women after Weight Loss. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Ou, J.; Liu, M.; Shi, L.; Li, Y.; Xiao, L.; Dong, H.; Zhang, F.; Xia, K.; Zhao, J. Altered Plasma Levels of Chemokines in Autism and Their Association with Social Behaviors. Psychiatry Res. 2016, 244, 300–305. [Google Scholar] [CrossRef]
- Zhou, X.; Tian, B.; Han, H.-B. Serum Interleukin-6 in Schizophrenia: A System Review and Meta-Analysis. Cytokine 2021, 141, 155441. [Google Scholar] [CrossRef]
- Roohi, E.; Jaafari, N.; Hashemian, F. On Inflammatory Hypothesis of Depression: What Is the Role of IL-6 in the Middle of the Chaos? J. Neuroinflamm. 2021, 18, 45. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rane, M. Interleukin-6 Signaling and Anti-Interleukin-6 Therapeutics in Cardiovascular Disease. Circ. Res. 2021, 128, 1728–1746. [Google Scholar] [CrossRef] [PubMed]
- Cainzos-Achirica, M.; Enjuanes, C.; Greenland, P.; McEvoy, J.W.; Cushman, M.; Dardari, Z.; Nasir, K.; Budoff, M.J.; Al-Mallah, M.H.; Yeboah, J.; et al. The Prognostic Value of Interleukin 6 in Multiple Chronic Diseases and All-Cause Death: The Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis 2018, 278, 217–225. [Google Scholar] [CrossRef]
- Eindor, A.; Tsai, K.; Jacobson, K. P125 MIG (CXCL9) and IL22 Are Key Biomarkers That Discriminates between Paediatric IBD Patients and Non-IBD Patients in a Novel Biomarker Model. J. Crohns Colitis 2024, 18, i418. [Google Scholar] [CrossRef]
- Jarlborg, M.; Gabay, C. Systemic Effects of IL-6 Blockade in Rheumatoid Arthritis beyond the Joints. Cytokine 2022, 149, 155742. [Google Scholar] [CrossRef]
- Schoepf, I.C.; Esteban-Cantos, A.; Thorball, C.W.; Rodés, B.; Reiss, P.; Rodríguez-Centeno, J.; Riebensahm, C.; Braun, D.L.; Marzolini, C.; Seneghini, M.; et al. Epigenetic Ageing Accelerates before Antiretroviral Therapy and Decelerates after Viral Suppression in People with HIV in Switzerland: A Longitudinal Study over 17 Years. Lancet Healthy Longev. 2023, 4, e211–e218. [Google Scholar] [CrossRef]
- Nicholson, T.; Dhaliwal, A.; Quinlan, J.I.; Allen, S.L.; Williams, F.R.; Hazeldine, J.; McGee, K.C.; Sullivan, J.; Breen, L.; Elsharkawy, A.M.; et al. Accelerated Aging of Skeletal Muscle and the Immune System in Patients with Chronic Liver Disease. Exp. Mol. Med. 2024, 56, 1667–1681. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zong, X.; Li, D.; He, Y.; Tang, J.; Hu, M.; Chen, X. Epigenetic Clock Analysis of Blood Samples in Drug-Naive First-Episode Schizophrenia Patients. BMC Psychiatry 2023, 23, 45. [Google Scholar] [CrossRef] [PubMed]
- Bejaoui, Y.; Humaira Amanullah, F.; Saad, M.; Taleb, S.; Bradic, M.; Megarbane, A.; Ait Hssain, A.; Abi Khalil, C.; El Hajj, N. Epigenetic Age Acceleration in Surviving versus Deceased COVID-19 Patients with Acute Respiratory Distress Syndrome Following Hospitalization. Clin. Epigenetics 2023, 15, 186. [Google Scholar] [CrossRef]
- Higgins-Chen, A.T.; Boks, M.P.; Vinkers, C.H.; Kahn, R.S.; Levine, M.E. Schizophrenia and Epigenetic Aging Biomarkers: Increased Mortality, Reduced Cancer Risk, and Unique Clozapine Effects. Biol. Psychiatry 2020, 88, 224–235. [Google Scholar] [CrossRef]
- Shinko, Y.; Okazaki, S.; Otsuka, I.; Horai, T.; Kim, S.; Tanifuji, T.; Hishimoto, A. Accelerated Epigenetic Age and Shortened Telomere Length Based on DNA Methylation in Nicolaides–Baraitser Syndrome. Mol. Genet. Genom. Med. 2022, 10, e1876. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.; O’Halloran, A.M.; Fallon, P.; Kenny, R.A.; McCrory, C. Metabolic Syndrome Accelerates Epigenetic Ageing in Older Adults: Findings from The Irish Longitudinal Study on Ageing (TILDA). Exp. Gerontol. 2023, 183, 112314. [Google Scholar] [CrossRef]
- Protsenko, E.; Yang, R.; Nier, B.; Reus, V.; Hammamieh, R.; Rampersaud, R.; Wu, G.W.Y.; Hough, C.M.; Epel, E.; Prather, A.A.; et al. “GrimAge,” an Epigenetic Predictor of Mortality, Is Accelerated in Major Depressive Disorder. Transl. Psychiatry 2021, 11, 193. [Google Scholar] [CrossRef] [PubMed]
- Lima, C.N.C.; Suchting, R.; Scaini, G.; Cuellar, V.A.; Favero-Campbell, A.D.; Walss-Bass, C.; Soares, J.C.; Quevedo, J.; Fries, G.R. Epigenetic GrimAge Acceleration and Cognitive Impairment in Bipolar Disorder. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2022, 62, 10–21. [Google Scholar] [CrossRef] [PubMed]
- García-delaTorre, P.; Rivero-Segura, N.A.; Sánchez-García, S.; Becerril-Rojas, K.; Sandoval-Rodriguez, F.E.; Castro-Morales, D.; Cruz-Lopez, M.; Vazquez-Moreno, M.; Rincón-Heredia, R.; Ramirez-Garcia, P.; et al. GrimAge Is Elevated in Older Adults with Mild COVID-19 an Exploratory Analysis. GeroScience 2024, 46, 3511–3524. [Google Scholar] [CrossRef]
- Wikström Shemer, D.; Mostafaei, S.; Tang, B.; Pedersen, N.L.; Karlsson, I.K.; Fall, T.; Hägg, S. Associations between Epigenetic Aging and Diabetes Mellitus in a Swedish Longitudinal Study. GeroScience 2024, 46, 5003–5014. [Google Scholar] [CrossRef]
- Miao, K.; Hong, X.; Cao, W.; Lv, J.; Yu, C.; Huang, T.; Sun, D.; Liao, C.; Pang, Y.; Hu, R.; et al. Association between Epigenetic Age and Type 2 Diabetes Mellitus or Glycemic Traits: A Longitudinal Twin Study. Aging Cell 2024, 23, e14175. [Google Scholar] [CrossRef]
- Föhr, T.; Hendrix, A.; Kankaanpää, A.; Laakkonen, E.K.; Kujala, U.; Pietiläinen, K.H.; Lehtimäki, T.; Kähönen, M.; Raitakari, O.; Wang, X.; et al. Metabolic Syndrome and Epigenetic Aging: A Twin Study. Int. J. Obes. 2024, 48, 778–787. [Google Scholar] [CrossRef]
- Aroke, E.N.; Wiggins, A.M.; Hobson, J.M.; Srinivasasainagendra, V.; Quinn, T.L.; Kottae, P.; Tiwari, H.K.; Sorge, R.E.; Goodin, B.R. The Pace of Biological Aging Helps Explain the Association between Insomnia and Chronic Low Back Pain. Mol. Pain 2023, 19, 17448069231210648. [Google Scholar] [CrossRef]
- Thomas, A.; Ryan, C.P.; Caspi, A.; Liu, Z.; Moffitt, T.E.; Sugden, K.; Zhou, J.; Belsky, D.W.; Gu, Y. Diet, Pace of Biological Aging, and Risk of Dementia in the Framingham Heart Study. Ann. Neurol. 2024, 95, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Whitman, E.T.; Ryan, C.P.; Abraham, W.C.; Addae, A.; Corcoran, D.L.; Elliott, M.L.; Hogan, S.; Ireland, D.; Keenan, R.; Knodt, A.R.; et al. A Blood Biomarker of the Pace of Aging Is Associated with Brain Structure: Replication across Three Cohorts. Neurobiol. Aging 2024, 136, 23–33. [Google Scholar] [CrossRef]
- Sugden, K.; Caspi, A.; Elliott, M.L.; Bourassa, K.J.; Chamarti, K.; Corcoran, D.L.; Hariri, A.R.; Houts, R.M.; Kothari, M.; Kritchevsky, S.; et al. Association of Pace of Aging Measured by Blood-Based DNA Methylation With Age-Related Cognitive Impairment and Dementia. Neurology 2022, 99, e1402–e1413. [Google Scholar] [CrossRef] [PubMed]
- Pidsley, R.; Zotenko, E.; Peters, T.J.; Lawrence, M.G.; Risbridger, G.P.; Molloy, P.; Van Djik, S.; Muhlhausler, B.; Stirzaker, C.; Clark, S.J. Critical Evaluation of the Illumina MethylationEPIC BeadChip Microarray for Whole-Genome DNA Methylation Profiling. Genome Biol. 2016, 17, 208. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated Methylation Analysis Pipeline for Illumina BeadChips. Bioinforma. Oxf. Engl. 2017, 33, 3982–3984. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A Flexible and Comprehensive Bioconductor Package for the Analysis of Infinium DNA Methylation Microarrays. Bioinforma. Oxf. Engl. 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- Zhu, Y.; Davis, S.; Stephens, R.; Meltzer, P.S.; Chen, Y. GEOmetadb: Powerful Alternative Search Engine for the Gene Expression Omnibus. Bioinformatics 2008, 24, 2798–2800. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A Bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed]
- Gumienny, R.; van Heeringen, S.; Bismeijer, T.; Ramdhani, H. GEOparse: Python Library to Access Gene Expression Omnibus Database (GEO). Available online: https://github.com/guma44/GEOparse (accessed on 25 November 2024).
- Ding, C.; Peng, H. Minimum Redundancy Feature Selection from Microarray Gene Expression Data. In Proceedings of the Computational Systems Bioinformatics. In Proceedings of the 2003 IEEE Bioinformatics Conference. CSB2003, Stanford, CA, USA, 11–14 August 2003; pp. 523–528. [Google Scholar]
- Cover, T.M. The Best Two Independent Measurements Are Not the Two Best. IEEE Trans. Syst. Man Cybern. 1974, SMC-4, 116–117. [Google Scholar] [CrossRef]
- Chen, J.; Liao, K.; Wan, Y.; Chen, D.Z.; Wu, J. DANets: Deep Abstract Networks for Tabular Data Classification and Regression. Proc. AAAI Conf. Artif. Intell. 2022, 36, 3930–3938. [Google Scholar] [CrossRef]
- Gorishniy, Y.; Rubachev, I.; Khrulkov, V.; Babenko, A. Revisiting Deep Learning Models for Tabular Data. In Proceedings of the Advances in Neural Information Processing Systems, Online, 6–14 December 2021; Curran Associates, Inc.: Red Hook, NY, USA, 2021; Volume 34, pp. 18932–18943. [Google Scholar]
- Joseph, M.; Raj, H. GANDALF: Gated Adaptive Network for Deep Automated Learning of Features. arXiv 2024. [Google Scholar] [CrossRef]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A Next-Generation Hyperparameter Optimization Framework. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; ACM: Anchorage AK USA, 2019; pp. 2623–2631. [Google Scholar]
- Bergstra, J.; Bardenet, R.; Bengio, Y.; Kégl, B. Algorithms for Hyper-Parameter Optimization. In Proceedings of the Advances in Neural Information Processing Systems, Granada, Spain, 12–14 December 2011; Curran Associates, Inc.: Red Hook, NY, USA, 2011; Volume 24. [Google Scholar]
- Lundberg, S.M.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; Curran Associates Inc.: Red Hook, NY, USA, 2017; pp. 4768–4777. [Google Scholar]
- Mann, H.B.; Whitney, D.R. On a Test of Whether One of Two Random Variables Is Stochastically Larger than the Other. Ann. Math. Stat. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- de Lima Camillo, L.P. Pyaging: A Python-Based Compendium of GPU-Optimized Aging Clocks. Bioinformatics 2024, 40, btae200. [Google Scholar] [CrossRef]
- Lin, Q.; Weidner, C.I.; Costa, I.G.; Marioni, R.E.; Ferreira, M.R.P.; Deary, I.J.; Wagner, W. DNA Methylation Levels at Individual Age-Associated CpG Sites Can Be Indicative for Life Expectancy. Aging 2016, 8, 394–401. [Google Scholar] [CrossRef]
- Yang, Z.; Wong, A.; Kuh, D.; Paul, D.S.; Rakyan, V.K.; Leslie, R.D.; Zheng, S.C.; Widschwendter, M.; Beck, S.; Teschendorff, A.E. Correlation of an Epigenetic Mitotic Clock with Cancer Risk. Genome Biol. 2016, 17, 205. [Google Scholar] [CrossRef]
- Zhang, Y.; Wilson, R.; Heiss, J.; Breitling, L.P.; Saum, K.-U.; Schöttker, B.; Holleczek, B.; Waldenberger, M.; Peters, A.; Brenner, H. DNA Methylation Signatures in Peripheral Blood Strongly Predict All-Cause Mortality. Nat. Commun. 2017, 8, 14617. [Google Scholar] [CrossRef]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic Clock for Skin and Blood Cells Applied to Hutchinson Gilford Progeria Syndrome and Ex Vivo Studies. Aging 2018, 10, 1758–1775. [Google Scholar] [CrossRef]
- Lu, A.T.; Seeboth, A.; Tsai, P.-C.; Sun, D.; Quach, A.; Reiner, A.P.; Kooperberg, C.; Ferrucci, L.; Hou, L.; Baccarelli, A.A.; et al. DNA Methylation-Based Estimator of Telomere Length. Aging 2019, 11, 5895–5923. [Google Scholar] [CrossRef]
- Zhang, Q.; Vallerga, C.L.; Walker, R.M.; Lin, T.; Henders, A.K.; Montgomery, G.W.; He, J.; Fan, D.; Fowdar, J.; Kennedy, M.; et al. Improved Precision of Epigenetic Clock Estimates across Tissues and Its Implication for Biological Ageing. Genome Med. 2019, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Franzen, J.; Stiehl, T.; Gobs, M.; Kuo, C.-C.; Nikolić, M.; Hapala, J.; Koop, B.E.; Strathmann, K.; Ritz-Timme, S.; et al. New Targeted Approaches for Epigenetic Age Predictions. BMC Biol. 2020, 18, 71. [Google Scholar] [CrossRef] [PubMed]
- de Lima Camillo, L.P.; Lapierre, L.R.; Singh, R. A Pan-Tissue DNA-Methylation Epigenetic Clock Based on Deep Learning. Npj Aging 2022, 8, 4. [Google Scholar] [CrossRef]
- Higgins-Chen, A.T.; Thrush, K.L.; Wang, Y.; Minteer, C.J.; Kuo, P.-L.; Wang, M.; Niimi, P.; Sturm, G.; Lin, J.; Moore, A.Z.; et al. A Computational Solution for Bolstering Reliability of Epigenetic Clocks: Implications for Clinical Trials and Longitudinal Tracking. Nat. Aging 2022, 2, 644–661. [Google Scholar] [CrossRef]
- Lu, A.T.; Binder, A.M.; Zhang, J.; Yan, Q.; Reiner, A.P.; Cox, S.R.; Corley, J.; Harris, S.E.; Kuo, P.-L.; Moore, A.Z.; et al. DNA Methylation GrimAge Version 2. Aging 2022, 14, 9484–9549. [Google Scholar] [CrossRef]
- McGreevy, K.M.; Radak, Z.; Torma, F.; Jokai, M.; Lu, A.T.; Belsky, D.W.; Binder, A.; Marioni, R.E.; Ferrucci, L.; Pośpiech, E.; et al. DNAmFitAge: Biological Age Indicator Incorporating Physical Fitness. Aging 2023, 15, 3904–3938. [Google Scholar] [CrossRef]
- Dec, E.; Clement, J.; Cheng, K.; Church, G.M.; Fossel, M.B.; Rehkopf, D.H.; Rosero-Bixby, L.; Kobor, M.S.; Lin, D.T.S.; Lu, A.T.; et al. Centenarian Clocks: Epigenetic Clocks for Validating Claims of Exceptional Longevity. GeroScience 2023, 45, 1817–1835. [Google Scholar] [CrossRef]
- Tong, H.; Dwaraka, V.B.; Chen, Q.; Luo, Q.; Lasky-Su, J.A.; Smith, R.; Teschendorff, A.E. Quantifying the Stochastic Component of Epigenetic Aging. Nat. Aging 2024, 4, 886–901. [Google Scholar] [CrossRef]
- Zhu, T.; Tong, H.; Du, Z.; Beck, S.; Teschendorff, A.E. An Improved Epigenetic Counter to Track Mitotic Age in Normal and Precancerous Tissues. Nat. Commun. 2024, 15, 4211. [Google Scholar] [CrossRef]
- Ndhlovu, L.C.; Bendall, M.L.; Dwaraka, V.; Pang, A.P.S.; Dopkins, N.; Carreras, N.; Smith, R.; Nixon, D.F.; Corley, M.J. Retro-Age: A Unique Epigenetic Biomarker of Aging Captured by DNA Methylation States of Retroelements. Aging Cell 2024, 23, e14288. [Google Scholar] [CrossRef] [PubMed]
- Tomusiak, A.; Floro, A.; Tiwari, R.; Riley, R.; Matsui, H.; Andrews, N.; Kasler, H.G.; Verdin, E. Development of an Epigenetic Clock Resistant to Changes in Immune Cell Composition. Commun. Biol. 2024, 7, 934. [Google Scholar] [CrossRef] [PubMed]
- Abid, A.; Abdalla, A.; Abid, A.; Khan, D.; Alfozan, A.; Zou, J. Gradio: Hassle-Free Sharing and Testing of ML Models in the Wild. arXiv 2019. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalyakulina, A.; Yusipov, I.; Trukhanov, A.; Franceschi, C.; Moskalev, A.; Ivanchenko, M. EpInflammAge: Epigenetic-Inflammatory Clock for Disease-Associated Biological Aging Based on Deep Learning. Int. J. Mol. Sci. 2025, 26, 6284. https://doi.org/10.3390/ijms26136284
Kalyakulina A, Yusipov I, Trukhanov A, Franceschi C, Moskalev A, Ivanchenko M. EpInflammAge: Epigenetic-Inflammatory Clock for Disease-Associated Biological Aging Based on Deep Learning. International Journal of Molecular Sciences. 2025; 26(13):6284. https://doi.org/10.3390/ijms26136284
Chicago/Turabian StyleKalyakulina, Alena, Igor Yusipov, Arseniy Trukhanov, Claudio Franceschi, Alexey Moskalev, and Mikhail Ivanchenko. 2025. "EpInflammAge: Epigenetic-Inflammatory Clock for Disease-Associated Biological Aging Based on Deep Learning" International Journal of Molecular Sciences 26, no. 13: 6284. https://doi.org/10.3390/ijms26136284
APA StyleKalyakulina, A., Yusipov, I., Trukhanov, A., Franceschi, C., Moskalev, A., & Ivanchenko, M. (2025). EpInflammAge: Epigenetic-Inflammatory Clock for Disease-Associated Biological Aging Based on Deep Learning. International Journal of Molecular Sciences, 26(13), 6284. https://doi.org/10.3390/ijms26136284