
Molecular and Clinical Aspects of Osteogenesis Imperfecta Type VI: A Case Series with Novel SERPINF1 Gene Variants

 ,
,  , , , ,
, , , , 
Abstract
1. Introduction
2. Results
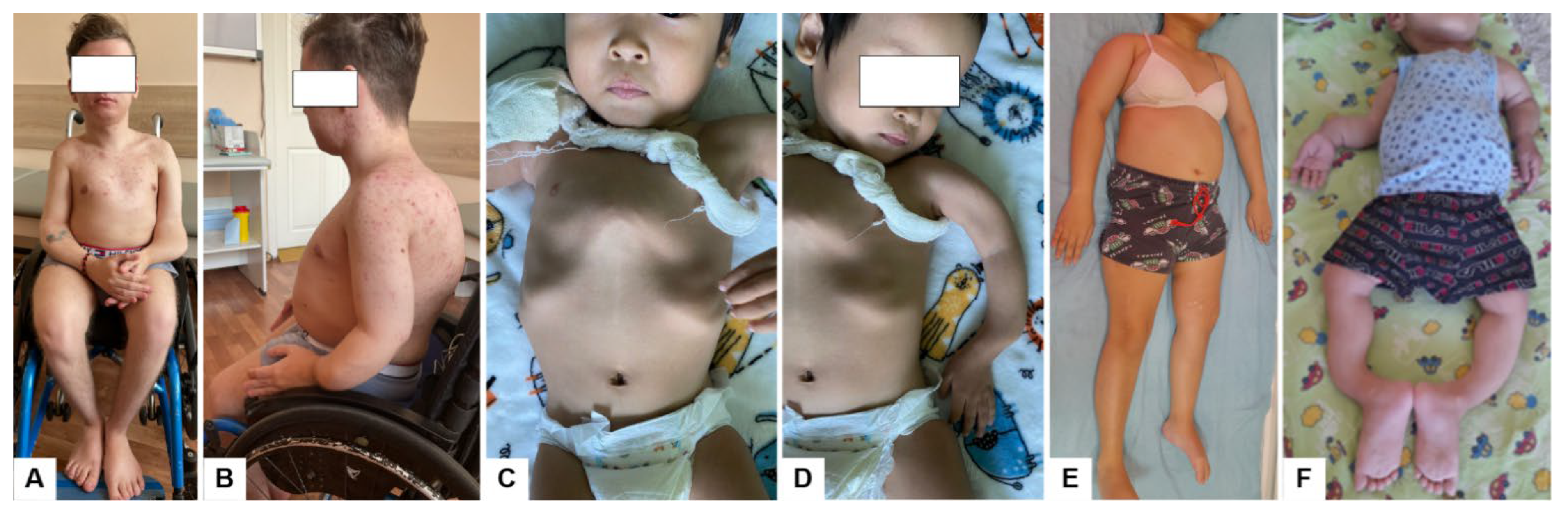
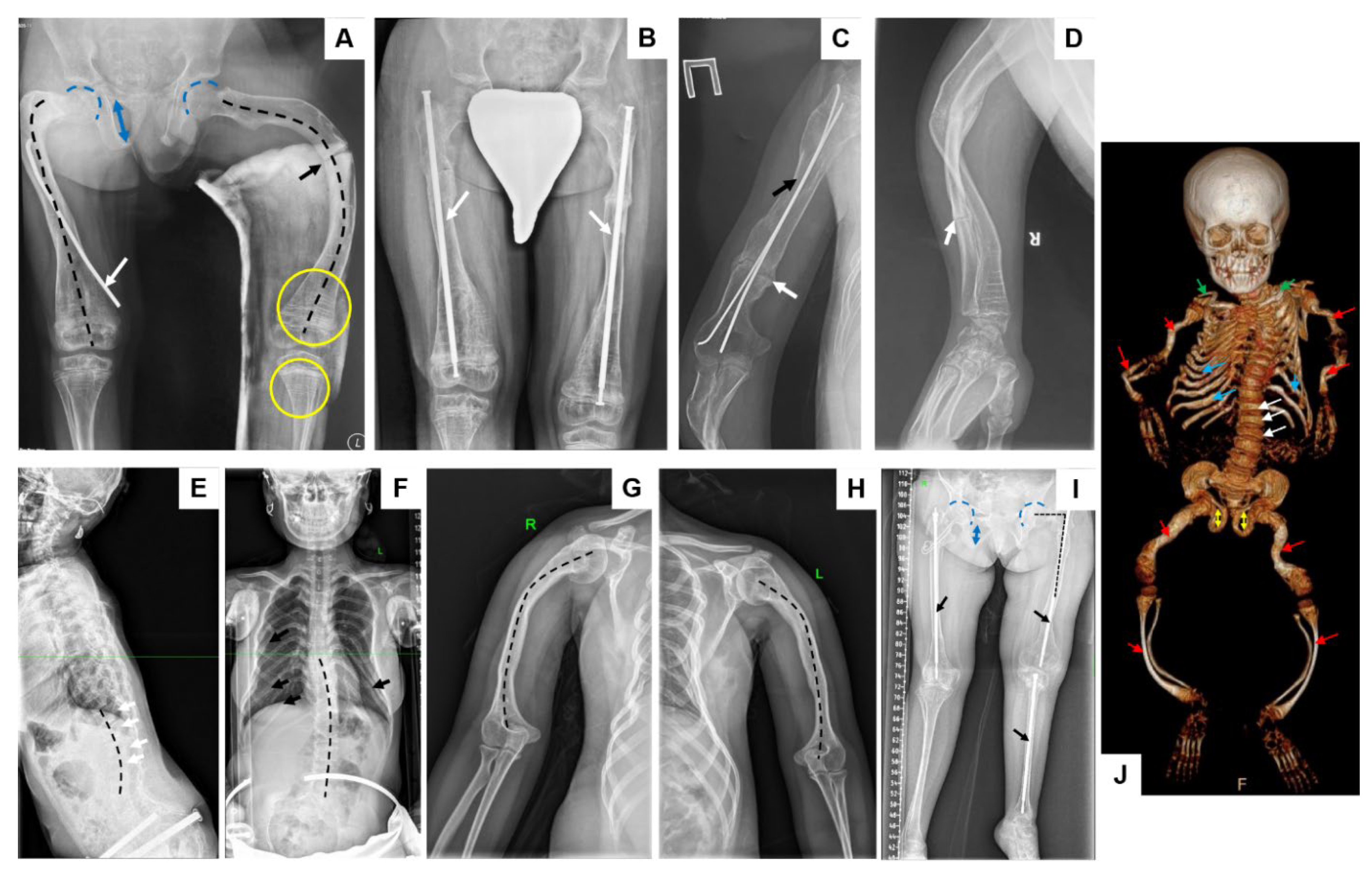
2.1. Patient Report
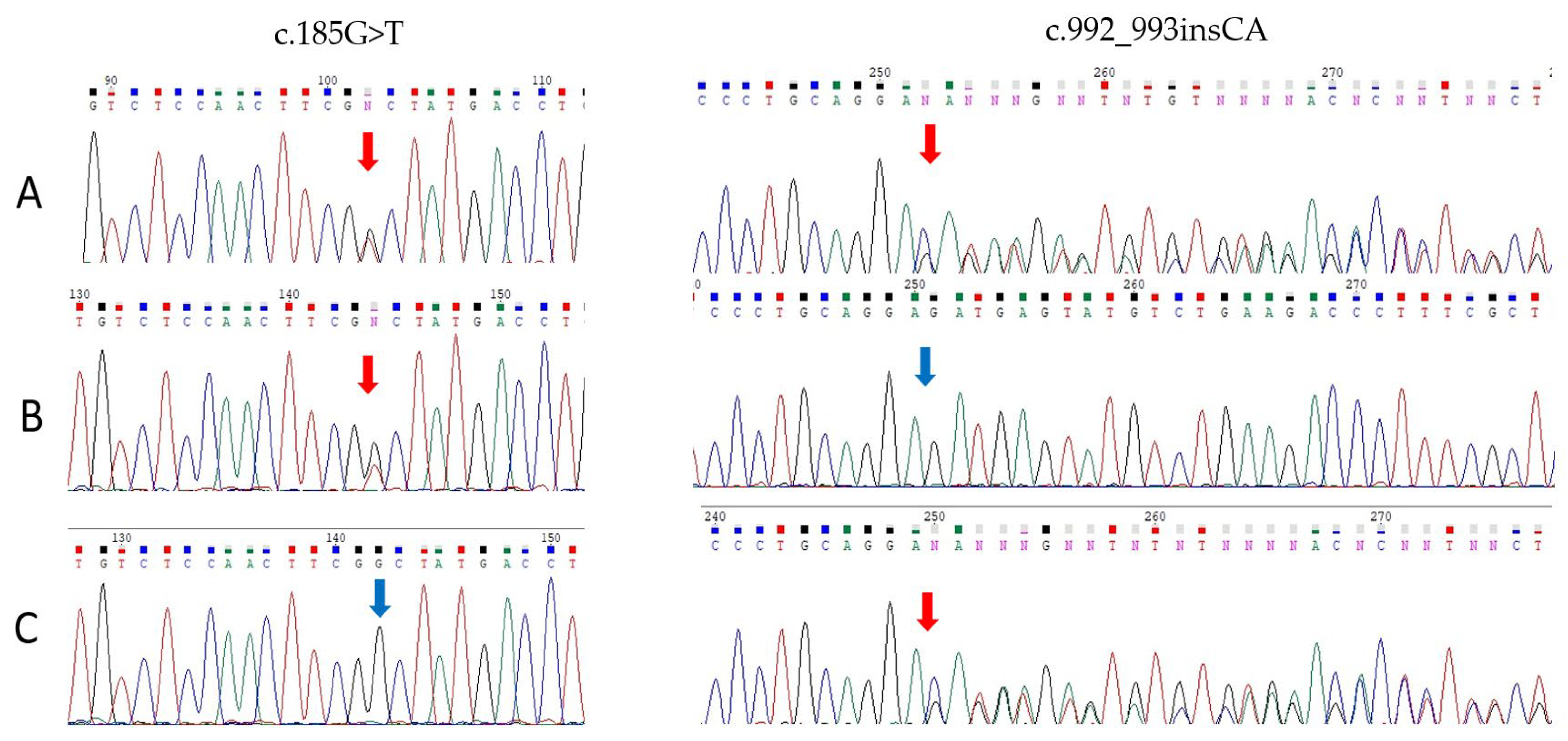
2.2. Molecular Genetic Analysis
2.3. RNA Analysis of the Variant c.992_993insCA
2.4. Frequency Assessment of Type VI Osteogenesis Imperfecta in the Republic of Tuva
3. Discussion
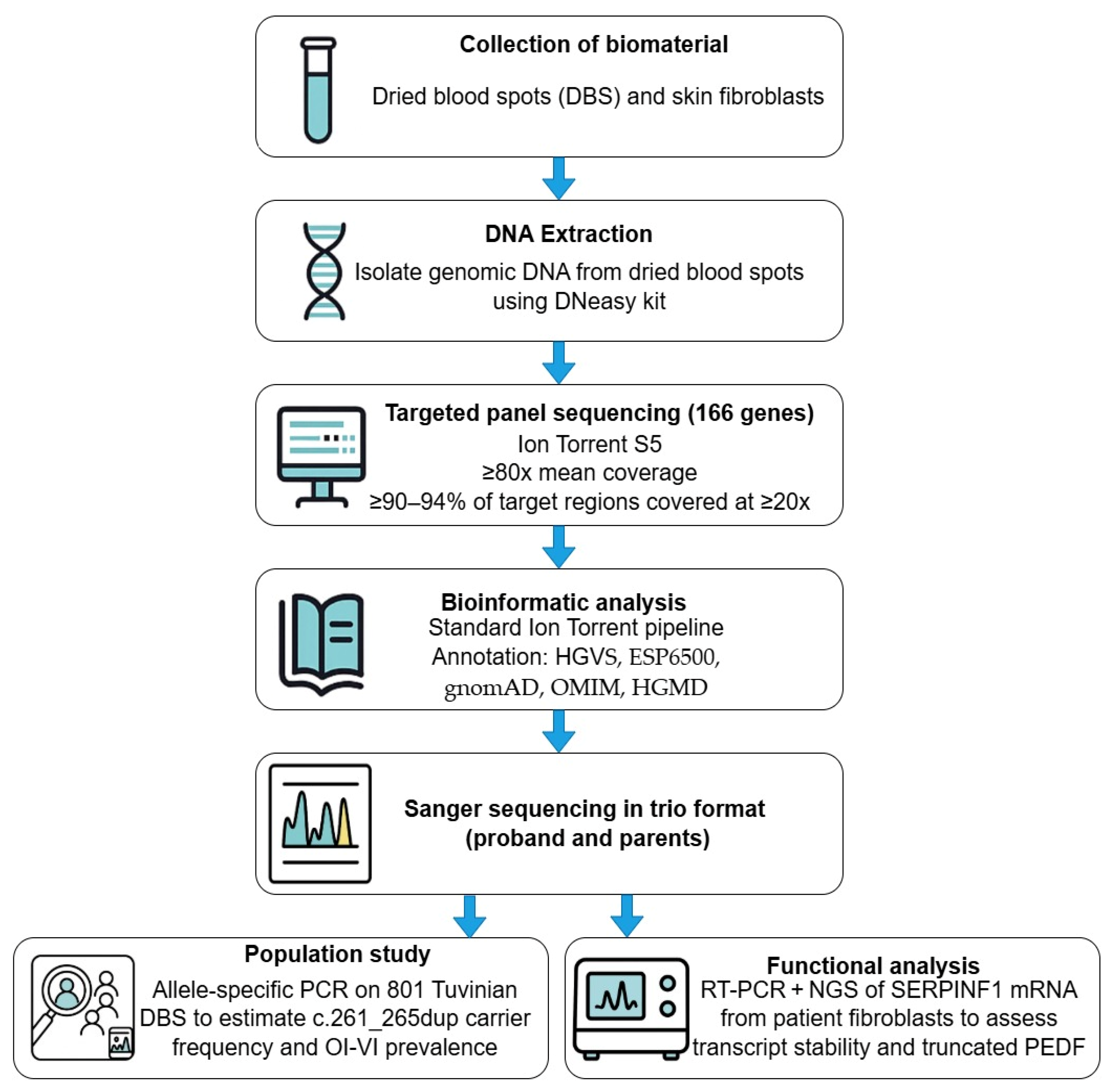
4. Materials and Methods
4.1. Clinical Evaluation
4.2. Radiographic Analysis
4.3. Genetic Sequencing
4.4. Population Study
- SERPINF1_261_265dup_RT_1_F: AACGTGCTCCTGTCTCCTCT (0.04 µM)
- SERPINF1_261_265dup_RT_1_R: GAGCACTCACCCAGCGAG (0.04 µM)
- SERPINF1_261_265dup_RT_2_F: CTGTCTCCTCTCAGTGTGGC (0.04 µM)
- SERPINF1_261_265dup_RT_2_R: GCTTCCTGCATCTGAGCACT (0.04 µM)
4.5. RNA Analysis of the Variant c.992_993insCA
Targeted Next-Generation Sequencing of RT-PCR Product
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krakow, D.; Byers, P.H.; De Paepe, A.; Marini, J.C.; Forlino, A.; Kozloff, K.M.; Montpetit, K.; Semler, O.; Bächinger, H.P.; Bishop, N.J.; et al. Osteogenesis imperfecta. Nat. Rev. Dis. Primers 2017, 3, 17052. [Google Scholar] [CrossRef]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [PubMed]
- Homan, E.P.; Rauch, F.; Grafe, I.; Lietman, C.; Doll, J.A.; Dawson, B.; Bertin, T.; Napierala, D.; Morello, R.; Gibbs, R.; et al. Mutations in SERPINF1 cause osteogenesis imperfecta type VI. J. Bone Miner. Res. 2011, 26, 2798–2803. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Semler, O.; Gilissen, C.; Li, Y.; Bolz, H.J.; Giunta, C.; Bergmann, C.; Rohrbach, M.; Koerber, F.; Zimmermann, K.; et al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am. J. Hum. Genet. 2011, 88, 362–371. [Google Scholar] [CrossRef]
- Venturi, G.; Gandini, A.; Monti, E.; Carbonare, L.D.; Corradi, M.; Vincenzi, M.; Valenti, M.T.; Valli, M.; Pelilli, E.; Boner, A.; et al. Lack of expression of SERPINF1, the gene coding for pigment epithelium-derived factor, causes progressively deforming osteogenesis imperfecta with normal type I collagen. J. Bone Miner. Res. 2012, 27, 723–728. [Google Scholar] [CrossRef]
- Glorieux, F.H.; Ward, L.M.; Rauch, F.; Lalic, L.; Roughley, P.J.; Travers, R. Osteogenesis imperfecta type VI: A form of brittle bone disease with a mineralization defect. J. Bone Miner. Res. 2002, 17, 30–38. [Google Scholar] [CrossRef]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next generation in gene variant databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef]
- Li, L.; Mao, B.; Li, S.; Xiao, J.; Wang, H.; Zhang, J.; Ren, X.; Wang, Y.; Wu, Y.; Cao, Y.; et al. Genotypic and phenotypic characterization of Chinese patients with osteogenesis imperfecta. Hum. Mutat. 2019, 40, 588–600. [Google Scholar] [CrossRef]
- Zhalsanova, I.Z.; Postrigan, A.E.; Valiakhmetov, N.R.; Kolesnikov, N.A.; Zhigalina, D.I.; Zarubin, A.A.; Petrova, V.V.; Minaycheva, L.I.; Seitova, G.N.; Skryabin, N.A.; et al. Case report: A novel homozygous variant of the SERPINF1 gene in rare osteogenesis imperfecta type VI. Int. J. Mol. Sci. 2023, 24, 6672. [Google Scholar] [CrossRef]
- Rohrbach, M.; Giunta, C. Recessive osteogenesis imperfecta: Clinical, radiological and molecular findings. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160C, 175–189. [Google Scholar] [CrossRef]
- Caparrós-Martin, J.A.; Valencia, M.; Pulido, V.; Martínez-Glez, V.; Rueda-Arenas, I.; Amr, K.; Farra, C.; Lapunzina, P.; Ruiz-Perez, V.L.; Temtamy, S.; et al. Clinical and molecular analysis in families with autosomal recessive osteogenesis imperfecta identifies mutations in five genes and suggests genotype-phenotype correlations. Am. J. Med. Genet. A 2013, 161, 1354–1369. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.Y.; Ki, C.-S.; Sohn, Y.B.; Kim, S.J.; Maeng, S.H.; Jin, D.-K. Osteogenesis imperfecta type VI with severe bony deformities caused by novel compound heterozygous mutations in SERPINF1. J. Korean Med. Sci. 2013, 28, 1107–1110. [Google Scholar] [CrossRef] [PubMed]
- Minillo, R.M.; Sobreira, N.; de Faria Soares, M.F.; Jurgens, J.; Ling, H.; Hetrick, K.N.; Doheny, K.F.; Valle, D.; Brunoni, D.; Perez, A.B. Novel deletion of SERPINF1 causes autosomal recessive osteogenesis imperfecta type VI in two Brazilian families. Mol. Syndromol. 2014, 5, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Selina, A.; Kandagaddala, M.; Kumar, V.; Abraham, S.S.C.; Danda, S.; Madhuri, V. SERPINF1 gene variants causing late-onset progressive deforming osteogenesis imperfecta: A study of 18 patients from India. Bone Rep. 2023, 18, 101690. [Google Scholar] [CrossRef]
- Hayat, A.; Hussain, S.; Bilal, M.; Kausar, M.; Almuzzaini, B.; Abbas, S.; Tanveer, A.; Khan, A.; Siddiqi, S.; Foo, J.N.; et al. Biallelic variants in four genes underlying recessive osteogenesis imperfecta. Eur. J. Med. Genet. 2020, 63, 103954. [Google Scholar] [CrossRef]
- Semler, O.; Netzer, C.; Hoyer-Kuhn, H.; Becker, J.; Eysel, P.; Schoenau, E. First use of the RANKL antibody denosumab in osteogenesis imperfecta type VI. Musculoskelet. Neuronal Interact. 2012, 12, 183–188. [Google Scholar]
- Wang, D.; Tang, X.; Shi, Q.; Wang, R.; Ji, T.; Tang, X.; Guo, W. Denosumab in pediatric bone disorders and the role of RANKL blockade: A narrative review. Transl. Pediatr. 2023, 12, 470–486. [Google Scholar] [CrossRef]
- Shao, H.; Schvartz, I.; Shaltiel, S. Secretion of pigment epithelium-derived factor: Mutagenic study. Eur. J. Biochem. 2003, 270, 822–831. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The extracellular matrix as a multitasking player in disease. FEBS J. 2019, 286, 2830–2869. [Google Scholar] [CrossRef]
- Merkuryeva, E.S. Clinical and Genetic Characteristics of Isolated and Syndromic Forms of Osteogenesis Imperfecta. Ph.D. Thesis, Medical Genetic Research Center, Moscow, Russia, 2024. [Google Scholar]
- Zytsar, M.V.; Bady-Khoo, M.S.; Danilchenko, V.Y.; Maslova, E.A.; Barashkov, N.A.; Morozov, I.V.; Bondar, A.A.; Posukh, O.L. High rates of three common GJB2 mutations c.516G>C, c.-23+1G>A, c.235delC in deaf patients from Southern Siberia are due to the founder effect. Genes 2020, 11, 833. [Google Scholar] [CrossRef]
- Markova, T.V. Hereditary Systemic Skeletal Dysplasias of Childhood: Clinical and Genetic Characteristics and Diagnostic Strategies. Ph.D. Thesis, Medical Genetic Research Center, Moscow, Russia, 2023. [Google Scholar]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Genomes Project Consortium; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; O’connor, T.D.; Jun, G.; Kang, H.M.; Abecasis, G.; Leal, S.M.; Gabriel, S.; Rieder, M.J.; Altshuler, D.; Shendure, J.; et al. Analysis of 6515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013, 493, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef]
- Signorell, A. DescTools: Tools for Descriptive Statistics. R Package Version 0.99.59. 2025. Available online: https://andrisignorell.github.io/DescTools/ (accessed on 2 June 2025).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Patient 1 | Patient 2 | Patient 3 * | Patient 4 * |
|---|---|---|---|---|
| Mutation | c.185G>T/c.992_993insCA | c.261_265dup (homozygous) | c.261_265dup (homozygous) | c.261_265dup (homozygous) |
| Ethnicity | Russian | Tuvan | Tuvan | Tuvan |
| Consanguinity | No | No | No | No |
| Age (years) | 23 | 3 | 16 | 12 |
| Sex | M | M | F | M |
| Age at first fracture (months) | 13 | 6 | 18 | 9 |
| Fracture sites 1 | 1, 2, 3, 4, 5, 6 | 2, 3, 4, 5, 6 | 1, 2, 3, 4, 5, 6 | 1, 2, 3, 4, 5, 6 |
| Fractures total (n) | 200 | 15 | 12 | 14 |
| Fractures/year | 8.69 | 3.0 | 0.8 | 1.27 |
| Scoliosis (grade) | IV | - | I | II |
| Severity of long bone deformities | Severe | Moderate | Moderate | Severe |
| Height (SD Score) | −5.52 | −2.73 | −2.96 | −7.71 |
| BMD (L1–L4) (g/cm2) | 0.592 | 0.874 | 0.874 | 0.256 |
| Z-score (L1–L4) | −3.1 | −1.7 | −1.7 | −4.6 |
| Blue sclera | No | No | No | No |
| Dentinogenesis imperfecta | No | No | No | No |
| Hearing loss | No | No | No | No |
| Age at initiation of bisphosphonates | 6 years | 1 year 8 months | 8 years | 6 years |
| Independent walking | No | No | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merkuryeva, E.S.; Nagornova, T.S.; Kenis, V.M.; Deviataikina, A.S.; Akimova, D.B.; Buklaev, D.S.; Dantsev, I.S.; Dulush, A.O.; Zakharova, E.Y.; Markova, T.V. Molecular and Clinical Aspects of Osteogenesis Imperfecta Type VI: A Case Series with Novel SERPINF1 Gene Variants. Int. J. Mol. Sci. 2025, 26, 6200. https://doi.org/10.3390/ijms26136200
Merkuryeva ES, Nagornova TS, Kenis VM, Deviataikina AS, Akimova DB, Buklaev DS, Dantsev IS, Dulush AO, Zakharova EY, Markova TV. Molecular and Clinical Aspects of Osteogenesis Imperfecta Type VI: A Case Series with Novel SERPINF1 Gene Variants. International Journal of Molecular Sciences. 2025; 26(13):6200. https://doi.org/10.3390/ijms26136200
Chicago/Turabian StyleMerkuryeva, Elena S., Tatyana S. Nagornova, Vladimir M. Kenis, Anna S. Deviataikina, Daria B. Akimova, Dmitry S. Buklaev, Ilya S. Dantsev, Aisluu O. Dulush, Ekaterina Y. Zakharova, and Tatiana V. Markova. 2025. "Molecular and Clinical Aspects of Osteogenesis Imperfecta Type VI: A Case Series with Novel SERPINF1 Gene Variants" International Journal of Molecular Sciences 26, no. 13: 6200. https://doi.org/10.3390/ijms26136200
APA StyleMerkuryeva, E. S., Nagornova, T. S., Kenis, V. M., Deviataikina, A. S., Akimova, D. B., Buklaev, D. S., Dantsev, I. S., Dulush, A. O., Zakharova, E. Y., & Markova, T. V. (2025). Molecular and Clinical Aspects of Osteogenesis Imperfecta Type VI: A Case Series with Novel SERPINF1 Gene Variants. International Journal of Molecular Sciences, 26(13), 6200. https://doi.org/10.3390/ijms26136200