
Primary Plasma Cell Leukemia: Recent Advances in Molecular Understanding and Treatment Approaches



Abstract
1. Introduction
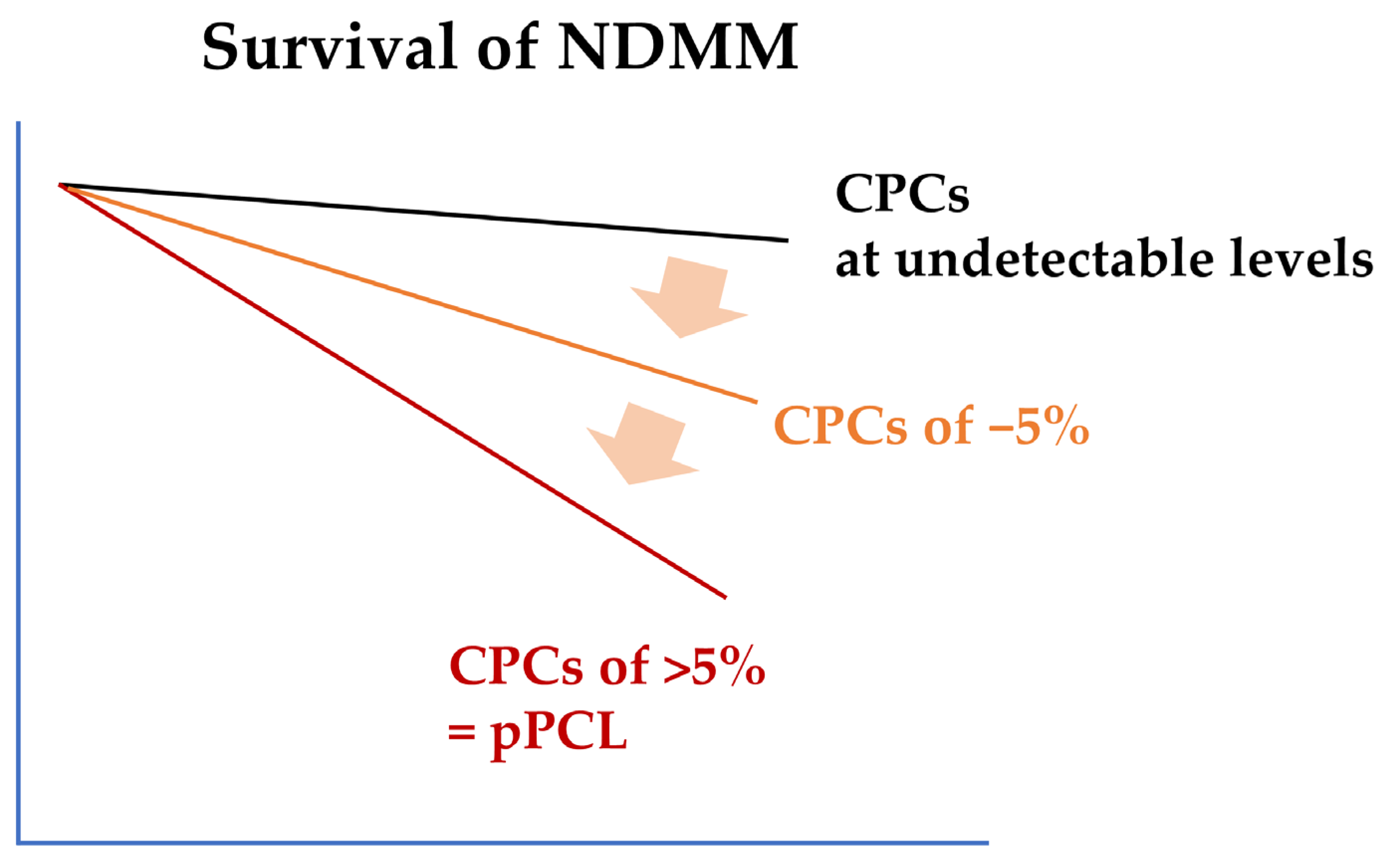
2. Diagnostic Criteria and Clinical Features of pPCL
3. Biologic Features of pPCL
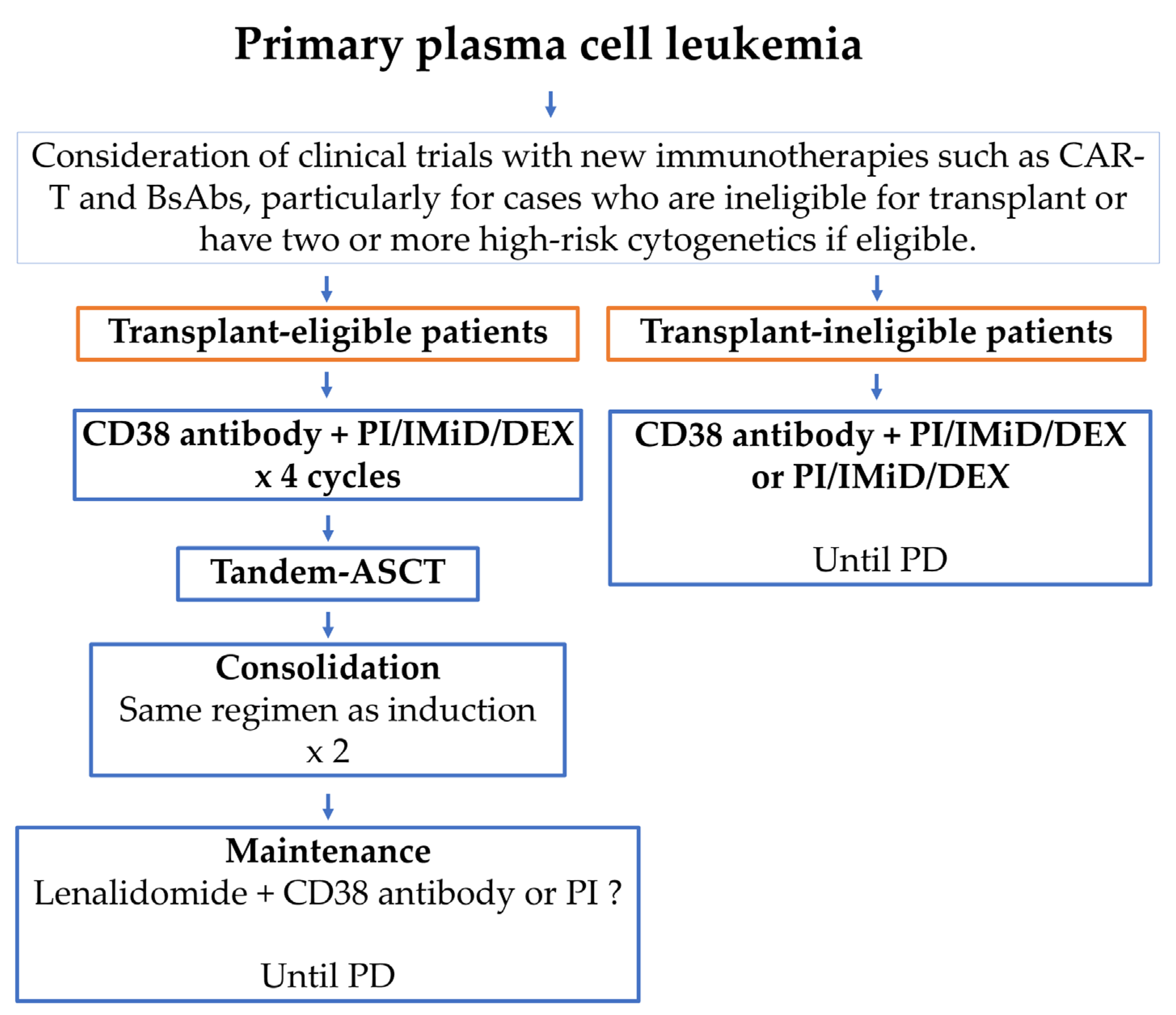
4. Treatment and Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADC | Antibody–drug conjugate |
| BCMA | B-cell maturation antigen |
| BsAb | Bispecific antibody |
| CAR-T | Chimeric antigen receptor T cell |
| CPC | Circulating plasma cell |
| CR | Complete remission |
| GIMEMA | Gruppo Italiano Malattle Ematologiche d’Adulto |
| FCM | Flow cytometry |
| IFM | Intergroupe Francophone du Mye′ lome |
| SCT | Hematopoietic stem cell transplantation |
| MM | Multiple myeloma |
| MRD | Minimal residual disease |
| OS | Overall survival |
| pPCL | Primary plasma cell leukemia |
| PFS | Progression-free survival |
| RIC | Reduced-intensity conditioning |
| TE | Transplantation-eligible |
| TIE | Transplantation-ineligible |
References
- Kyle, R.A.; Maldonado, J.E.; Bayrd, E.D. Plasma cell leukemia. Report on 17 cases. Arch. Intern. Med. 1974, 133, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Palumbo, A.; Delasalle, K.B.; Alexanian, R. Primary plasma cell leukaemia. Br. J. Haematol. 1994, 88, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.; Lokhorst, H.M.; Anderson, K.C.; Richardson, P.G. How I treat plasma cell leukemia. Blood 2012, 120, 2376–2389. [Google Scholar] [CrossRef] [PubMed]
- Fernández de Larrea, C.; Kyle, R.; Rosiñol, L.; Paiva, B.; Engelhardt, M.; Usmani, S.; Caers, J.; Gonsalves, W.; Schjesvold, F.; Merlini, G.; et al. Primary plasma cell leukemia: Consensus definition by the International Myeloma Working Group according to peripheral blood plasma cell percentage. Blood Cancer J. 2021, 11, 192. [Google Scholar] [CrossRef]
- Shah, R.A.; Mohite, S.; Baladandayuthapani, V.; Thomas, S.K.; Weber, D.M.; Wang, M.; Alexanian, R.; Qazilbash, M.H.; Popat, U.R.; Champlin, R.E.; et al. Circulating Plasma Cells by Routine Complete Blood Count Identify Patients with Similar Outcome as Plasma Cell Leukemia. Blood 2013, 122, 5356. [Google Scholar] [CrossRef]
- Bertamini, L.; Oliva, S.; Rota-Scalabrini, D.; Paris, L.; Morè, S.; Corradini, P.; Ledda, A.; Gentile, M.; De Sabbata, G.; Pietrantuono, G.; et al. High Levels of Circulating Tumor Plasma Cells as a Key Hallmark of Aggressive Disease in Transplant-Eligible Patients with Newly Diagnosed Multiple Myeloma. J. Clin. Oncol. 2022, 40, 3120–3131. [Google Scholar] [CrossRef]
- Garcés, J.-J.; Cedena, M.-T.; Puig, N.; Burgos, L.; Perez, J.J.; Cordon, L.; Flores-Montero, J.; Sanoja-Flores, L.; Calasanz, M.-J.; Ortiol, A.; et al. Circulating Tumor Cells for the Staging of Patients with Newly Diagnosed Transplant-Eligible Multiple Myeloma. J. Clin. Oncol. 2022, 40, 3151–3161. [Google Scholar] [CrossRef]
- Jelinek, T.; Bezdekova, R.; Zihala, D.; Sevcikova, T.; Anilkumar Sithara, A.; Pospisilova, L.; Sevcikova, S.; Polackova, P.; Stork, M.; Knechtova, Z.; et al. More Than 2% of Circulating Tumor Plasma Cells Defines Plasma Cell Leukemia-like Multiple Myeloma. J. Clin. Oncol. 2023, 41, 1383–1392. [Google Scholar] [CrossRef]
- Van de Donk, N. How We Manage Newly Diagnosed Multiple Myeloma with Circulating Tumor Cells. J. Clin. Oncol. 2023, 41, 1342–1349. [Google Scholar] [CrossRef]
- Sant, M.; Allemani, C.; Tereanu, C.; De Angelis, R.; Capocaccia, R.; Visser, O.; Marcos-Gragera, R.; Maynadié, M.; Simonetti, A.; Lutz, J.M.; et al. Incidence of hematologic malignancies in Europe by morphologic subtype: Results of the HAEMACARE project. Blood 2010, 116, 3724–3734. [Google Scholar] [CrossRef]
- Gonsalves, W.I.; Rajkumar, S.V.; Go, R.S.; Dispenzieri, A.; Gupta, V.; Singh, P.P.; Buadi, F.K.; Lacy, M.Q.; Kapoor, P.; Dingli, D.; et al. Trends in survival of patients with primary plasma cell leukemia: A population-based analysis. Blood 2014, 124, 907–912. [Google Scholar] [CrossRef] [PubMed]
- García-Sanz, R.; Orfão, A.; González, M.; Tabernero, M.D.; Bladé, J.; Moro, M.J.; Fernández-Calvo, J.; Sanz, M.A.; Pérez-Simón, J.A.; Rasillo, A.; et al. Primary Plasma Cell Leukemia: Clinical, Immunophenotypic, DNA Ploidy, and Cytogenetic Characteristics. Blood 1999, 93, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Avet-Loiseau, H.; Daviet, A.; Brigaudeau, C.; Callet-Bauchu, E.; Terré, C.; Lafage-Pochitaloff, M.; Désangles, F.; Ramond, S.; Talmant, P.; Bataille, R. Cytogenetic, interphase, and multicolor fluorescence in situ hybridization analyses in primary plasma cell leukemia: A study of 40 patients at diagnosis, on behalf of the Intergroupe Francophone du Myélome and the Groupe Français de Cytogénétique Hématologique. Blood 2001, 97, 822–825. [Google Scholar] [CrossRef] [PubMed]
- Rojas, E.A.; Gutiérrez, N.C. Genomics of Plasma Cell Leukemia. Cancers 2022, 14, 1594. [Google Scholar] [CrossRef]
- Mina, R.; Joseph, N.S.; Kaufman, J.L.; Gupta, V.A.; Heffner, L.T.; Hofmeister, C.C.; Boise, L.H.; Dhodapkar, M.V.; Gleason, C.; Nooka, A.K.; et al. Survival outcomes of patients with primary plasma cell leukemia (pPCL) treated with novel agents. Cancer 2019, 125, 416–423. [Google Scholar] [CrossRef]
- Nandakumar, B.; Kumar, S.K.; Dispenzieri, A.; Buadi, F.K.; Dingli, D.; Lacy, M.Q.; Hayman, S.R.; Kapoor, P.; Leung, N.; Fonder, A.; et al. Clinical Characteristics and Outcomes of Patients with Primary Plasma Cell Leukemia in the Era of Novel Agent Therapy. Mayo Clin. Proc. 2021, 96, 677–687. [Google Scholar] [CrossRef]
- Van de Donk, N.; Usmani, S.Z.; Yong, K. CAR T-cell therapy for multiple myeloma: State of the art and prospects. Lancet Haematol. 2021, 8, e446–e461. [Google Scholar] [CrossRef]
- Sidana, S.; Patel, K.K.; Peres, L.C.; Bansal, R.; Kocoglu, M.H.; Shune, L.; Atrash, S.; Smith, K.; Midha, S.; Ferreri, C.; et al. Safety and efficacy of standard-of-care ciltacabtagene autoleucel for relapsed/refractory multiple myeloma. Blood 2025, 145, 85–97. [Google Scholar] [CrossRef]
- Granell, M.; Calvo, X.; Garcia-Guiñón, A.; Escoda, L.; Abella, E.; Martínez, C.M.; Teixidó, M.; Gimenez, M.T.; Senín, A.; Sanz, P.; et al. Prognostic impact of circulating plasma cells in patients with multiple myeloma: Implications for plasma cell leukemia definition. Haematologica 2017, 102, 1099–1104. [Google Scholar] [CrossRef]
- Ravi, P.; Kumar, S.K.; Roeker, L.; Gonsalves, W.; Buadi, F.; Lacy, M.Q.; Go, R.S.; Dispenzieri, A.; Kapoor, P.; Lust, J.A.; et al. Revised diagnostic criteria for plasma cell leukemia: Results of a Mayo Clinic study with comparison of outcomes to multiple myeloma. Blood Cancer J. 2018, 8, 116. [Google Scholar] [CrossRef]
- Jung, S.-H.; Kim, K.; Yoon, S.E.; Moon, J.H.; Kim, D.; Kim, H.J.; Kim, M.K.; Kim, K.H.; Lee, H.J.; Lee, J.H.; et al. Validation of the revised diagnostic criteria for primary plasma cell leukemia by the Korean Multiple Myeloma Working Party. Blood Cancer J. 2022, 12, 157. [Google Scholar] [CrossRef] [PubMed]
- Katodritou, E.; Kastritis, E.; Dalampira, D.; Delimpasi, S.; Spanoudakis, E.; Labropoulou, V.; Ntanasis-Stathopoulos, I.; Gkioka, A.I.; Giannakoulas, N.; Kanellias, N.; et al. Improved survival of patients with primary plasma cell leukemia with VRd or daratumumab-based quadruplets: A multicenter study by the Greek myeloma study group. Am. J. Hematol. 2023, 98, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Li, A.Y.; Kamangar, F.; Holtzman, N.G.; Rapoport, A.P.; Kocoglu, M.H.; Atanackovic, D.; Badros, A.Z. A Clinical Perspective on Plasma Cell Leukemia: A Single-Center Experience. Cancers 2024, 16, 2149. [Google Scholar] [CrossRef] [PubMed]
- Evans, L.A.; Jevremovic, D.; Nandakumar, B.; Dispenzieri, A.; Buadi, F.K.; Dingli, D.; Lacy, M.Q.; Hayman, S.R.; Kapoor, P.; Leung, N.; et al. Utilizing multiparametric flow cytometry in the diagnosis of patients with primary plasma cell leukemia. Am. J. Hematol. 2020, 95, 637–642. [Google Scholar] [CrossRef]
- Ramsingh, G.; Mehan, P.; Luo, J.; Vij, R.; Morgensztern, D. Primary plasma cell leukemia: A Surveillance, Epidemiology, and End Results database analysis between 1973 and 2004. Cancer 2009, 115, 5734–5739. [Google Scholar] [CrossRef]
- Fernández de Larrea, C.; Kyle, R.A.; Durie, B.G.; Ludwig, H.; Usmani, S.; Vesole, D.H.; Hajek, R.; San Miguel, J.F.; Sezer, O.; Sonneveld, P.; et al. Plasma cell leukemia: Consensus statement on diagnostic requirements, response criteria and treatment recommendations by the International Myeloma Working Group. Leukemia 2013, 27, 780–791. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Nair, B.; Qu, P.; Hansen, E.; Zhang, Q.; Petty, N.; Waheed, S.; Shaughnessy, J.D.; Alsayed, Y.; Heuck, C.J.; et al. Primary plasma cell leukemia: Clinical and laboratory presentation, gene-expression profiling and clinical outcome with Total Therapy protocols. Leukemia 2012, 26, 2398–2405. [Google Scholar] [CrossRef]
- Musto, P.; Statuto, T.; Valvano, L.; Grieco, V.; Nozza, F.; Vona, G.; Bochicchio, G.B.; La Rocca, F.; D’Auria, F. An update on biology, diagnosis and treatment of primary plasma cell leukemia. Expert. Rev. Hematol. 2019, 12, 245–253. [Google Scholar] [CrossRef]
- Visram, A.; Suska, A.; Jurczyszyn, A.; Gonsalves, W.I. Practical management and assessment of primary plasma cell leukemia in the novel agent era. Cancer Treat. Res. Commun. 2021, 28, 100414. [Google Scholar] [CrossRef]
- Musto, P.; Engelhardt, M.; van de Donk, N.W.C.J.; Gay, F.; Terpos, E.; Einsele, H.; Fernández de Larrea, C.; Sgherza, N.; Bolli, N.; Katodritou, E.; et al. European Myeloma Network Group review and consensus statement on primary plasma cell leukemia. Ann. Oncol. 2025, 36, 361–374. [Google Scholar] [CrossRef]
- Chang, H.; Qi, X.; Yeung, J.; Reece, D.; Xu, W.; Patterson, B. Genetic aberrations including chromosome 1 abnormalities and clinical features of plasma cell leukemia. Leuk. Res. 2009, 33, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Cazaubiel, T.; Leleu, X.; Perrot, A.; Manier, S.; Buisson, L.; Maheo, S.; Do Souto Ferreira, L.; Lannes, R.; Pavageau, L.; Hulin, C.; et al. Primary Plasma Cell Leukemia displaying t(11;14) have specific genomic, transcriptional and clinical feature. Blood 2022, 139, 2666–2672. [Google Scholar] [CrossRef]
- Hanamura, I. Multiple myeloma with high-risk cytogenetics and its treatment approach. Int. J. Hematol. 2022, 115, 762–777. [Google Scholar] [CrossRef]
- Van de Donk, N.; Minnema, M.C.; van der Holt, B.; Schjesvold, F.; Wu, K.L.; Broijl, A.; Roeloffzen, W.W.H.; Gadisseur, A.; Pietrantuono, G.; Pour, L.; et al. Treatment of primary plasma cell leukaemia with carfilzomib and lenalidomide-based therapy (EMN12/HOVON-129): Final analysis of a non-randomised, multicentre, phase 2 study. Lancet Oncol. 2023, 24, 1119–1133. [Google Scholar] [CrossRef]
- Schinke, C.; Boyle, E.M.; Ashby, C.; Wang, Y.; Lyzogubov, V.; Wardell, C.; Qu, P.; Hoering, A.; Deshpande, S.; Ryan, K.; et al. Genomic analysis of primary plasma cell leukemia reveals complex structural alterations and high-risk mutational patterns. Blood Cancer J. 2020, 10, 70. [Google Scholar] [CrossRef]
- Kraj, M.; Kopeć-Szlęzak, J.; Pogłód, R.; Kruk, B. Flow cytometric immunophenotypic characteristics of 36 cases of plasma cell leukemia. Leuk. Res. 2011, 35, 169–176. [Google Scholar] [CrossRef]
- Guikema, J.E.; Vellenga, E.; Abdulahad, W.H.; Hovenga, S.; Bos, N.A. CD27-triggering on primary plasma cell leukaemia cells has anti-apoptotic effects involving mitogen activated protein kinases. Br. J. Haematol. 2004, 124, 299–308. [Google Scholar] [CrossRef]
- Paiva, B.; Paino, T.; Sayagues, J.-M.; Garayoa, M.; San-Segundo, L.; Martín, M.; Mota, I.; Sanchez, M.-L.; Bárcena, P.; Aires-Mejia, I.; et al. Detailed characterization of multiple myeloma circulating tumor cells shows unique phenotypic, cytogenetic, functional, and circadian distribution profile. Blood 2013, 122, 3591–3598. [Google Scholar] [CrossRef]
- Hofste Op Bruinink, D.; Kuiper, R.; van Duin, M.; Cupedo, T.; van der Velden, V.H.J.; Hoogenboezem, R.; van der Holt, B.; Beverloo, H.B.; Valent, E.T.; Vermeulen, M.; et al. Identification of High-Risk Multiple Myeloma with a Plasma Cell Leukemia-like Transcriptomic Profile. J. Clin. Oncol. 2022, 40, 3132–3150. [Google Scholar] [CrossRef]
- Almodovar Diaz, A.A.; Alouch, S.; Dispenzieri, A.; Yadav, U.; Buadi, F.; Dingli, D.; Muchtar, E.; Leung, N.; Kourelis, T.; Warsame, R.M.; et al. Impact of Multiple High-Risk Cytogenetic Abnormalities on the Survival Outcomes of Patients with Primary Plasma Cell Leukemia. Blood 2024, 144, 3315. [Google Scholar] [CrossRef]
- Xia, Y.; Shen, N.; Zhang, R.; Wu, Y.; Shi, Q.; Li, J.; Chen, L.; Xu, M.; Jin, Y. High-risk multiple myeloma predicted by circulating plasma cells and its genetic characteristics. Front. Oncol. 2023, 13, 1083053. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Otero, P.; Paiva, B.; San-Miguel, J.F. Roadmap to cure multiple myeloma. Cancer Treat. Rev. 2021, 100, 102284. [Google Scholar] [CrossRef] [PubMed]
- Garcés, J.-J.; Bretones, G.; Burgos, L.; Valdes-Mas, R.; Puig, N.; Cedena, M.-T.; Alignani, D.; Rodriguez, I.; Puente, D.Á.; Álvarez, M.-G.; et al. Circulating tumor cells for comprehensive and multiregional non-invasive genetic characterization of multiple myeloma. Leukemia 2020, 34, 3007–3018. [Google Scholar] [CrossRef]
- Rojas, E.A.; Corchete, L.A.; Mateos, M.V.; García-Sanz, R.; Misiewicz-Krzeminska, I.; Gutiérrez, N.C. Transcriptome analysis reveals significant differences between primary plasma cell leukemia and multiple myeloma even when sharing a similar genetic background. Blood Cancer J. 2019, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, M.F.; Hall, A.; Walker, K.; Sherborne, A.; De Tute, R.M.; Newnham, N.; Roberts, S.; Ingleson, E.; Bowles, K.; Garg, M.; et al. Daratumumab, Cyclophosphamide, Bortezomib, Lenalidomide, and Dexamethasone as Induction and Extended Consolidation Improves Outcome in Ultra-High-Risk Multiple Myeloma. J. Clin. Oncol. 2023, 41, 3945–3955. [Google Scholar] [CrossRef]
- Musto, P.; Simeon, V.; Martorelli, M.C.; Petrucci, M.T.; Cascavilla, N.; Di Raimondo, F.; Caravita, T.; Morabito, F.; Offidani, M.; Olivieri, A.; et al. Lenalidomide and low-dose dexamethasone for newly diagnosed primary plasma cell leukemia. Leukemia 2014, 28, 222–225. [Google Scholar] [CrossRef]
- Musto, P. Progress in the Treatment of Primary Plasma Cell Leukemia. J. Clin. Oncol. 2016, 34, 2082–2084. [Google Scholar] [CrossRef]
- Gundesen, M.T.; Lund, T.; Moeller, H.E.H.; Abildgaard, N. Plasma Cell Leukemia: Definition, Presentation, and Treatment. Curr. Oncol. Rep. 2019, 21, 8. [Google Scholar] [CrossRef]
- Saba, L.; Landau, K.S.; Liang, H.; Fu, C.-L.; Chaulagain, C.P. Real world analysis on the determinants of survival in primary plasma cell leukemia in the United States. Leukemia 2024, 38, 435–437. [Google Scholar] [CrossRef]
- Dhakal, B.; Patel, S.; Girnius, S.; Bachegowda, L.; Fraser, R.; Davila, O.; Kanate, A.S.; Assal, A.; Hanbali, A.; Bashey, A.; et al. Hematopoietic cell transplantation utilization and outcomes for primary plasma cell leukemia in the current era. Leukemia 2020, 34, 3338–3347. [Google Scholar] [CrossRef]
- Musto, P.; Wäsch, R. Plasma cell leukemia: Another piece of the puzzle. Haematologica 2023, 108, 941–944. [Google Scholar] [CrossRef] [PubMed]
- Tuazon, S.A.; Holmberg, L.A.; Nadeem, O.; Richardson, P.G. A clinical perspective on plasma cell leukemia; current status and future directions. Blood Cancer J. 2021, 11, 23. [Google Scholar] [CrossRef]
- Royer, B.; Minvielle, S.; Diouf, M.; Roussel, M.; Karlin, L.; Hulin, C.; Arnulf, B.; Macro, M.; Cailleres, S.; Brion, A.; et al. Bortezomib, Doxorubicin, Cyclophosphamide, Dexamethasone Induction Followed by Stem Cell Transplantation for Primary Plasma Cell Leukemia: A Prospective Phase II Study of the Intergroupe Francophone du Myélome. J. Clin. Oncol. 2016, 34, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Elmaleh, H.; Li, H.; Raza, S.; Mazzoni, S.; Samaras, C.J.; Faiman, B.M.; Khouri, J.; Valent, J.; Anwer, F.; Williams, L.S. Impact of T-Cell Redirecting Therapies on Survival Outcomes in Ultra-High-Risk Multiple Myeloma and Plasma Cell Leukemia. Blood 2024, 144, 7092. [Google Scholar] [CrossRef]
- Li, C.; Cao, W.; Que, Y.; Wang, Q.; Xiao, Y.; Gu, C.; Wang, D.; Wang, J.; Jiang, L.; Xu, H.; et al. A phase I study of anti-BCMA CAR T cell therapy in relapsed/refractory multiple myeloma and plasma cell leukemia. Clin. Transl. Med. 2021, 11, e346. [Google Scholar] [CrossRef]
- Bernardi, C.; Beauverd, Y.; Tran, T.A.; Maulini, M.; Mappoura, M.; Morin, S.; Simonetta, F.; Cairoli, A.; Auner, H.W.; Samii, K.; et al. Anti-BCMA and GPRC5D bispecific antibodies in relapsed/refractory primary plasma cell leukemia: A case report. Front. Immunol. 2024, 15, 1495233. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, S.; Yan, W.; Liu, Y.; Lyu, R.; Du, C.; Sui, W.; Qiu, L.-G.; An, G. A Phase 2 Study of CART-ASCT-CART2 Sandwich Regimen Treatment in Patients with Primary Plasma Cell Leukemia. Blood 2024, 144, 3389. [Google Scholar] [CrossRef]
- Moreau, P.; Chanan-Khan, A.A.; Roberts, A.W.; Agarwal, A.B.; Facon, T.; Kumar, S.; Touzeau, C.; Cordero, J.; Ross, J.; Munasinghe, W.; et al. Venetoclax Combined with Bortezomib and Dexamethasone for Patients with Relapsed/Refractory Multiple Myeloma. Blood 2016, 128, 975. [Google Scholar] [CrossRef]
- Kumar, S.; Vij, R.; Kaufman, J.L.; Mikhael, J.; Facon, T.; Pegourie, B.; Benboubker, L.; Gasparetto, C.; Amiot, M.; Moreau, P.; et al. Venetoclax Monotherapy for Relapsed/Refractory Multiple Myeloma: Safety and Efficacy Results from a Phase I Study. Blood 2016, 128, 488. [Google Scholar] [CrossRef]
- Jelinek, T.; Mihalyova, J.; Kascak, M.; Duras, J.; Popkova, T.; Benkova, K.; Richterova, P.; Plonkova, H.; Zuchnicka, J.; Broskevicova, L.; et al. Single-agent venetoclax induces MRD-negative response in relapsed primary plasma cell leukemia with t(11;14). Am. J. Hematol. 2019, 94, E35–E37. [Google Scholar] [CrossRef]
- Gonsalves, W.I.; Buadi, F.K.; Kumar, S.K. Combination therapy incorporating Bcl-2 inhibition with Venetoclax for the treatment of refractory primary plasma cell leukemia with t (11;14). Eur. J. Haematol. 2018, 100, 215–217. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Cytogenetic Abnormality | EMN12/HOVON129 n = 61, % | IFM n = 32, % | GIMEMA n = 23, % | ||
|---|---|---|---|---|---|
| Age, years | Overall (52–74) | 52–63 (n = 36) | 69–74 (n = 25) | 70 years old and younger | 44–80 |
| t(4;14) | 6 | 9 | 4 | 6 | 13 |
| t(14;16) | 11 | 19 | 4 | 16 | 30 |
| t(14;20) | N/A | N/A | N/A | N/A | 4 |
| t(11;14) | 32 | 26 | 55 | 50 | 30 |
| Hyperdiploidy | 8 | 13 | 17 | 9 | N/A |
| 1q ≥ 3 copies | 51 | 61 | 52 | 53 | 43 |
| del(1p) | 26 | 36 | 0 | 16 (homozygous deletion of CDKN2C) | N/A |
| del(13q) | 54 | 71 | 52 | 59 | 70 |
| del(17p) | 31 | 47 | 18 | 28 | 30 |
| del(17p), t(4;14), or t(14;16) | 40 | 59 | 27 | N/A | N/A |
| ≥2 high-risk cytogenetics (del(17p), t(4;14), or t(14;16)) | 24 | 34 | 14 | N/A | 38 (del(17p), t(4;14), t(14;16), t(14;20), 1q gain) |
| Clinical characteristics | |||||
| IgG | 40 | 31 | 56 | 37 | N/A |
| IgA | 11 | 19 | 0 | 12 | N/A |
| IgM | 1 | 0 | 4 | 2 | N/A |
| IgD | 6 | 6 | 8 | 5 | N/A |
| Light chain only | 39 | 44 | 32 | 32 | N/A |
| EMD other than PB | 14 | 17 | 12 | 12 | 8 |
| ISS stage 2 | 21 | 25 | 17 | 43 | 17 |
| ISS stage 3 | 65 | 64 | 74 | 43 | 61 |
| Study Name or Group (Years) | Patients | Treatments | Actual Transplant (s) (Number) | Results | |
|---|---|---|---|---|---|
| Response, % | Median PFS/OS (months) | ||||
| GIMEMA [46] (2009–2011) | N = 23 TE 15, TIE 8 | Induction Lenalidomide + dexamethasone | Single ASCT 4 Double ASCT 4 ASCT/RIC-allo 1 | Overall ORR 73 (CR 13, VGPR 26) | Transplant-eligible (n = 15) PFS/OS 21/not reached Transplant-ineligible (n = 8) PFS/OS 10/12 |
| IFM [53] (2010–2013) | N = 40 All patients were TE Median age 57 years (range, 27 to 71) | Induction Bortezomib + dexamethasone + doxorubicin/cyclophosphamide (PAD/VCd) | ASCT/RIC-allo 16 ASCT/ASCT 9 (One patient received a syngeneic transplant conditioned with high-dose melphalan.) | Overall ORR 69 (CR 10, VGPR 26) After induction (n = 39) ORR 69 (sCR 0, CR 10, VGPR 26, PR 23) Refractory disease 25, early death 5 in induction After 1st ASCT (n = 26) ORR 92 (sCR 4, CR 34, VGPR 38, PR 16, PD 8) | Overall PFS/OS 15/36 ASCT/ASCT (n = 7) (From the second transplant) PFS/OS not reached/not reached ASCT/RIC-allo (n = 17) (From the second transplant) PFS/OS 11/28 |
| EMN12/HOVON129 [34] (2015–2021) | N = 61 TE 36 TIE 25 | TE (n = 36) Induction 4 cycles of carfilzomib + lenalidomide + dexamethasone (KRd) Consolidation 4 cycles of KRd in ASCT/ASCT Maintenance KR in ASCT/ASCT and ASCT/RIC-allo (until PD) | 1st ASCT 24 ASCT/ASCT 12 ASCT/RIC-allo 5 | After induction (n = 36) ORR 83 (sCR 3, CR 11, VGPR 61, PR 8, PD 8) After 1st ASCT (n = 24) ORR 96 (sCR 13, CR 21, VGPR 63, PD 1) After 2nd ASCT (n = 12) ORR 100 (sCR 8, CR 17, VGPR 75) After RIC-allo (n = 5) ORR 100 (sCR 40, CR 40, VGPR 20) | Overall cohort of TE PFS/OS 15/28 (Early mortality rate 8% at 6 months) After 1st ASCT (n = 24) PFS/OS 26/not reached (From date of first ASCT) After 2nd ASCT (n = 12) PFS/OS at 2 years 58%/82% (From the second transplant) After RIC-allo (n = 5) PFS/OS at 2 years 60%/53% (From the second transplant) |
| TIE (n = 25) Induction 8 cycles of carfilzomib + lenalidomide + dexamethasone (KRd) Maintenance KR until PD | None | After induction cycle 1–4 (n = 25) ORR 80 (sCR 12, CR 12, VGPR 44, PR 12); 2 early death, 2 excessive toxicity After induction cycle 5–8 (n = 19) ORR 95 (sCR 21, CR 21, VGPR 47, PR 5, PD 1) | PFS/OS 13/24 (Early mortality rate 16% at 6 months) | ||
| NCT Number | Study Title | Candidates and Targets of CAR-T | Study Phase and Treatment Flow | Brief Summary | Comments |
|---|---|---|---|---|---|
| NCT05979363 | A Study of Bortezomib, Lenalidomide and Dexamethasone (VRd) Followed by BCMA CAR-T Therapy in Transplant-Ineligible Patients With Primary Plasma Cell Leukemia | Newly diagnosed pPCL Anti-BCMA CAR-T Cells | Phase 2 VRd x3→CAR-T →VR x3 →VR maintenance | A single-arm, open-label study to evaluate the efficacy and safety of VRD-based regimen combined with BCMA CAR-T in transplant-ineligible patients with primary plasma cell leukemia. | CART without ASCT in patients (age ≤ 75 years old). |
| NCT05870917 | A Study of VRD-based Regimen Combined With CART-ASCT-CART2 Treatment in Patients With Primary Plasma Cell Leukemia | Newly diagnosed pPCL Anti-BCMA CAR-T Cells | Phase 2 VRd x3→1st CAR-T →VR x3→2nd CAR-T →R maintenance | A single-arm, open-label study to evaluate the efficacy and safety of a VRD-based regimen (Bortezomib, Lenalidomide, and Dexamethasone) combined with CART-ASCT-CART2 in Chinese patients with newly diagnosed primary plasma cell leukemia. | CART-ASCT-CART2 sandwich regimen in transplant-eligible patients (age ≤ 65 years old). [57] |
| NCT05838131 | A Clinical Trial to Explore the Safety and Efficacy of CT071 Injection in Patients With Relapsed/Refractory Multiple Myeloma or Primary Plasma Cell Leukemia | Relapsed/refractory MM and relapsed/refractory pPCL Anti-GPRC5D CAR-T Cells (CT071) | Phase 1/2 study | A single-arm, open-label, dose-finding, first-in-human clinical trial. The main aim of this study is to preliminarily evaluate the safety and tolerability of CT071 after infusion, and explore the dose range of CT071 in patients with relapsed/refractory multiple myeloma or primary plasma cell leukemia, so as to determine the possible recommended therapeutic dose (RD). | Shortened manufacturing process to around 30 h in vein-to-vein time. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanamura, I.; Karnan, S.; Ota, A.; Takami, A. Primary Plasma Cell Leukemia: Recent Advances in Molecular Understanding and Treatment Approaches. Int. J. Mol. Sci. 2025, 26, 6166. https://doi.org/10.3390/ijms26136166
Hanamura I, Karnan S, Ota A, Takami A. Primary Plasma Cell Leukemia: Recent Advances in Molecular Understanding and Treatment Approaches. International Journal of Molecular Sciences. 2025; 26(13):6166. https://doi.org/10.3390/ijms26136166
Chicago/Turabian StyleHanamura, Ichiro, Sivasundaram Karnan, Akinobu Ota, and Akiyoshi Takami. 2025. "Primary Plasma Cell Leukemia: Recent Advances in Molecular Understanding and Treatment Approaches" International Journal of Molecular Sciences 26, no. 13: 6166. https://doi.org/10.3390/ijms26136166
APA StyleHanamura, I., Karnan, S., Ota, A., & Takami, A. (2025). Primary Plasma Cell Leukemia: Recent Advances in Molecular Understanding and Treatment Approaches. International Journal of Molecular Sciences, 26(13), 6166. https://doi.org/10.3390/ijms26136166