Abstract
T-cadherin (CDH13) is an atypical, glycosyl-phosphatidylinositol-anchored cadherin with functions ranging from axon guidance and vascular patterning to adipokine signaling and cell-fate specification. Originally identified as a homophilic cue for migrating neural crest cells, projecting axons, and growing blood vessels, it later emerged as a dual metabolic receptor for cardioprotective high-molecular-weight adiponectin and atherogenic low-density lipoproteins. We recently showed that mesenchymal stem/stromal cells lacking T-cadherin are predisposed to adipogenesis, underscoring its role in lineage choice. Emerging evidence indicates that CDH13 expression and function are fine-tuned by non-coding RNAs (ncRNAs). MiR-199b-5p, miR-377-3p, miR-23a/27a/24-2, and the miR-142 family directly bind CDH13 3′-UTR or its epigenetic regulators, affecting transcription or accelerating decay. Long non-coding RNAs (lncRNAs), including antisense transcripts CDH13-AS1/AS2, brain-restricted FEDORA, and context-dependent LINC00707 and UPAT, either sponge these miRNAs or recruit DNMT/TET enzymes to the CDH13 promoter. Circular RNAs (circRNAs), i.e.circCDH13 and circ_0000119, can add a third level of complexity by sequestering miRNA repressors or boosting DNMT1. Collectively, this ncRNA circuitry regulates T-cadherin across cardiovascular, metabolic, oncogenic, and neurodegenerative conditions. This review integrates both experimentally validated data and in silico predictions to map the ncRNA-CDH13 crosstalk between health and disease, opening new avenues for biomarker discovery and RNA-based therapeutics.
1. Introduction
T-cadherin (also named cadherin-13, H-cadherin) is encoded by the CDH13 gene. The CDH13 gene is highly conserved across species, underscoring its evolutionary importance. Human T-cadherin was initially mapped to chromosome 16q24 [1], yet its location was later refined to 16q24.2 [2]. In humans, the CDH13 gene consists of 1,169,627 base pairs and comprises 14 exons. It produces a 3711-base pair transcript that encodes a 713-amino acid pre-proprotein. This pre-protein features a carboxy-terminal glycosylphosphatidylinositol (GPI)-tethered protein comprising a pro-peptide sequence and five extracellular calcium-binding domains (EC1–EC5) [3,4].
T-cadherin belongs to the cadherin superfamily, whose intricate structural diversity underpins an array of various biological functions [5]. “Classical” cadherins are well-known for mediating cell–cell adhesion, typically through homophilic binding between cadherin molecules. The extracellular domains of “classical” cadherins, which are transmembrane glycoproteins, are characterized by the presence of five extracellular Ca2+-binding domains, transmembranes, and extracellular parts. Stable cell–cell adhesion in organs and tissues results from interactions between cadherin cytoplasmic domains, catenins, and actin cytoskeleton [6]. Yet, even E-cadherin, the first discovered member of the cadherin superfamily, is now considered far more than a simple molecular “glue”, as it has been linked to various signaling cascades that drive embryonic development, tissue morphogenesis, and homeostasis [6].
T-cadherin is a non-canonical member of the cadherin superfamily. Although T-cadherin possesses a hallmark extracellular part of “classical” cadherins (five Ca2+-binding domains), it lacks both transmembrane and cytoplasmic parts, and is tethered to the plasma membrane via a glycosyl-phosphatidylinositol (GPI) anchor. Moreover, T-cadherin lacks the signature His-Ala-Val (HAV) motif that mediates the homophilic binding of “classical” cadherins, underscoring its distinctive structural and functional profile [6]. T-cadherin ensures only weak homophilic binding and provides negative guidance cues in the developing nervous system, as well as in physiological angiogenesis and tumor-driven neovascularization [7]. This GPI anchor endows T-cadherin with rapid lateral mobility within the plasma membrane and transient interactions with diverse signaling partners [5,6,7].
Mounting evidence indicates that T-cadherin participates in multiple intracellular signaling cascades [3]. Consistent with this, T-cadherin is involved in several intracellular signaling pathways that regulate apoptosis, proliferation, differentiation, migration, and tissue regeneration [3]. Essential T-cadherin-dependent signaling pathways have been identified, including Grp78/BiP/integrin-β3 [8] and PI3K/mTOR [9], which trigger pro-survival signaling and promote endothelial cell proliferation and migration. T-cadherin contributes to angiogenesis, modulating endothelial cell polarization and promoting de-adhesion via homophilic T-cadherin-mediated interaction and activation of RhoA-ROCK and Rac signaling [3]. Furthermore, it suppresses endothelial cell migration, capillary sprouting, and capillary-like tube formation in in vitro, ex vivo, and in vivo models. Under oxidative stress conditions, the overexpression of T-cadherin in cultured endothelial cells protects them from apoptosis [10].
Over the past three decades, multiple studies have underscored the relevance of T-cadherin in tumor biology. CDH13 often restrains cancer cell proliferation, invasion, and overall tumor growth, yet paradoxically supports tumor progression by promoting neovascularization [3].
Many biological functions are mediated through T-cadherin, which serves as a receptor for two metabolically important ligands, adiponectin and LDL, which may compete for receptor binding in both physiological and pathological contexts [7]. The interaction between adiponectin and T-cadherin is critically important for localizing adiponectin within organs and tissues, thereby facilitating adiponectin’s protective roles against vascular atherosclerosis and myocardial ischemia [11,12,13]. Tanaka et al. demonstrated that adiponectin promotes muscle regeneration specifically through binding to T-cadherin [14]. In addition, Obata et al. reported that the adiponectin/T-cadherin system enhances exosome biogenesis and secretion, leading to a decrease in cellular ceramide levels in cultured endothelial cells and in the aorta in vivo [15]. Plasma adiponectin levels can dramatically decrease in metabolic syndrome, while elevated LDL may bind to T-cadherin, correlating with the LDL-mediated atherogenic impact on the cardiovascular system [16]. We recently demonstrated that mesenchymal stem/stromal cells lacking full-length T-cadherin spontaneously undergo adipogenic differentiation and produce adipocytes with conspicuously large lipid droplets, a propensity that persists in adipogenic conditions [17]. In ligand-comparison experiments, we found that the absence of T-cadherin rendered cells more susceptible to adiponectin’s suppressive effects and LDL’s stimulatory effects on adipogenesis, suggesting that T-cadherin may serve as an important metabolic sensor [17].
The atypical molecular structure of T-cadherin raises compelling questions about how T-cadherin transmits signals and orchestrates such a broad range of biological functions, with the underlying mechanisms likely extending far beyond simple protein–protein interactions between T-cadherin and other membrane receptors or signaling molecules. One promising area of investigation involves non-coding RNAs (ncRNAs), which may regulate the expression and activity of both the T-cadherin protein and its mRNA, as well as the functional activity of its targets and interaction networks. Uncovering the mechanisms governing T-cadherin expression and CDH13 gene activity offers valuable insights into T-cadherin’s involvement in a wide range of physiological and pathological processes.
Thus, the present review integrates growing evidence that microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs) form an intricate, multilayered network converging on the atypical member of the cadherin superfamily, CDH13. We outline how these ncRNAs govern CDH13/T-cadherin, from promoter methylation and mRNA stability to ligand presentation, and illustrate that their dysregulation is manifested through cardiovascular, metabolic, oncogenic, and neurological pathways. By assembling both experimentally validated findings and bioinformatically predicted interactions, we highlight the ncRNA/CDH13 axis as a nexus and a promising source of new biomarkers and therapeutic targets.
2. T-Cadherin/CDH13 Expression Landscapes in Development, Adult Tissues, and Disease
T-cadherin is most prominently expressed in the nervous and cardiovascular systems [3,7,18]. Its expression pattern changes across embryogenesis and in adults, both in health and disease.
T-cadherin is among the earliest cadherins detected in the developing nervous system (E8.5–9.5 in mouse) and nascent vasculature (≈E11.5) [19]. Being differentially expressed in somite sclerotomes, T-cadherin acts as a negative guidance cue defining pathways for migrating neural crest cells and growing axons of motor neurons, as well as ensuring precise synapse formation. In adults, T-cadherin remains widespread throughout the central nervous system (CNS), being robust in the human adult cerebral cortex, medulla, thalamus, and midbrain, but absent from the spinal cord. T-cadherin is expressed at levels even higher than in the developing brain (cerebral cortex, medulla oblongata, and inferior olivary nucleus), but not in the spinal cord, with its expression being even higher than in the developing brain [20].
During cardiovascular development, T-cadherin/CDH13 localizes to the forming vascular plexus, coincident with intense vessel sprouting [19]. In adult tissues, T-cadherin is highly enriched in the heart and large arteries (aorta, carotid, iliac, and renal arteries), specifically detected in cardiomyocytes, endothelial cells, vascular smooth muscle cells (VSMCs), and perivascular cells [21]. T-cadherin expression rises further in endothelial cells, VSMCs, and pericytes within atherosclerotic plaques and after arterial injury, particularly during neointima formation [22]. In contrast, T-cadherin is modestly expressed in the lungs, skeletal muscle, and kidneys, and is virtually absent in the liver or pancreas [21].
In the skin, strong T-cadherin expression in the basal layer of keratinocytes is lost upon malignant transformation, paralleling its downregulation observed in many carcinomas (breast, lung, and cutaneous squamous cell carcinomas), pituitary adenomas, B-cell lymphomas, and nasopharyngeal carcinoma [23].
T-cadherin is abundantly present in the basal layer of keratinocytes in normal skin and its expression is lost upon malignant transformation as well as in numerous human cancers such as breast and lung carcinomas, cutaneous squamous cell carcinomas, pituitary adenomas, malignant B cell lymphomas and nasopharyngeal carcinoma, osteosarcoma, ovarian cancer and endometrial cancer, and gallbladder cancer [3,18]. Conversely, certain mesenchymal and neural malignancies (osteosarcoma, NF1-deficient astrocytoma, and subsets of hepatocellular carcinoma) retain or even upregulate T-cadherin [3].
Compared to the nervous and cardiovascular systems, adipose tissue displays lower overall T-cadherin levels, yet it is clearly present in the stromal-vascular fraction, notably in mesenchymal stem/stromal cells and adipocyte progenitors, suggesting its role in cellular self-renewal and tissue homeostasis [7]. Table 1 summarizes T-cadherin’s key structural features, its tissue-specific expression, and functional roles in development and adulthood, both in health and disease.

Table 1.
Structural and functional features of T-cadherin.
3. Non-Coding RNAs: A General Overview
Non-coding RNAs (ncRNAs) are a novel class of RNA molecules that do not encode proteins. The non-coding transcripts are now recognized for their critical roles in epigenetic regulation, including gene silencing, DNA methylation, chromatin remodeling, and gene regulation. NcRNAs regulate a wide range of biological processes, making them promising therapeutic targets as well as biomarkers [42,43]. Dysregulated expression of ncRNAs is associated with various pathological conditions, including cancer.
Regulatory non-coding RNAs are generally classified into two main categories: small non-coding RNAs (sncRNAs) (less than 200 nucleotides) and long non-coding RNAs (lncRNAs) (exceeding 200 nucleotides). In the present review, we narrow our focus down to three major ncRNA categories: miRNAs, lncRNAs, and circRNAs, with a particular focus laid on T-cadherin/(CDH13) expression and function [43,44].
NcRNAs can originate from both intronic and exonic regions of protein-coding and non-coding genes, as well as from intergenic areas, transposable elements, and gene promoter regions [44,45,46]. Some lncRNAs are transcribed from their dedicated promoters, while others are synthesized from introns of the neighboring protein-coding loci [47,48]. CircRNAs are derived from back-splicing of lncRNAs of pre-mRNA [49]. In most cases, sncRNAs are excised from longer RNA transcripts by RNAase III-family nucleases, whose double-stranded RNA-binding and catalytic domains guide and cleave the substrate [50]. However, approximately one-third of intronic miRNAs possess independent promoters and can be transcribed separately from their host genes [51].
MiRNAs are short, single-stranded RNA molecules that regulate gene expression by binding to target mRNAs at their 3′ untranslated regions (3′-UTRs) to suppress stable translation [52]. MiRNAs maintain tissue homeostasis and support the differentiated state of cells in various organs and tissues [53]. Additionally, they participate in intercellular communication as components of exosomes, microvesicles, apoptotic bodies, lipoproteins, and ribonucleoproteins [54].
The biological functions of lncRNAs include the regulation of the chromatin architecture, transcription, splicing (particularly by antisense lncRNAs), translation, protein localization, and various RNA processing events, as well as RNA editing, localization, and stability [55]. Numerous lncRNAs orchestrate cell differentiation and development in both animals and plants. They are also involved in regulating critical biological processes, including the p53-mediated DNA damage response, cytokine expression, endotoxic shock, inflammation, neuropathic pain, and cholesterol biosynthesis, to name just a few [45].
Circular RNAs (circRNAs) are long, non-coding endogenous RNA molecules with a covalently closed continuous loop structure that lacks both a 5′ cap and a 3′ poly(A) tail [56]. CircRNAs function as molecular sponges for miRNAs, sequestering them and thereby preventing the degradation of their target messenger RNAs. Emerging evidence also suggests that circRNAs can exert both positive and negative regulatory effects on the transcription of their parental genes [57,58,59]. CircRNAs have been shown to regulate gene expression by interacting with the RNA polymerase II (Pol II) complex in the nucleus or by directly interacting with RNA-binding proteins in the cytoplasm, forming RNA–protein complexes that modulate mRNA transcription. Additionally, circRNAs can suppress gene expression by competing with the pre-mRNA splicing machinery, functioning as “mRNA traps” that sequester translation initiation sites or compromise the integrity of mature linear RNAs, ultimately generating fragmented, non-translatable transcripts or inactive protein products. These mechanisms collectively contribute to the reduced expression of target proteins [60].
Recent studies highlight the growing interest in non-coding RNAs (ncRNAs) encoded within intronic or exonic regions of genes, as these molecules may regulate cellular processes through mechanisms independent of the proteins encoded by the same loci [61]. Despite the well-characterized functions of the T-cadherin protein in health and disease, the mechanisms underlying its regulation and the functional roles of the gene locus remain relatively unexplored. The CDH13 gene also contains non-coding RNAs within its structure, which will be discussed in detail in the following sections.
4. Non-Coding RNAs Expressed from the CDH13 Gene
The miRBase reference database includes numerous human pre-miRNAs; approximately half are derived from intergenic non-coding pri-miRNA transcripts, while the remaining are excised from introns of protein-coding transcripts [62]. A small fraction (about 6%) of human mature miRNAs, annotated in miRbase, originates from multiple pre-miRNAs encoded at different genomic loci. The precise location of promoters has not yet been mapped for most miRNA genes. Some miRNAs reside within the introns of protein-coding genes and therefore share the host gene’s promoter. However, it has been shown that miRNA genes frequently have multiple transcription start sites [63], and the promoters of intronic miRNAs may sometimes differ from those of their host genes [64].
The CDH13 gene contains transcripts of several miRNAs. One of them, miR-3182, is located within an intron of the CDH13 gene (chr16:83,508,346-83,508,408) [65]. This miRNA functionally interacts with its targets in a tissue-specific manner, including targets that are also associated with the CDH13 gene itself. These interactions were examined using the network-wide association study (NetWAS), which evaluates the functional association between gene pairs based on tissue-specific posterior probabilities. This analysis identified 18 targets of intronic miR-3182, which interact with the CDH13 gene in the cardiac muscle network, and received high NetWAS scores. Among them, in cardiac muscle and endothelial cells, are genes such as SUB1, CCND2, ARHGEF37, MXRA7, DEFB107A, DAB2IP, and MAGEB10 expressed in cardiac muscle and endothelial cells [66]. While this bioinformatic analysis provides new insight into the regulatory network of the cardiovascular-associated pathways, it still requires further experimental verification.
Gene set enrichment analysis (GSEA) further revealed that the targets of miR-3182 are enriched in biological processes related to modulating the frequency, rate, or strength of cardiomyocyte contraction. These associations point to a broad range of cardiovascular conditions, including cardiac hypertrophy, heart failure, angiogenesis, and endothelial dysfunction, where RAS (rat sarcoma family of small GTP-binding proteins) signaling is central to pathogenesis [67,68].
MiR-3182 is not the only miRNA expressed from the CDH13 gene. A novel serum miRNA in sepsis patients with varying clinical outcomes was discovered and characterized to be the previously unannotated miR-8058. This miRNA was identified through bioinformatic analysis and subsequently validated by RT-PCR. Although the functions and targets of miR-8058 remain unknown, it was suggested that several miRNAs identified in the study, including miR-8058, may contribute to the pathogenesis of sepsis and hold promise as prognostic biomarkers for diagnostic applications [69,70]. The genomic location of miR-8058 was determined using bioinformatic mapping [71].
In addition to miRNAs, the CDH13 gene also gives rise to lncRNAs. In search of molecular mechanisms underlying sex-related differences in the frequency of depression, Issler et al. examined the lncRNA expression patterns in several limbic brain regions. They highlighted one human-specific lncRNA, RP11-298D21.1, which was named FEDORA, and which was differentially expressed across females and males [72]. FEDORA is an antisense lncRNA transcribed from the CDH13 locus. FEDORA was reported to modulate synaptic plasticity by promoting an increased myelin thickness in oligodendrocytes. While its expression may also be altered in neurons, the mechanisms underlying this regulation remain unclear.
Gene ontology analysis revealed the opposing regulatory patterns in Neuro-FEDORA and Oligo-FEDORA transcriptional profiles, highlighting divergent patterns of expression: high in oligodendrocytes and low in neurons. In human studies, FEDORA was significantly upregulated in the cortical brain regions of women, but not men, diagnosed with major depressive disorder, as compared to healthy controls in two independent cohorts. In a mouse model, FEDORA was shown to induce depression-like behavioral abnormalities, yet exclusively in female mice, associated with cell-type-specific changes in synaptic function, myelination, and gene expression. The authors proposed that FEDORA may exert these effects, in part, through its host gene Cdh13, which contributes to divergent gene regulatory patterns across different neural cell types [72].
Another lncRNA, named RP11-543N12.1 and also known as CDH13-AS1 (CDH13 antisense RNA 1), was shown to be transcribed from the CDH13 gene [73]. LncRNAs can compete with endogenous miRNAs for common miRNA-binding sites on target mRNAs, thereby relieving miRNA-mediated repression of gene expression [74]. This competitive mechanism was demonstrated for CDH13-AS1 and miR-324-3p, further supported by predictions from the miRDB4.0 and PITA databases, and followed by validation through dual-luciferase reporter assays. The analysis showed that miR-324-3p is a direct target of CDH13-AS1, and its expression was significantly upregulated when CDH13-AS1 was directly bound to the 3′ untranslated region (3′-UTR) of miR-324-3p. Moreover, the authors demonstrated that CDH13-AS1 targeted miR-324-3p to suppress proliferation and promote apoptosis in an Alzheimer’s disease cell model in vitro, suggesting that CDH13-AS1 and miR-324-3p may be potential biomarkers and therapeutic targets for Alzheimer’s disease. A similar effect was observed in SH-SY5Y cells: upregulated CDH13-AS1 and miR-324-3p led to downregulated proliferation and induced apoptosis. Conversely, silencing CDH13-AS1 and miR-324-3p increased proliferation and suppressed apoptosis in these cells [73].
Towards that end, miR-324-3p is considered a multifunctional miRNA involved in the proliferation and apoptosis of nasopharyngeal carcinoma cells, and its low expression was associated with poor overall survival and recurrence-free survival of patients. It was demonstrated that miR-324-3p can modulate the growth and survival of nasopharyngeal carcinoma cells by upregulating SMAD7 expression [75].
Another illuminating example involves CDH13-AS2, an lncRNA transcribed from the CDH13 gene, which targets its own mRNA. CDH13-AS2 was predicted to bind the 3′-UTR of CDH13 using LncRRIsearch and RNAup servers. This binding interaction was further validated by dCas13-mediated RNA immunoprecipitation. Weighted correlation network analysis (WGCNA) of transcriptomic data from patients with CAD revealed a positive correlation between the expression of the CDH13 gene and its antisense lncRNA CDH13-AS2 in arterial tissues. Functional validation using CRISPR/Cas9-mediated knockout of either CDH13 or CDH13-AS2 in human umbilical vein endothelial cells (HUVECs) resulted in synergistically pro-atherogenic effects, including impaired proliferation and migration, increased apoptosis, and enhanced monocyte adhesion. These findings were consistent with the observed downregulation of CDH13 in arterial tissues from patients with atherosclerosis. Accordingly, these in vitro data show that Cdh13 and Apoe double-knockout mice (Cdh13-/-/Apoe-/-) exhibit increased aortic lesions under a high-fat diet compared to Apoe-/- controls. Target prediction tools (TargetScan, miRWalk, and scanMiRApp) identified several endothelial-enriched miRNAs, including miR-let-7, miR-30, and miR-125, with predicted binding sites in the 3′UTR of CDH13, some of which overlap with disease-associated variants. A dual luciferase RNAi assay confirmed the direct binding of miR-let-7, while CRISPR/dCas9-mediated activation of CDH13-AS2 reduced this binding and increased CDH13 expression [76].
Interestingly, the CDH13 gene also harbors a circular RNA known as CircCDH13 (hsa_circ_0040646) [77], which is formed through back-splicing of exons 9 and 10 of the CDH13 gene [78]. CircCDH13 was found to be significantly upregulated in chondrocytes of osteoarthritic cartilage and contributed to the disease progression by promoting chondrocyte apoptosis, enhancing extracellular matrix (ECM) catabolism, and suppressing ECM anabolism. In vitro and in vivo experiments, employing gain-of-function and loss-of-function approaches, including RNA pulldown, luciferase assays, and adeno-associated virus-mediated delivery of CircCDH13 in a mouse model of destabilization in medial meniscus-induced osteoarthritis, defined the important role of CircCDH13 in osteoarthritis. Mechanistically, CircCDH13 promotes osteoarthritis progression by acting as a molecular sponge for miR-296-3p, thereby modulating the miR-296-3p-PTEN signaling axis. Specifically, CircCDH13 overexpression enhanced apoptosis through the upregulation of PTEN signaling, which is mediated by miR-296-3p sequestration.
In addition to chondrocytes from osteoarthritic tissue, the elevated expression of CircCDH13 was detected in chondrocytes treated with IL-1β and TNF-α, ultimately linking CircCDH13 expression and function to inflammation [78]. CircCDH13 knockdown resulted in the suppression of matrix metalloproteinase 13 (MMP13) and ADAMTS5 (a disintegrin and metalloproteinase with thrombospondin motifs 5), along with the induction of collagen type II alpha 1 (COL2A1) and aggrecan. Overall, silencing CircCDH13 alleviated osteoarthritis symptoms and restored cartilage homeostasis. These findings highlight CircCDH13 as a critical regulator of osteoarthritis pathogenesis and a promising therapeutic target [77].
Non-coding RNAs transcribed from the CDH13 gene (Figure 1) may contribute to its own regulation and affect the related signaling pathways. However, CDH13 expression is governed not only by internal regulatory elements but also by a complex interplay of external factors. Interactions between CDH13 and a whole range of ncRNAs (miRNAs, lncRNAs, and circRNAs), originating from both the CDH13 locus and other genomic regions, form an intricate regulatory network capable of modulating CDH13 activity in both enhancing and suppressive directions. Acting in concert, these ncRNAs orchestrate context-dependent modulation of CDH13/T-cadherin across cardiovascular, metabolic, neurological, and inflammatory settings.
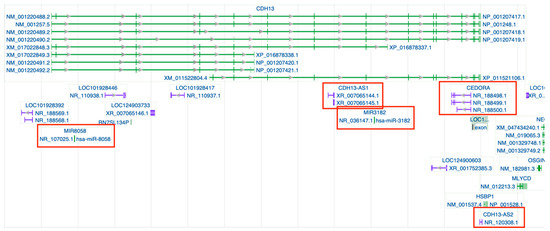
Figure 1.
Genomic regions and transcripts of CDH13 indexed in the NCBI Gene ID database https://www.ncbi.nlm.nih.gov/gene/1012#top (accessed on 26 May 2025). Gray arrows within introns mark splice direction. Gene transcripts are shown in green, whereas non-coding RNA loci appear in purple; those highlighted in red are the specific ncRNAs discussed in this review. A complete graphical key is available in the NCBI legend (https://www.ncbi.nlm.nih.gov/tools/sviewer/legends/) (accessed on 26 May 2025).
In the following sections, we explore the non-coding RNAs that directly regulate CDH13 gene expression, elucidate their mechanisms of action, and unveil the biological implications of these regulatory interactions.
5. Regulation of CDH13/T-Cadherin Expression via Non-Coding RNAs
T-cadherin expression is orchestrated by multiple factors ranging from transcriptional and post-transcriptional regulation to epigenetic control. Earlier studies demonstrated the elaborate regulation of T-cadherin by growth factors and hormones. Specifically, estradiol, progesterone, EGF, and dexamethasone added to the culture medium of human osteosarcoma cells significantly increased T-cadherin levels, as was shown by Western blotting and qRT-PCR assays [79]. In contrast, in our lab, we demonstrated that EGF, FGF-2, PDGF, and IGF-1 downregulated T-cadherin expression in smooth muscle cells in vitro, while 8-Br-cGMP, forskolin, and TGFβ increased T-cadherin [80,81].
Epigenetic control emerges to play an important role in CDH13 regulation: hypermethylation of the CDH13 promoter correlated with CDH13 silencing and oncogenic transformation across many tumor types [18,82,83,84]. Direct regulation of CDH13 expression by transcription factors was reported previously for melanoma cells. Bioinformatic analysis with Genomatix revealed consensus binding sequences for BRN2 (a member of the POU family of transcription factors) in the proximal promoter region of CDH13. BRN2 is present in normal melanocytes, where it coordinates complex transcriptional events required for normal melanocytic development and homeostasis. Yet, BRN2 expression was elevated in melanoma tissue samples and melanoma cell lines, correlating with the enhanced invasive and metastatic behavior. Ectopic BRN2 expression in BRN2-deficient/T-cadherin-positive melanoma cells led to suppression of CDH13 promoter activity, whereas BRN2 knockdown in BRN2-positive/T-cadherin-negative cells resulted in re-expression of CDH13 mRNA and protein [85].
Recent studies revealed that the expression of CDH13 can be regulated at a post-translational level, specifically by miRNAs. A prominent example is miR-199b-5p, an intragenic miRNA encoded in the dynamin 1 (DNM1) locus on chromosome 9. The mature 5p strand encoded by hsa-miR-199b is highly conserved across multiple species, and bioinformatic analyses predicted that its transcription is independent of its host DNM1 gene [86]. In gain-of-function assays, transfecting HL-1 cardiomyocytes with a miR-199b mimic markedly suppressed CDH13 mRNA levels [87].
Suppression of miR-199b was reported in several types of cancer and predicted poor prognosis in hepatocarcinoma, where it directly targets HIF-1α, in breast cancer, where it targets HER2, and in prostate cancer [88,89,90]. The downregulation of miR-199b is strongly linked to imatinib resistance in chronic myeloid leukemia (CML) patients [91], while the loss of miR-199b expression contributes to the acquired chemoresistance in ovarian cancer [92]. Conversely, overexpression of miR-199b-5p in medulloblastoma stem cells depletes CD133+ tumor-initiating cells by repressing Notch signaling [93].
Clinically, low miR-199b correlates with myeloproliferation and poor overall survival in acute myeloid leukemia (AML), and its expression can be restored by histone deacetylases (HDAC) inhibitors, promoting cancer cell apoptosis [86]. Although none of the cited studies directly explored a connection between hsa-miR-199b and CDH13 expression, T-cadherin itself is widely viewed as a tumor suppressor in numerous malignancies (Table 1) [3,18,26]. Conversely, in other cancers, T-cadherin upregulated expression has been associated with elevated tumor aggressiveness, pronounced tumor vascularization, and an increased propensity for metastasis [7,94]. Clarifying whether miR-199b regulates CDH13 expression during oncogenesis and whether manipulating this miRNA could yield therapeutic benefit represents an intriguing direction for future cancer research.
The pertinent literature indicates that the adiponectin–T-cadherin axis is prominent for adiponectin-mediated effects in skeletal muscles and the cardiovascular system. Adiponectin, a unique adipokine primarily produced by white adipocytes, exerts numerous protective effects: it improves insulin sensitivity, reduces inflammation and cell proliferation, and suppresses both atherogenesis and tumorigenesis [95,96]. T-cadherin operates as a specific receptor for high molecular weight (HMW) adiponectin, the most metabolically active form of adiponectin [11]. Experimental studies demonstrated that T-cadherin sequesters circulating adiponectin to the cardiac and skeletal muscle tissues. T-cadherin expression is a prerequisite for adiponectin’s protective effects, as demonstrated in mouse models of cardiac hypertrophy and ischemia-reperfusion injury [12,24,25]. In T-cadherin-deficient mice, adiponectin was dramatically elevated in the bloodstream, but failed to associate with cardiomyocytes, and these animals sustained infarct sizes after ischemia-reperfusion comparable to those in adiponectin-knockout mice. Adenoviral delivery of adiponectin rescued the cardiac phenotype in adiponectin-knockout mice, yet with no effect in T-cadherin-knockout mice, underscoring the essential role of T-cadherin in adiponectin-mediated cardioprotection [12,24]. A similar scenario has been reported for skeletal muscles [24].
Towards this end, decreased T-cadherin levels were demonstrated in human myocardium in non-ischemic dilated cardiomyopathy (NI-DCM) and chronic heart failure (HF) [97]. While systemic levels of adiponectin were high in these patients, adiponectin failed to exert protective effects, most probably due to the diminished T-cadherin content, which is an additional indicator of HF severity. Beyond sarcomeric proteins, in silico analyses indicated that the adiponectin axis, comprising adiponectin and its receptors, is a putative target for several miRNAs, including miR-199b-5p. Transfecting HL-1 cells (muscle cell line) with a synthetic miR-199b-5p mimic further confirmed this interaction, producing a noticeable reduction in T-cadherin expression [87].
Several independent research groups have shown that miR-377-3p accelerates cell-cycle progression, promoting proliferation, whereas suppressing miR-377-3p results in cell-cycle arrest and apoptosis. For example, in human hepatocellular carcinoma cell lines (Hep3B, HepG2, LM3, Li-7, and HuH-7), miR-377-3p was shown to suppress the transcription factor EGR1, thereby attenuating p53 activation [98]. Interestingly, CDH13 was identified as a direct target of miR-377-3p in both colorectal cancer and an Alzheimer’s disease model, resulting in its downregulated expression [99,100].
Of note, adiponectin has attracted considerable attention in oncology, as low circulating levels of this adipokine correlate with a high incidence and poor prognosis of several types of cancer. This association is particularly strong for digestive tract malignancies, such as esophageal adenocarcinoma, gastric, liver, and colorectal cancers [101,102,103,104,105,106]. Studies in colorectal cancer cell lines reinforce this link: in HCT116, HT29, and LoVo, adiponectin suppressed proliferation, migration, and clonogenicity [103,104]. It was suggested that adiponectin may inhibit proliferation, migration, and clonogenicity by modulating cell metabolism as well as cell cycle progression [105].
With respect to miR-377-3p, bioinformatic screening performed by Di Palo et al. predicted that this miRNA targets the 3′-UTR of T-cadherin mRNA in colorectal adenocarcinoma (HT-29 and Caco-2) cell lines, downregulating its expression [99]. A dual-luciferase reporter assay confirmed the direct binding, and gain- or loss-of-function experiments showed that a miR-377 mimic markedly reduced T-cadherin protein level, whereas a miR-377 inhibitor restored its expression. An analysis of TCGA data via the ENCORI platform further revealed an inverse correlation between miR-377 and CDH13 expression in colorectal cancer samples. Functionally, miR-377-3p was demonstrated to accelerate G1-S transition, enhance migration and invasion, and drive epithelial–mesenchymal transition in colorectal cancer through GSK-3β upregulation and NF-κB activation [107]. Together, these data further underscore a miR-377/T-cadherin/adiponectin axis that may contribute to colorectal carcinogenesis and offer a potential avenue for biomarker discovery and therapeutic intervention aimed at restoring T-cadherin expression. Reinforcing this link, miR-377 has recently been shown to aggravate adipose-tissue inflammation and insulin resistance, in which the adiponectin–T-cadherin pathway is similarly dysregulated [108].
With regard to tumors originating from the CNS, T-cadherin was shown to function as a negative regulator of cell proliferation. While T-cadherin expression was elevated during physiological growth arrest of normal astrocytes in vitro, in several cell lines, T-cadherin re-expression decreased cell growth, while enhancing substrate attachment, homophilic adhesion, and reducing motility. T-cadherin re-expression resulted in G2-phase arrest in glioma cells, which coincided with the upregulation of the cyclin-dependent kinase inhibitor p21, but was independent of p53 status [109]. Moreover, TGW and NH-12 neuroblastoma cell lines transfected with a T-cadherin cDNA-expressing vector lost their mitogenic potential and proliferative response to epidermal growth factor (EGF) [20].
Interestingly, miR-377-3p can override T-cadherin-mediated regulation. In an in vitro Alzheimer’s disease model, Liu et al. showed that miR-377 serves as a potent negative regulator of T-cadherin. β-Amyloid-treated SH-SY5Y neuroblastoma cells displayed a robust rise in CDH13 mRNA and protein levels, accompanied by a pronounced drop in endogenous miR-377. A markedly reduced CDH13 expression was detected upon transfection with miR-377 mimics, whereas anti-miRNA-377 produced the opposite effect, correlating with T-cadherin protein and mRNA expression levels. Functionally, the elevated miR-377 enhanced cell proliferation and reduced apoptosis in the β-amyloid-treated neurons. These findings confirm a negative regulatory relationship between miR-377 and T-cadherin in β-amyloid-challenged neurons [100].
Non-coding RNAs typically act within complex regulatory networks, which are interconnected molecular components that interact with one another and with their target genes to orchestrate gene expression. miRNAs that regulate CDH13 expression and function form part and parcel of this dynamic system. For example, H19, an lncRNA, is among the most abundant and evolutionarily conserved transcripts in mammalian development, with expression in both embryonic and extraembryonic cell lineages. The oncogenic role of H19 is now well recognized: aberrant H19 expression correlates with tumor growth, proliferation, invasion, metastasis, treatment resistance, and poor patient outcomes in several cancers [110]. For example, high-grade gliomas display elevated H19 levels, whereas siRNA-mediated knockdown of H19 suppresses glioma-cell invasion. miR-675 is embedded within the H19 first exon [111]. High H19 expression demonstrated a positive association with miR-675, and silencing H19 concomitantly inhibited miR-675 expression. Bioinformatic screening with miRanda and PicTar revealed a perfect seed-match between miR-675 and the 3′-UTR of CDH13, while luciferase assay using CDH13 3′-UTR constructs confirmed that miR-675 directly targets CDH13. Re-expression of miR-675 reversed the anti-invasive effect of H19 knockdown, demonstrating that miR-675 modulates CDH13 by binding its 3′-UTR [112].
Moreover, H19 functions as a competing endogenous RNA that sequesters miR-138 and miR-200a and antagonizes their functions. Inhibition of H19 has been shown to repress MMP9, vimentin, E-cadherin, and CDH13 expression in retinoblastoma [113], colorectal, and gastric cancers [114], thereby contributing to epithelial to mesenchymal transition progression [76]. H19 is not the only lncRNA that modulates CDH13 expression. Using LncRRIsearch and RNAup for bioinformatic analysis, CDH13-AS2, a lncRNA transcribed from the CDH13 locus, was identified as a candidate binder of the CDH13 3′-UTR. dCas13-based RNA immunoprecipitation subsequently verified this interaction, showing that CDH13-AS2 negatively regulates CDH13. Weighted correlation network analysis of human CAD datasets (STARNET and GTEx) further revealed a positive correlation between CDH13 and CDH13-AS2 expression in arterial tissue. CRISPR/Cas9 knockout of CDH13 or CDH13-AS2 in HUVECs produced concordant atherogenic phenotypes, suppressed proliferation and migration, coupled with an increased apoptosis and monocyte adhesion. In silico screens (TargetScan, miRWalk, and scanMiRApp) mapped several endothelial miRNAs to the CDH13 3′-UTR. Several disease-associated variants were mapped to the predicted binding sites of a few miRNAs, including mir-let7. The dual luciferase-based RNA interference assay confirmed the binding of mir-let7 on 3′UTR of CDH13, while dCas9-driven activation of CDH13-AS2 weakened the binding of mir-let7 and enhanced CDH13 expression. Collectively, these data position CDH13 and CDH13-AS2 as co-regulated, while their inhibition may contribute to CAD. Functionally, CDH13-AS2 stabilizes CDH13 mRNA, partly by preventing mir-let7-mediated CDH13 mRNA decay, highlighting the therapeutic potential of targeting the CDH13-AS2/let-7/CDH13 axis [76].
Therefore, the above-mentioned data indicate that CDH13/T-cadherin is controlled by an intricate ncRNA circuitry, including miRNAs, antisense and intergenic lncRNAs, and circRNAs, that operates alongside classic transcription-factor and promoter-methylation cues to fine-tune its expression. Disruption of this ncRNA–CDH13 axis spreads through cardiovascular, metabolic, oncogenic, and neurodegenerative pathways, underscoring its value as both a mechanistic understanding and a promising therapeutic target.
6. Non-Coding RNAs That Indirectly Regulate CDH13
Non-coding RNAs can modulate CDH13 expression not only through direct mRNA binding but also through engaging the epigenetic machinery that controls CDH13 transcription. A key pathway involves the opposing activities of two enzymes: DNMT3B and TET1. DNMT3B, a DNA methyltransferase that is frequently overexpressed in hepatocellular carcinoma, breast, prostate, colorectal, and lung cancers and predicts poor clinical outcome. TET1, a DNA demethylase that is often downregulated in the same cancer types, including breast, liver, lung, pancreatic, and prostate malignancies [115,116].
An increase in DNMT3B activity and/or a decrease in TET1 activity leads to promoter hypermethylation, consequently suppressing CDH13 (T-cadherin) expression (Figure 2) [117,118,119].
Certain miRNAs can modulate DNMT3B and TET1 activity. Simultaneous overexpression of the miR-23a/27a/24-2 cluster is an independent predictor of postoperative recurrence and poor survival at the early stage of non-small cell lung cancer (NSCLC). Mechanistically, each miRNA in the cluster targets the 3′-UTR of TET1 and FOXO3, the DNMT3B repressor: direct binding suppresses TET1, while relieving the FOXO3-mediated restraint of DNMT3B shifts the TET1/DNMT3B balance toward gene promoter hypermethylation. Immunohistochemical and Western blot analyses in xenografts confirmed the downstream effects, including the loss of p16 and T-cadherin (CDH13) expression along with DNMT3B upregulation. Accordingly, in vitro and in vivo studies have shown that the enforced miRNAs cluster expression heightens Wnt/β-catenin signaling and enhances stemness, tumorigenicity, and metastasis, whereas inhibition of all three miRNAs reverses those traits. Thus, the miR-23a/27a/24-2 cluster epigenetically silences multiple tumor suppressor genes, including CDH13, establishing a feed-forward circuit accelerating NSCLC progression and offering a rationale for multi-miRNA blockade as an adjuvant strategy to prevent early relapse [119].
A similar DNMT1-related mechanism, triggering hypermethylation of the CDH13 promoter and suppressing T-cadherin protein expression, was demonstrated for miR-142-3p [120]. This miRNA drives a positive-feedback circuit, in which it silences DNMT1 repressors and stabilizes DNMT1 mRNA, thereby shifting the DNMT1/TET balance towards net DNA methylation. Elevated DNMT1 then hypermethylates CpG-rich promoters of the key tumor suppressors, including CDH13, hampering their transcription and disrupting cell–cell adhesion across multiple malignancies (lung, gastric, and breast). Forced miR-142-3p expression drives proliferation, migration, and chemoresistance in lung cancer and hepatoma lines, whereas miR-142-3p knockdown or pharmacological DNMT1 inhibition restores CDH13 expression and sensitivity to apoptosis induction. Functionally, the overexpression of miR-142-3p results in invasive growth, whereas its inhibition, or DNMT1 knockdown, suppresses metastasis. Thus, the miR-142-3p/DNMT1/CDH13 cascade represents a convergent epigenetic switch that can be exploited in two ways. Therapeutically, by antagonizing miR-142-3p, or pharmacologically, by suppressing DNMT1, could restore CDH13 expression and inhibit tumor progression [120].
MiRNAs are not the only non-coding RNAs that can regulate CDH13 through promoter methylation. In ovarian cancer, circ_0000119 and DNMT1 have been reported to be upregulated, whereas miR-142-5p is reported to be depleted. Functional assays show that circ_0000119 overexpression accelerates cell proliferation, migration, invasion, and in vivo tumor growth, whereas its silencing produces the opposite effects. Functionally, circ_0000119 acts as a sponge for miR-142-5p, thereby releasing DNMT1 from miRNA-mediated repression. The consequent rise in DNMT1 hypermethylates the CpG-rich CDH13 promoter, thus blocking T-cadherin tumor-suppressive activity. miR-142-5p, in turn, can interact with hsa_circ_0000119 and DNMT1 3′-UTR. Silencing of DNMT1 can reverse the inhibition of hsa_circ_0000119 and circ_0000119-mediated repression of miR-142-5p and CDH13. Clinically, CDH13 promoters are more heavily methylated in ovarian tumors than in normal tissue, and a DNA-methyltransferase inhibitor can reactivate CDH13 expression in cancer cells and its anti-tumor functions [121].
Another non-coding RNA that represses CDH13 is the long non-coding RNA UPAT (ubiquitin-like with PHD and RING finger domains 1 (UHRF1) protein-associated transcript). UPAT is overexpressed in several malignancies, including NSCLC and colorectal cancer [122,123], where its levels correlate with tumor size and tumor-node metastasis stage. In NSCLC cells, UPAT markedly promotes proliferation and G1-to-S phase transition, counteracting the growth arrest normally enforced by T-cadherin via the cyclin-dependent kinase-inhibitor-1 pathway. UPAT acts via a more intricate mechanism than the circRNAs described above: UPAT upregulates UHRF1 (ubiquitin-like with PHD and RING finger domains 1), preventing its ubiquitin-mediated degradation. The accumulated UHRF1 recruits DNMT1 to replicating DNA, enhances CpG methylation across the CDH13 promoter, and inhibits T-cadherin expression. Silencing UPAT, therefore, restores T-cadherin, thereby restraining tumor growth [122].
It is noteworthy that T-cadherin/CDH13 itself is capable of regulating miRNAs. A recent study highlighted a compelling interplay among T-cadherin, miR-101-3p, ATP-binding cassette transporter A1 (ABCA1), and C1q/TNF-related protein 15 (CTRP15) in atherosclerosis progression [124]. CTRP15 (myonectin) is a myokine within the highly conserved CTRP family of adiponectin paralogues that links skeletal-muscle activity to lipid homeostasis in liver and adipose tissue in response to the energy-state changes, thereby outlining a novel myonectin-mediated metabolic circuit [125]. CTRPs engage three receptors (adiponectin receptors (AdipoR1 and AdipoR2), and T-cadherin), yet CTRP15 overexpression in THP-1 macrophages selectively elevated T-cadherin (protein and mRNA) without altering AdipoR1 or AdipoR2. Functionally, CTRP15 promotes the reverse-cholesterol transport and reduces atherosclerotic lesions in ApoE−/− mice. Moreover, CTRP15 enhanced cholesterol efflux from macrophages and increased ATP-binding cassette transporter A1 (ABCA1) expression via the T-cadherin/miR-101-3p axis. miR-101-3p itself is a pleiotropic miRNA implicated in atherosclerosis, cardiovascular disease and metabolic disorders [126,127].
Bioinformatic analyses with miRDB and TargetScan revealed a putative miR-101-3p binding site within the ABCA1 3′-UTR. CTRP15 upregulates ABCA1 and cholesterol efflux by downregulating miR-101-3p. Importantly, siRNA-mediated inhibition of T-cadherin abolished the CTRP15-induced decrease in miR-101-3p, demonstrating that T-cadherin is essential for CTRP15-dependent miR-101-3p suppression, ABCA1 induction, enhanced cholesterol efflux, and subsequent attenuation of atherosclerotic progression. This may indicate the existence of as-yet-undiscovered regulatory mechanisms between T-cadherin and miRNAs [124].
Collectively, a diverse set of miRNAs, circRNAs and lncRNAs modulate CDH13 indirectly by shifting the DNMT vs. TET balance toward (or away from) promoter hypermethylation, thereby silencing or rescuing T-cadherin transcription. Given that this ncRNA epigenetic circuitry governs CDH13 expression across cardiovascular and metabolic disorders and cancer, targeting these key ncRNA pathways offers a promising strategy to reestablish T-cadherin/CDH13 protective functions in disease.

Figure 2.
Several miRNAs directly bind the CDH13 3′-UTR and repress T-cadherin synthesis, notably miR-377-3p, miR-675, miR-199b-5p, and the let-7 family [76,87,99]. The miR-23a/27a/24-2 cluster silences CDH13 through an indirect route: it suppresses the demethylase TET1, while elevating the methyl-transferase DNMT3B, shifting the balance toward promoter hypermethylation and transcriptional repression [119]. Beyond miRNAs, circRNA circ_0000119 and lncRNA UPAT also converge on DNMT1/3-dependent methylation, reinforcing CDH13 repression [121,122]. Conversely, T-cadherin can influence the non-coding landscape itself: for example, it can lower miR-101-3p, thereby lifting inhibition of the cholesterol transporter ABCA1 [124]. Together, these layers of direct binding and epigenetic crosstalk intertwine CDH13 into a dynamic ncRNA network that fine-tunes its expression in embryogenesis and disease.
7. Bioinformatically Predicted Non-Coding RNAs Regulating CDH13/T-Cadherin
Bioinformatically predicted interactions between non-coding RNAs (ncRNAs) and CDH13/T-cadherin collectively draw the compelling conceptual landscape for future research aimed at elucidating physiological and pathological mechanisms. Yet, due to the low binding specificity of ncRNAs to their targets, various databases ultimately yield thousands of predicted partners, with most of them still awaiting experimental confirmation [128]. Even so, a subset of high-confidence candidates clearly warrants closer scrutiny.
One miRNA of interest is miR-495-3p, which has been implicated in the pathogenesis of gastric cancer [129]. In their study, Wang et al., retrieved miRNAs predicted to target dysregulated cadherins from miRDB, and cross-checked these miRNA–cadherin pairs in miRTarBase, followed by refining the list with TCGA (The Cancer Genome Atlas) miRNA-seq data and retaining only miRNAs differentially expressed between gastric cancer and normal tissue. miR-495-3p emerged from this pipeline as a putative regulator of several cadherins, including CDH13, but its own expression is reduced in gastric cancer. miR-495-3p may represent an attractive yet still unverified component of the CDH13 regulatory network, and direct binding, along with functional assays, are warranted to define its impact on T-cadherin expression.
Another candidate is miR-584-5p, described as a marker of pathological states in coronary artery disease (CAD), which was demonstrated to target CDH13 [130]. In a Turkish cohort, whole-blood miRNA profiling of angiographically verified CAD patients versus controls revealed several miRNA candidates, where miR-584-5p appeared to be one of the most dysregulated miRNAs. These microarray findings were further validated by qRT-PCR, followed by diagnostic and clinical correlation assays. Ingenuity pathway analysis defined CDH13 as the sole CAD-related predicted target of miR-584-5p. These data highlight miR-584-5p, previously recognized as a tumor-suppressor miRNA, as a potential CAD biomarker and implicate this miRNA and CDH13 in the disease pathogenesis.
A group of miRNAs (miR-181d, miR-30a-3p, miR-30c, miR-30d, miR-30e-3p, miR-370, miR-493-5p, and miR-532-5p) has been associated with ovarian carcinoma progression and predicted to target CDH13. Their expression was quantified by TaqMan RT-qPCR in formalin-fixed, paraffin-embedded ovarian tumor samples and in six normal human ovarian surface-epithelial (HOSE) cell lines. Subsequent RT-qPCR analyses of ovarian carcinoma tissues revealed that in Her2/neu-negative tumors, CDH13 was silenced through miRNA-mediated mechanisms, whereas in Her2/neu-positive tumors, these miRNAs were suppressed and CDH13 was repressed via promoter methylation. Overall, these data indicated the CDH13 downregulation across ovarian carcinoma subtypes. The above-mentioned miRNA levels also correlated with key clinicopathologic parameters. Although direct binding to CDH13 calls for experimental confirmation, TargetScan predicted interaction sites for each of these miRNAs [131,132].
Ducoli and colleagues combined RNA-seq, CAGE-seq, and ChIRP-seq to map the lncRNA landscape of human dermal blood (BEC) and lymphatic endothelial cells (LEC) [133]. LET1R, an endothelial-specific lncRNA, was predicted to be involved in regulating cell proliferation and migration. Bioinformatic mapping placed LET1R-binding motifs within promoters, exons, or introns of 1607 protein-coding genes; T-cadherin (CDH13) was among the predicted targets and appeared to be repressed by LET1R. Filtering for genes expressed in endothelial and lymphatic cells, and intersecting this list with the 255 transcripts altered after LET1R antisense-oligonucleotide knockdown, yielded 44 overlapping genes (12 upregulated and 32 downregulated). Among the downregulated set was CDH13, suggesting that LETR1 may repress T-cadherin in endothelial cells. These bioinformatic data position LETR1 as a novel lncRNA that potentially modulates the CDH13 axis in the endothelium and contributes to the emerging network of non-coding RNAs that define T-cadherin biology [133].
Another lncRNA of interest is LINC00707. Transforming growth factor-β (TGF-β) signaling stands at the nexus of tumor-suppressive and tumor-promoting pathways: in early lesions, it blocks proliferation, whereas in advanced cancers, it drives epithelial-to-mesenchymal transition (EMT), migration, and metastasis through a Smad-dependent transcriptional program [134,135]. Genome-wide profiling of TGF-β responses revealed LINC00707 as a direct Smad/MAPK target that is rapidly repressed when TGF-β is activated [136]. Mechanistically, TGF-β lowers the occupancy of the transcription factor KLF6 at the LINC00707 promoter, suppressing its transcription. Loss of LINC00707 then releases cytoplasmic Smad proteins, allowing their nuclear accumulation and activation of the mesenchymal gene program. Although being oncogenic in lung adenocarcinoma and hepatocarcinoma, where its overexpression correlates with increased proliferation, invasion, and a poor prognosis, LINC00707 operates as a negative modulator of TGF-β signaling in other settings. Gene ontology analysis yielding 263 differentially expressed genes (99 upregulated and 164 downregulated) highlighted processes classically associated with a mesenchymal switch: extracellular-matrix assembly, cell–cell adhesion, and cell migration (FN1, COL3A1, SERPINE2, and CDH13). CDH13 was one of the transcripts most consistently elevated when LINC00707 was silenced, yet remained low in cells that ectopically overexpressed LINC00707, indicating that lncRNA normally limits T-cadherin expression. Defining the exact mechanism of LINC00707-mediated repression of CDH13 and determining whether manipulating this lncRNA can promote mesenchymal-to-epithelial transition and restore CDH13 function remains an intriguing question for future study [136].
A recent bioinformatic analysis applied to transcriptomic profiling during oocyte maturation in spotted scats used RNA-seq and small-RNA-seq to map circRNA–miRNA–mRNA circuits during IGF3-induced ovarian maturation. Gene ontology (GO) enrichment analysis showed that host genes of differentially expressed circRNAs and target genes of differentially expressed miRNAs were enriched for various processes with a high degree of overlap. Among 176 differentially expressed circRNAs and 52 miRNAs, pathway enrichment highlighted cell adhesion, ECM remodeling, and oocyte meiosis programs. CDH13 (T-cadherin) appeared consistently among the top Kyoto Encyclopedia of Genes and Genomes (KEGG)-enriched categories, specifically within the “cell-adhesion molecules” and “ECM–receptor interaction” pathways. A circRNA–miRNA–mRNA regulatory network reconstruction identified a single circRNA (Lachesis_group5:6245955|6270787) that is co-expressed in the ovary and is predicted to sponge three IGF3-responsive miRNAs (novel_miR_622, novel_miR_980, and novel_miR_64), thereby affecting a cluster of maturation-related genes that includes CDH13. These in silico findings imply that sponging these miRNAs could modulate CDH13 function during oocyte meiosis and extracellular matrix remodeling throughout teleost ovarian development [137].
In silico screens across diverse datasets converge on a certain list of miRNAs, lncRNAs, and circRNAs that are predicted to bind CDH13 or its promoter in tissue-specific and disease-related contexts. While these candidates outline a rich regulatory network linking T-cadherin to gastric and ovarian cancer, coronary artery disease, and endothelial and developmental pathways, definitive binding assays and functional studies are warranted to move them from bioinformatically predicted interactions to biological facts.
The growing body of evidence underscores the importance of non-coding RNA interactions with the CDH13 in both normal and pathological contexts. These findings highlight the regulatory potential of ncRNAs in modulating CDH13 expression and are summarized in Table 2.
Table 2.
Non-coding RNAs interacting with CDH13.
8. Conclusions
Mounting evidence positions non-coding RNAs (ncRNAs) as pivotal regulators of CDH13 gene/T-cadherin at all levels of gene/mRNA/protein expression control, from chromatin architecture to mRNA turnover and protein synthesis. ncRNAs can modulate CDH13 gene/T-cadherin expression and functions through a variety of pathways. MiRNAs, lncRNAs, and circRNAs can bind directly to the CDH13 transcript, compete for the shared regulatory elements, or reshape promoter methylation, thereby adjusting CDH13 gene/T-cadherin output with exquisite precision. Intriguingly, the CDH13 locus itself encodes several of these regulatory molecules, for example, miR-3182, the antisense lncRNAs FEDORA and CDH13-AS1/-AS2, and CircCDH13, each displaying tissue-restricted expression patterns and establishing feedback loops on the same signaling circuits in which CDH13 gene/T-cadherin participates.
Collectively, this multilayered ncRNA network fine-tunes physiological programs, such as vascular remodeling, metabolic homeostasis, and neuroplasticity, while its dysregulation contributes to the development of cardiovascular disease, tumor progression, inflammation, and neurodegeneration. These insights make these ncRNAs both informative biomarkers and attractive therapeutic entry points in conditions when CDH13 gene activity is disrupted.
Yet the landscape remains only partially mapped. Further studies are warranted, including (i) delineating ncRNA-induced chromatin remodeling at the CDH13 promoter, (ii) constructing cell-type resolved maps of ncRNA/CDH13 interactomes in vivo, and (iii) converting these insights into clinically viable ncRNA delivery or inhibition strategies. Concerted application of high-resolution multiomics, predictive bioinformatics, and precision functional assays will open new vistas for understanding CDH13 gene/T-cadherin-related mechanisms in health and disease. Integrating these tools will clarify how ncRNA circuits modulate T-cadherin signaling and pave the way for novel therapies aimed at restoring the CDH13 expression.
Author Contributions
Conceptualization, formal analysis, writing—original draft preparation: K.R., A.M., P.K., and D.R.; resources, supervision, project administration, funding acquisition: K.R., V.S., E.S., V.T., K.R., and A.M. made an equal contribution to the review writing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by grant No. 23-11-00205 https://rscf.ru/en/project/23-11-00205/ (accessed on 26 May 2025) and performed under the State Assignment # 03p-23/110-03 of Lomonosov Moscow State University.
Institutional Review Board Statement
Not applicable.
Acknowledgments
The authors are grateful to Anastasia Sharapkova (Rosetta Stone MSU) for her careful reading of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lee, S.W. H–Cadherin, a Novel Cadherin with Growth Inhibitory Functions and Diminished Expression in Human Breast Cancer. Nat. Med. 1996, 2, 776–782. [Google Scholar] [CrossRef] [PubMed]
- Kremmidiotis, G.; Baker, E.; Crawford, J.; Eyre, H.J.; Nahmias, J.; Callen, D.F. Localization of Human Cadherin Genes to Chromosome Regions Exhibiting Cancer-Related Loss of Heterozygosity. Genomics 1998, 49, 467–471. [Google Scholar] [CrossRef]
- Philippova, M.; Joshi, M.B.; Kyriakakis, E.; Pfaff, D.; Erne, P.; Resink, T.J. A Guide and Guard: The Many Faces of T-Cadherin. Cell. Signal. 2009, 21, 1035–1044. [Google Scholar] [CrossRef]
- Ranscht, B.; Bronner-Fraser, M. T-Cadherin Expression Alternates with Migrating Neural Crest Cells in the Trunk of the Avian Embryo. Development 1991, 111, 15–22. [Google Scholar] [CrossRef] [PubMed]
- George, S.J.; Beeching, C.A. Cadherin:Catenin Complex: A Novel Regulator of Vascular Smooth Muscle Cell Behaviour. Atherosclerosis 2006, 188, 1–11. [Google Scholar] [CrossRef]
- Hulpiau, P.; van Roy, F. Molecular Evolution of the Cadherin Superfamily. Int. J. Biochem. Cell Biol. 2009, 41, 349–369. [Google Scholar] [CrossRef]
- Rubina, K.A.; Semina, E.V.; Kalinina, N.I.; Sysoeva, V.Y.; Balatskiy, A.V.; Tkachuk, V.A. Revisiting the Multiple Roles of T-Cadherin in Health and Disease. Eur. J. Cell Biol. 2021, 100, 151183. [Google Scholar] [CrossRef] [PubMed]
- Philippova, M.; Ivanov, D.; Joshi, M.B.; Kyriakakis, E.; Rupp, K.; Afonyushkin, T.; Bochkov, V.; Erne, P.; Resink, T.J. Identification of Proteins Associating with Glycosylphosphatidylinositol- Anchored T-Cadherin on the Surface of Vascular Endothelial Cells: Role for Grp78/BiP in T-Cadherin-Dependent Cell Survival. Mol. Cell Biol. 2008, 28, 4004–4017. [Google Scholar] [CrossRef]
- Philippova, M.; Joshi, M.B.; Pfaff, D.; Kyriakakis, E.; Maslova, K.; Erne, P.; Resink, T.J. T-Cadherin Attenuates Insulin-Dependent Signalling, eNOS Activation, and Angiogenesis in Vascular Endothelial Cells. Cardiovasc. Res. 2012, 93, 498–507. [Google Scholar] [CrossRef][Green Version]
- Joshi, M.B.; Philippova, M.; Ivanov, D.; Allenspach, R.; Erne, P.; Resink, T.J. T-Cadherin Protects Endothelial Cells from Oxidative Stress-Induced Apoptosis. FASEB J. 2005, 19, 1737–1739. [Google Scholar] [CrossRef]
- Hug, C.; Wang, J.; Ahmad, N.S.; Bogan, J.S.; Tsao, T.-S.; Lodish, H.F. T-Cadherin Is a Receptor for Hexameric and High-Molecular-Weight Forms of Acrp30/Adiponectin. Proc. Natl. Acad. Sci. USA 2004, 101, 10308–10313. [Google Scholar] [CrossRef] [PubMed]
- Denzel, M.S.; Scimia, M.-C.; Zumstein, P.M.; Walsh, K.; Ruiz-Lozano, P.; Ranscht, B. T-Cadherin Is Critical for Adiponectin-Mediated Cardioprotection in Mice. J. Clin. Investig. 2010, 120, 4342–4352. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Kihara, S.; Ouchi, N.; Nishida, M.; Arita, Y.; Kumada, M.; Ohashi, K.; Sakai, N.; Shimomura, I.; Kobayashi, H.; et al. Adiponectin Reduces Atherosclerosis in Apolipoprotein E-Deficient Mice. Circulation 2002, 106, 2767–2770. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kita, S.; Nishizawa, H.; Fukuda, S.; Fujishima, Y.; Obata, Y.; Nagao, H.; Masuda, S.; Nakamura, Y.; Shimizu, Y.; et al. Adiponectin Promotes Muscle Regeneration through Binding to T-Cadherin. Sci. Rep. 2019, 9, 16. [Google Scholar] [CrossRef]
- Obata, Y.; Kita, S.; Koyama, Y.; Fukuda, S.; Takeda, H.; Takahashi, M.; Fujishima, Y.; Nagao, H.; Masuda, S.; Tanaka, Y.; et al. Adiponectin/T-Cadherin System Enhances Exosome Biogenesis and Decreases Cellular Ceramides by Exosomal Release. JCI Insight 2018, 3, e99680. [Google Scholar] [CrossRef]
- Rubina и др. T-Cadherin: A Missing Puzzle Between Cancer and Obesity. Available online: https://juniperpublishers.com/ijcsmb/IJCSMB.MS.ID.555726.php (accessed on 23 June 2025).
- Sysoeva, V.; Semina, E.; Klimovich, P.; Kulebyakin, K.; Dzreyan, V.; Sotskaya, E.; Shchipova, A.; Popov, V.; Shilova, A.; Brodsky, I.; et al. T-Cadherin Modulates Adipogenic Differentiation in Mesenchymal Stem Cells: Insights into Ligand Interactions. Front. Cell Dev. Biol. 2024, 12, 1446363. [Google Scholar] [CrossRef]
- Andreeva, A.V.; Kutuzov, M.A. Cadherin 13 in Cancer. Genes Chromosomes Cancer 2010, 49, 775–790. [Google Scholar] [CrossRef]
- Rubina, K.A.; Smutova, V.A.; Semenova, M.L.; Poliakov, A.A.; Gerety, S.; Wilkinson, D.; Surkova, E.I.; Semina, E.V.; Sysoeva, V.Y.; Tkachuk, V.A. Detection of T-Cadherin Expression in Mouse Embryos. Acta Naturae 2015, 7, 87–94. [Google Scholar] [CrossRef]
- Takeuchi, T.; Misaki, A.; Liang, S.B.; Tachibana, A.; Hayashi, N.; Sonobe, H.; Ohtsuki, Y. Expression of T-Cadherin (CDH13, H-Cadherin) in Human Brain and Its Characteristics as a Negative Growth Regulator of Epidermal Growth Factor in Neuroblastoma Cells. J. Neurochem. 2000, 74, 1489–1497. [Google Scholar] [CrossRef]
- Ivanov, D.; Philippova, M.; Antropova, J.; Gubaeva, F.; Iljinskaya, O.; Tararak, E.; Bochkov, V.; Erne, P.; Resink, T.; Tkachuk, V. Expression of Cell Adhesion Molecule T-Cadherin in the Human Vasculature. Histochem. Cell Biol. 2001, 115, 231–242. [Google Scholar] [CrossRef]
- Kudrjashova, E.; Bashtrikov, P.; Bochkov, V.; Parfyonova, Y.; Tkachuk, V.; Antropova, J.; Iljinskaya, O.; Tararak, E.; Erne, P.; Ivanov, D.; et al. Expression of Adhesion Molecule T-Cadherin Is Increased during Neointima Formation in Experimental Restenosis. Histochem. Cell Biol. 2002, 118, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Rubina, K.; Sysoeva, V.; Semina, E.; Kalinina, N.; Yurlova, E.; Khlebnikova, A.; Molochkov, V.; Rubina, K.; Sysoeva, V.; Semina, E.; et al. Malignant Transformation in Skin Is Associated with the Loss of T-Cadherin Expression in Human Keratinocytes and Heterogeneity in T-Cadherin Expression in Tumor Vasculature. In Tumor Angiogenesis; IntechOpen: London, UK, 2012; ISBN 978-953-51-0009-6. [Google Scholar]
- Parker-Duffen, J.L.; Nakamura, K.; Silver, M.; Kikuchi, R.; Tigges, U.; Yoshida, S.; Denzel, M.S.; Ranscht, B.; Walsh, K. T-Cadherin Is Essential for Adiponectin-Mediated Revascularization. J. Biol. Chem. 2013, 288, 24886–24897. [Google Scholar] [CrossRef]
- Tsugawa-Shimizu, Y.; Fujishima, Y.; Kita, S.; Minami, S.; Sakaue, T.-A.; Nakamura, Y.; Okita, T.; Kawachi, Y.; Fukada, S.; Namba-Hamano, T.; et al. Increased Vascular Permeability and Severe Renal Tubular Damage after Ischemia-Reperfusion Injury in Mice Lacking Adiponectin or T-Cadherin. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E179–E190. [Google Scholar] [CrossRef]
- Hebbard, L.; Ranscht, B. Multifaceted Roles of Adiponectin in Cancer. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Rubina, K.A.; Surkova, E.I.; Semina, E.V.; Sysoeva, V.Y.; Kalinina, N.I.; Poliakov, A.A.; Treshalina, H.M.; Tkachuk, V.A. T-Cadherin Expression in Melanoma Cells Stimulates Stromal Cell Recruitment and Invasion by Regulating the Expression of Chemokines, Integrins and Adhesion Molecules. Cancers 2015, 7, 1349–1370. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Ma, L.; Liu, Y.; Xiong, H.; Shi, D. Decoding the Enigmatic Role of T-Cadherin in Tumor Angiogenesis. Front. Immunol. 2025, 16, 1564130. [Google Scholar] [CrossRef]
- Sternberg, J.; Wankell, M.; Subramaniam, V.N.; Hebbard, L.W.; Sternberg, J.; Wankell, M.; Subramaniam, V.N.; Hebbard, L.W. The Functional Roles of T-Cadherin in Mammalian Biology. Aimsmoles 2017, 4, 62–81. [Google Scholar] [CrossRef]
- Fukuda, S.; Kita, S.; Miyashita, K.; Iioka, M.; Murai, J.; Nakamura, T.; Nishizawa, H.; Fujishima, Y.; Morinaga, J.; Oike, Y.; et al. Identification and Clinical Associations of 3 Forms of Circulating T-Cadherin in Human Serum. J. Clin. Endocrinol. Metab. 2021, 106, 1333–1344. [Google Scholar] [CrossRef]
- Yang, Y.R.; Kabir, M.H.; Park, J.H.; Park, J.-I.; Kang, J.S.; Ju, S.; Shin, Y.J.; Lee, S.M.; Lee, J.; Kim, S.; et al. Plasma Proteomic Profiling of Young and Old Mice Reveals Cadherin-13 Prevents Age-Related Bone Loss. Aging 2020, 12, 8652–8668. [Google Scholar] [CrossRef]
- Frismantiene, A.; Dasen, B.; Pfaff, D.; Erne, P.; Resink, T.J.; Philippova, M. T-Cadherin Promotes Vascular Smooth Muscle Cell Dedifferentiation via a GSK3β-Inactivation Dependent Mechanism. Cell. Signal. 2016, 28, 516–530. [Google Scholar] [CrossRef]
- Kyriakakis, E.; Frismantiene, A.; Dasen, B.; Pfaff, D.; Rivero, O.; Lesch, K.-P.; Erne, P.; Resink, T.J.; Philippova, M. T-Cadherin Promotes Autophagy and Survival in Vascular Smooth Muscle Cells through MEK1/2/Erk1/2 Axis Activation. Cell. Signal. 2017, 35, 163–175. [Google Scholar] [CrossRef]
- Otsuka, I.; Watanabe, Y.; Hishimoto, A.; Boku, S.; Mouri, K.; Shiroiwa, K.; Okazaki, S.; Nunokawa, A.; Shirakawa, O.; Someya, T.; et al. Association Analysis of the Cadherin13 Gene with Schizophrenia in the Japanese Population. Neuropsychiatr. Dis. Treat. 2015, 11, 1381–1393. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Priya, I.; Arora, M.; Singh, H.; Sharma, I.; Sharma, S.; Mahajan, R.; Kapoor, N. Association of the CDH13 Gene Variant Rs9940180 with Schizophrenia Risk in North Indian Population. Am. J. Transl. Res. 2023, 15, 6476–6485. [Google Scholar] [PubMed]
- Kiser, D.P.; Popp, S.; Schmitt-Böhrer, A.G.; Strekalova, T.; van den Hove, D.L.; Lesch, K.-P.; Rivero, O. Early-Life Stress Impairs Developmental Programming in Cadherin 13 (CDH13)-Deficient Mice. Progress. Neuro-Psychopharmacol. Biol. Psychiatry 2019, 89, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Mossink, B.; van Rhijn, J.-R.; Wang, S.; Linda, K.; Vitale, M.R.; Zöller, J.E.M.; van Hugte, E.J.H.; Bak, J.; Verboven, A.H.A.; Selten, M.; et al. Cadherin-13 Is a Critical Regulator of GABAergic Modulation in Human Stem-Cell-Derived Neuronal Networks. Mol. Psychiatry 2022, 27, 1–18. [Google Scholar] [CrossRef]
- Sanders, S.J.; He, X.; Willsey, A.J.; Ercan-Sencicek, A.G.; Samocha, K.E.; Cicek, A.E.; Murtha, M.T.; Bal, V.H.; Bishop, S.L.; Dong, S.; et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015, 87, 1215–1233. [Google Scholar] [CrossRef]
- Asherson, P.; Zhou, K.; Anney, R.J.L.; Franke, B.; Buitelaar, J.; Ebstein, R.; Gill, M.; Altink, M.; Arnold, R.; Boer, F.; et al. A High-Density SNP Linkage Scan with 142 Combined Subtype ADHD Sib Pairs Identifies Linkage Regions on Chromosomes 9 and 16. Mol. Psychiatry 2008, 13, 514–521. [Google Scholar] [CrossRef]
- Neale, B.M.; Medland, S.E.; Ripke, S.; Asherson, P.; Franke, B.; Lesch, K.-P.; Faraone, S.V.; Nguyen, T.T.; Schäfer, H.; Holmans, P.; et al. Meta-Analysis of Genome-Wide Association Studies of Attention-Deficit/Hyperactivity Disorder. J. Am. Acad. Child Adolesc. Psychiatry 2010, 49, 884–897. [Google Scholar] [CrossRef]
- Treutlein, J.; Cichon, S.; Ridinger, M.; Wodarz, N.; Soyka, M.; Zill, P.; Maier, W.; Moessner, R.; Gaebel, W.; Dahmen, N.; et al. Genome-Wide Association Study of Alcohol Dependence. Arch. Gen. Psychiatry 2009, 66, 773–784. [Google Scholar] [CrossRef]
- Sarı, E.; Erbaş, O. Non-Coding RNA and Functions. JEBMS 2021, 2, 128–132. [Google Scholar] [CrossRef]
- Hombach, S.; Kretz, M. Non-Coding RNAs: Classification, Biology and Functioning. Adv. Exp. Med. Biol. 2016, 937, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and Their Integrated Networks. J. Integr. Bioinform. 2019, 16, 20190027. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.-L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long Non-Coding RNAs: Definitions, Functions, Challenges and Recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef]
- Chellini, L.; Frezza, V.; Paronetto, M.P. Dissecting the Transcriptional Regulatory Networks of Promoter-Associated Noncoding RNAs in Development and Cancer. J. Exp. Clin. Cancer Res. 2020, 39, 51. [Google Scholar] [CrossRef]
- Robinson, E.K.; Covarrubias, S.; Carpenter, S. The How and Why of lncRNA Function: An Innate Immune Perspective. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194419. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Bhandari, N.; Kumar, P.; Bhandari, N. lncRNAs: Role in Regulation of Gene Expression. In Gene Expression; IntechOpen: Lundon, UK, 2022; ISBN 978-1-80355-622-2. [Google Scholar]
- Wang, X.; Li, H.; Lu, Y.; Cheng, L. Regulatory Effects of Circular RNAs on Host Genes in Human Cancer. Front. Oncol. 2021, 10, 586163. [Google Scholar] [CrossRef]
- Lau, P.-W.; Guiley, K.Z.; De, N.; Potter, C.S.; Carragher, B.; MacRae, I.J. The Molecular Architecture of Human Dicer. Nat. Struct. Mol. Biol. 2012, 19, 436–440. [Google Scholar] [CrossRef] [PubMed]
- Schanen, B.C.; Li, X. Transcriptional Regulation of Mammalian miRNA Genes. Genomics 2011, 97, 1–6. [Google Scholar] [CrossRef]
- MacFarlane, L.-A.; Murphy, P.R. MicroRNA: Biogenesis, Function and Role in Cancer. Curr. Genom. 2010, 11, 537–561. [Google Scholar] [CrossRef]
- Hammond, S.M. An Overview of microRNAs. Adv. Drug Deliv. Rev. 2015, 87, 3–14. [Google Scholar] [CrossRef]
- Maia, J.; Caja, S.; Strano Moraes, M.C.; Couto, N.; Costa-Silva, B. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front. Cell Dev. Biol. 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.J.; Aguilar, R.; Kesner, B.; Lee, H.-G.; Kriz, A.J.; Chu, H.-P.; Lee, J.T. Jpx RNA Regulates CTCF Anchor Site Selection and Formation of Chromosome Loops. Cell 2021, 184, 6157–6173.e24. [Google Scholar] [CrossRef]
- Granados-Riveron, J.T.; Aquino-Jarquin, G. The Complexity of the Translation Ability of circRNAs. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2016, 1859, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA Biogenesis Competes with Pre-mRNA Splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef]
- Belousova, E.A.; Filipenko, M.L.; Kushlinskii, N.E. Circular RNA: New Regulatory Molecules. Bull. Exp. Biol. Med. 2018, 164, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA Circles Function as Efficient microRNA Sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Ao, X.; Yu, W.; Zhang, L.; Wang, Y.; Chang, W. The Role of Non-Coding RNAs in Alzheimer’s Disease: From Regulated Mechanism to Therapeutic Targets and Diagnostic Biomarkers. Front. Aging Neurosci. 2021, 13, 654978. [Google Scholar] [CrossRef]
- Xiao, J. (Ed.) Circular RNAs: Biogenesis and Functions; Advances in Experimental Medicine and Biology; Springer: Singapore, 2018; ISBN 9789811314254. [Google Scholar]
- de Rie, D.; Abugessaisa, I.; Alam, T.; Arner, E.; Arner, P.; Ashoor, H.; Åström, G.; Babina, M.; Bertin, N.; Burroughs, A.M.; et al. An Integrated Expression Atlas of miRNAs and Their Promoters in Human and Mouse. Nat. Biotechnol. 2017, 35, 872–878. [Google Scholar] [CrossRef]
- Ozsolak, F.; Poling, L.L.; Wang, Z.; Liu, H.; Liu, X.S.; Roeder, R.G.; Zhang, X.; Song, J.S.; Fisher, D.E. Chromatin Structure Analyses Identify miRNA Promoters. Genes. Dev. 2008, 22, 3172–3183. [Google Scholar] [CrossRef] [PubMed]
- Ha, M.; Kim, V.N. Regulation of microRNA Biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Martin, J.; Han, C.; Gordon, L.A.; Terry, A.; Prabhakar, S.; She, X.; Xie, G.; Hellsten, U.; Chan, Y.M.; Altherr, M.; et al. The Sequence and Analysis of Duplication-Rich Human Chromosome 16. Nature 2004, 432, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Xie, J.; Sun, H. Three miRNAs Cooperate with Host Genes Involved in Human Cardiovascular Disease. Hum. Genom. 2019, 13, 40. [Google Scholar] [CrossRef]
- Chien, K.R.; Olson, E.N. Converging Pathways and Principles in Heart Development and Disease: CV@CSH. Cell 2002, 110, 153–162. [Google Scholar] [CrossRef]
- Ramos-Kuri, M.; Meka, S.H.; Salamanca-Buentello, F.; Hajjar, R.J.; Lipskaia, L.; Chemaly, E.R. Molecules Linked to Ras Signaling as Therapeutic Targets in Cardiac Pathologies. Biol. Res. 2021, 54, 23. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, P.; Chen, W.; Jie, D.; Dan, F.; Jia, Y.; Xie, L. Characterization and Identification of Novel Serum microRNAs in Sepsis Patients with Different Outcomes. Shock 2013, 39, 480–487. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Grocock, R.J.; van Dongen, S.; Bateman, A.; Enright, A.J. miRBase: MicroRNA Sequences, Targets and Gene Nomenclature. Nucleic Acids Res. 2006, 34, D140–D144. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.S.; Tyagi, S.; Nancarrow, D.J.; Boyle, G.M.; Cook, A.L.; Whiteman, D.C.; Parsons, P.G.; Schmidt, C.; Sturm, R.A.; Hayward, N.K. Characterization of the Melanoma miRNAome by Deep Sequencing. PLoS ONE 2010, 5, e9685. [Google Scholar] [CrossRef] [PubMed]
- Issler, O.; van der Zee, Y.Y.; Ramakrishnan, A.; Xia, S.; Zinsmaier, A.K.; Tan, C.; Li, W.; Browne, C.J.; Walker, D.M.; Salery, M.; et al. The Long Noncoding RNA FEDORA Is a Cell Type- and Sex-Specific Regulator of Depression. Sci. Adv. 2022, 8, eabn9494. [Google Scholar] [CrossRef]
- Cai, M.; Wang, Y.-W.; Xu, S.-H.; Qiao, S.; Shu, Q.-F.; Du, J.-Z.; Li, Y.-G.; Liu, X.-L. Regulatory Effects of the Long Non-coding RNA RP11-543N12.1 and microRNA-324-3p Axis on the Neuronal Apoptosis Induced by the Inflammatory Reactions of Microglia. Int. J. Mol. Med. 2018, 42, 1741–1755. [Google Scholar] [CrossRef]
- Wang, L.-K.; Chen, X.-F.; He, D.-D.; Li, Y.; Fu, J. Dissection of Functional lncRNAs in Alzheimer’s Disease by Construction and Analysis of lncRNA–mRNA Networks Based on Competitive Endogenous RNAs. Biochem. Biophys. Res. Commun. 2017, 485, 569–576. [Google Scholar] [CrossRef]
- Xu, J.; Ai, Q.; Cao, H.; Liu, Q. MiR-185-3p and miR-324-3p Predict Radiosensitivity of Nasopharyngeal Carcinoma and Modulate Cancer Cell Growth and Apoptosis by Targeting SMAD7. Med. Sci. Monit. 2015, 21, 2828–2836. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, S.; Li, L.; Diagel, A.; Moggio, A.; Li, Z.; Song, X.; Sager, H.; Boon, R.; Maegdefessel, L.; et al. The Orchestration of Coding and Non-Coding Genes at the CDH13 Locus in Coronary Artery Disease. Eur. Heart J. 2024, 45, ehae666.3831. [Google Scholar] [CrossRef]
- Zhou, Z.; Ma, J.; Lu, J.; Chen, A.; Zhu, L. Circular RNA CircCDH13 Contributes to the Pathogenesis of Osteoarthritis via CircCDH13/miR-296-3p/PTEN Axis. J. Cell Physiol. 2021, 236, 3521–3535. [Google Scholar] [CrossRef]
- Mao, X.; Cao, Y.; Guo, Z.; Wang, L.; Xiang, C. Biological Roles and Therapeutic Potential of Circular RNAs in Osteoarthritis. Mol. Ther.-Nucleic Acids 2021, 24, 856–867. [Google Scholar] [CrossRef]
- Bromhead, C.; Miller, J.H.; McDonald, F.J. Regulation of T-Cadherin by Hormones, Glucocorticoid and EGF. Gene 2006, 374, 58–67. [Google Scholar] [CrossRef]
- Kuzmenko, Y.S.; Kern, F.; Bochkov, V.N.; Tkachuk, V.A.; Resink, T.J. Density- and Proliferation Status-Dependent Expression of T-Cadherin, a Novel Lipoprotein-Binding Glycoprotein: A Function in Negative Regulation of Smooth Muscle Cell Growth? FEBS Lett. 1998, 434, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Kuzmenko, Y.S.; Stambolsky, D.; Kern, F.; Bochkov, V.N.; Tkachuk, V.A.; Resink, T.J. Characteristics of Smooth Muscle Cell Lipoprotein Binding Proteins (P105/P130) as T-Cadherin and Regulation by Positive and Negative Growth Regulators. Biochem. Biophys. Res. Commun. 1998, 246, 489–494. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, L.; Yang, J.; Li, B.; Wang, J. CDH13 Promoter Methylation Regulates Cisplatin Resistance of Non-Small Cell Lung Cancer Cells. Oncol. Lett. 2018, 16, 5715–5722. [Google Scholar] [CrossRef]
- Shiu, B.-H.; Lu, W.-Y.; Tantoh, D.M.; Chou, M.-C.; Nfor, O.N.; Huang, C.-C.; Liaw, Y.-P. Interactive Association between Dietary Fat and Sex on CDH13 Cg02263260 Methylation. BMC Med. Genom. 2021, 14, 13. [Google Scholar] [CrossRef]
- Pu, W.; Geng, X.; Chen, S.; Tan, L.; Tan, Y.; Wang, A.; Lu, Z.; Guo, S.; Chen, X.; Wang, J. Aberrant Methylation of CDH13 Can Be a Diagnostic Biomarker for Lung Adenocarcinoma. J. Cancer 2016, 7, 2280–2289. [Google Scholar] [CrossRef]
- Ellmann, L.; Joshi, M.B.; Resink, T.J.; Bosserhoff, A.K.; Kuphal, S. BRN2 Is a Transcriptional Repressor of CDH13 (T-Cadherin) in Melanoma Cells. Lab. Investig. 2012, 92, 1788–1800. [Google Scholar] [CrossRef] [PubMed]
- Favreau, A.J.; McGlauflin, R.E.; Duarte, C.W.; Sathyanarayana, P. miR-199b, a Novel Tumor Suppressor miRNA in Acute Myeloid Leukemia with Prognostic Implications. Exp. Hematol. Oncol. 2015, 5, 4. [Google Scholar] [CrossRef]
- Ragusa, R.; Di Molfetta, A.; Mercatanti, A.; Pitto, L.; Amodeo, A.; Trivella, M.G.; Rizzo, M.; Caselli, C. Changes in Adiponectin System after Ventricular Assist Device in Pediatric Heart Failure. JHLT Open 2024, 3, 100041. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Song, B.; Song, W.; Liu, J.; Sun, A.; Wu, D.; Yu, H.; Lian, J.; Chen, L.; Han, J. Underexpressed microRNA-199b-5p Targets Hypoxia-Inducible Factor-1α in Hepatocellular Carcinoma and Predicts Prognosis of Hepatocellular Carcinoma Patients. J. Gastroenterol. Hepatol. 2011, 26, 1630–1637. [Google Scholar] [CrossRef] [PubMed]
- Shang, W.; Chen, X.; Nie, L.; Xu, M.; Chen, N.; Zeng, H.; Zhou, Q. MiR199b Suppresses Expression of Hypoxia-Inducible Factor 1α (HIF-1α) in Prostate Cancer Cells. Int. J. Mol. Sci. 2013, 14, 8422–8436. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Zhao, Y.; Guo, B. MiR-199b-5p Targets HER2 in Breast Cancer Cells. J. Cell Biochem. 2013, 114, 1457–1463. [Google Scholar] [CrossRef]
- Joshi, D.; Chandrakala, S.; Korgaonkar, S.; Ghosh, K.; Vundinti, B.R. Down-Regulation of miR-199b Associated with Imatinib Drug Resistance in 9q34.1 Deleted BCR/ABL Positive CML Patients. Gene 2014, 542, 109–112. [Google Scholar] [CrossRef]
- Liu, M.X.; Siu, M.K.Y.; Liu, S.S.; Yam, J.W.P.; Ngan, H.Y.S.; Chan, D.W. Epigenetic Silencing of microRNA-199b-5p Is Associated with Acquired Chemoresistance via Activation of JAG1-Notch1 Signaling in Ovarian Cancer. Oncotarget 2014, 5, 944–958. [Google Scholar] [CrossRef]
- Garzia, L.; Andolfo, I.; Cusanelli, E.; Marino, N.; Petrosino, G.; De Martino, D.; Esposito, V.; Galeone, A.; Navas, L.; Esposito, S.; et al. MicroRNA-199b-5p Impairs Cancer Stem Cells through Negative Regulation of HES1 in Medulloblastoma. PLoS ONE 2009, 4, e4998. [Google Scholar] [CrossRef]
- Wyder, L.; Vitaliti, A.; Schneider, H.; Hebbard, L.W.; Moritz, D.R.; Wittmer, M.; Ajmo, M.; Klemenz, R. Increased Expression of H/T-Cadherin in Tumor-Penetrating Blood Vessels. Cancer Res. 2000, 60, 4682–4688. [Google Scholar]
- Whitehead, J.P.; Richards, A.A.; Hickman, I.J.; Macdonald, G.A.; Prins, J.B. Adiponectin—A Key Adipokine in the Metabolic Syndrome. Diabetes Obes. Metab. 2006, 8, 264–280. [Google Scholar] [CrossRef] [PubMed]
- Achari, A.E.; Jain, S.K. Adiponectin, a Therapeutic Target for Obesity, Diabetes, and Endothelial Dysfunction. Int. J. Mol. Sci. 2017, 18, 1321. [Google Scholar] [CrossRef]
- Baltrūnienė, V.; Rinkūnaitė, I.; Bogomolovas, J.; Bironaitė, D.; Kažukauskienė, I.; Šimoliūnas, E.; Ručinskas, K.; Puronaitė, R.; Bukelskienė, V.; Grabauskienė, A.V. The Role of Cardiac T-Cadherin in the Indicating Heart Failure Severity of Patients with Non-Ischemic Dilated Cardiomyopathy. Medicina 2020, 56, 27. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, K.; Wang, Z.; Fa, Z. MicroRNA-377-3p Promotes Cell Proliferation and Inhibits Cell Cycle Arrest and Cell Apoptosis in Hepatocellular Carcinoma by Affecting EGR1-Mediated P53 Activation. Pathol.-Res. Pract. 2022, 234, 153855. [Google Scholar] [CrossRef] [PubMed]
- Di Palo, A.; Siniscalchi, C.; Polito, R.; Nigro, E.; Russo, A.; Daniele, A.; Potenza, N. microRNA-377-3p Downregulates the Oncosuppressor T-cadherin in Colorectal Adenocarcinoma Cells. Cell Biol. Int. 2021, 45, 1797–1803. [Google Scholar] [CrossRef]
- Liu, F.-F.; Zhang, Z.; Chen, W.; Gu, H.-Y.; Yan, Q.-J. Regulatory Mechanism of microRNA-377 on CDH13 Expression in the Cell Model of Alzheimer’s Disease. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2801–2808. [Google Scholar] [CrossRef]
- Dalamaga, M.; Diakopoulos, K.N.; Mantzoros, C.S. The Role of Adiponectin in Cancer: A Review of Current Evidence. Endocr. Rev. 2012, 33, 547–594. [Google Scholar] [CrossRef]
- Hefetz-Sela, S.; Scherer, P.E. Adipocytes: Impact on Tumor Growth and Potential Sites for Therapeutic Intervention. Pharmacol. Ther. 2013, 138, 197–210. [Google Scholar] [CrossRef]
- Otake, S.; Takeda, H.; Suzuki, Y.; Fukui, T.; Watanabe, S.; Ishihama, K.; Saito, T.; Togashi, H.; Nakamura, T.; Matsuzawa, Y.; et al. Association of Visceral Fat Accumulation and Plasma Adiponectin with Colorectal Adenoma: Evidence for Participation of Insulin Resistance. Clin. Cancer Res. 2005, 11, 3642–3646. [Google Scholar] [CrossRef]
- Ishikawa, M.; Kitayama, J.; Kazama, S.; Hiramatsu, T.; Hatano, K.; Nagawa, H. Plasma Adiponectin and Gastric Cancer. Clin. Cancer Res. 2005, 11, 466–472. [Google Scholar] [CrossRef]
- Otani, K.; Ishihara, S.; Yamaguchi, H.; Murono, K.; Yasuda, K.; Nishikawa, T.; Tanaka, T.; Kiyomatsu, T.; Hata, K.; Kawai, K.; et al. Adiponectin and Colorectal Cancer. Surg. Today 2017, 47, 151–158. [Google Scholar] [CrossRef]
- Polito, R.; Nigro, E.; Fei, L.; DE Magistris, L.; Monaco, M.L.; D’Amico, R.; Naviglio, S.; Signoriello, G.; Daniele, A. Adiponectin Is Inversely Associated With Tumour Grade in Colorectal Cancer Patients. Anticancer Res. 2020, 40, 3751–3757. [Google Scholar] [CrossRef]
- Liu, W.-Y.; Yang, Z.; Sun, Q.; Yang, X.; Hu, Y.; Xie, H.; Gao, H.-J.; Guo, L.-M.; Yi, J.-Y.; Liu, M.; et al. miR-377-3p Drives Malignancy Characteristics via Upregulating GSK-3β Expression and Activating NF-κB Pathway in hCRC Cells. J. Cell Biochem. 2018, 119, 2124–2134. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Wu, Y.; Deng, Z.; Zhou, Y.; Song, T.; Yang, Y.; Zhang, X.; Xu, T.; Xia, M.; Cai, A.; et al. MiR-377 Promotes White Adipose Tissue Inflammation and Decreases Insulin Sensitivity in Obesity via Suppression of Sirtuin-1 (SIRT1). Oncotarget 2017, 8, 70550–70563. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wu, Y.; Hedrick, N.; Gutmann, D.H. T-Cadherin-Mediated Cell Growth Regulation Involves G2 Phase Arrest and Requires P21(CIP1/WAF1) Expression. Mol. Cell Biol. 2003, 23, 566–578. [Google Scholar] [CrossRef]
- Li, L.; Gao, Y.; Yu, B.; Zhang, J.; Ma, G.; Jin, X. Role of LncRNA H19 in Tumor Progression and Treatment. Mol. Cell Probes 2024, 75, 101961. [Google Scholar] [CrossRef] [PubMed]
- Keniry, A.; Oxley, D.; Monnier, P.; Kyba, M.; Dandolo, L.; Smits, G.; Reik, W. The H19 lincRNA Is a Developmental Reservoir of miR-675 That Suppresses Growth and Igf1r. Nat. Cell Biol. 2012, 14, 659–665. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, Y.; Luan, W.; Wang, P.; Tao, T.; Zhang, J.; Qian, J.; Liu, N.; You, Y. Long Non-Coding RNA H19 Promotes Glioma Cell Invasion by Deriving miR-675. PLoS ONE 2014, 9, e86295. [Google Scholar] [CrossRef]
- Li, L.; Chen, W.; Wang, Y.; Tang, L.; Han, M. Long Non-Coding RNA H19 Regulates Viability and Metastasis, and Is Upregulated in Retinoblastoma. Oncol. Lett. 2018, 15, 8424–8432. [Google Scholar] [CrossRef]
- Liang, W.-C.; Fu, W.-M.; Wong, C.-W.; Wang, Y.; Wang, W.-M.; Hu, G.-X.; Zhang, L.; Xiao, L.-J.; Wan, D.C.-C.; Zhang, J.-F.; et al. The lncRNA H19 Promotes Epithelial to Mesenchymal Transition by Functioning as miRNA Sponges in Colorectal Cancer. Oncotarget 2015, 6, 22513–22525. [Google Scholar] [CrossRef]
- Lin, R.-K.; Hsu, H.-S.; Chang, J.-W.; Chen, C.-Y.; Chen, J.-T.; Wang, Y.-C. Alteration of DNA Methyltransferases Contributes to 5′CpG Methylation and Poor Prognosis in Lung Cancer. Lung Cancer 2007, 55, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, Y.; Bai, F.; Zhang, J.-Y.; Ma, S.-H.; Liu, J.; Xu, Z.-D.; Zhu, H.-G.; Ling, Z.-Q.; Ye, D.; et al. Tumor Development Is Associated with Decrease of TET Gene Expression and 5-Methylcytosine Hydroxylation. Oncogene 2013, 32, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Masalmeh, R.H.A.; Taglini, F.; Rubio-Ramon, C.; Musialik, K.I.; Higham, J.; Davidson-Smith, H.; Kafetzopoulos, I.; Pawlicka, K.P.; Finan, H.M.; Clark, R.; et al. De Novo DNA Methyltransferase Activity in Colorectal Cancer Is Directed towards H3K36me3 Marked CpG Islands. Nat. Commun. 2021, 12, 694. [Google Scholar] [CrossRef]
- Maćkowska, N.; Drobna-Śledzińska, M.; Witt, M.; Dawidowska, M. DNA Methylation in T-Cell Acute Lymphoblastic Leukemia: In Search for Clinical and Biological Meaning. Int. J. Mol. Sci. 2021, 22, 1388. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Tao, S.; Li, Q.; Deng, B.; Tan, Q.-Y.; Jin, H. The miR-23a/27a/24-2 Cluster Promotes Postoperative Progression of Early-Stage Non-Small Cell Lung Cancer. Mol. Ther.-Oncolytics 2022, 24, 205–217. [Google Scholar] [CrossRef]
- Zareifar, P.; Ahmed, H.M.; Ghaderi, P.; Farahmand, Y.; Rahnama, N.; Esbati, R.; Moradi, A.; Yazdani, O.; Sadeghipour, Y. miR-142-3p/5p Role in Cancer: From Epigenetic Regulation to Immunomodulation. Cell Biochem. Funct. 2024, 42, e3931. [Google Scholar] [CrossRef]
- Ma, G.; Li, Y.; Meng, F.; Sui, C.; Wang, Y.; Cheng, D. Hsa_circ_0000119 Promoted Ovarian Cancer Development via Enhancing the Methylation of CDH13 by Sponging miR-142-5p. J. Biochem. Mol. Toxicol. 2023, 37, e23264. [Google Scholar] [CrossRef]
- Wang, H.; Cao, D.; Wu, F. Long Noncoding RNA UPAT Promoted Cell Proliferation via Increasing UHRF1 Expression in Non-Small Cell Lung Cancer. Oncol. Lett. 2018, 16, 1491–1498. [Google Scholar] [CrossRef]
- Taniue, K.; Kurimoto, A.; Sugimasa, H.; Nasu, E.; Takeda, Y.; Iwasaki, K.; Nagashima, T.; Okada-Hatakeyama, M.; Oyama, M.; Kozuka-Hata, H.; et al. Long Noncoding RNA UPAT Promotes Colon Tumorigenesis by Inhibiting Degradation of UHRF1. Proc. Natl. Acad. Sci. USA 2016, 113, 1273–1278. [Google Scholar] [CrossRef]
- Tan, W.-H.; Peng, Z.-L.; You, T.; Sun, Z.-L. CTRP15 Promotes Macrophage Cholesterol Efflux and Attenuates Atherosclerosis by Increasing the Expression of ABCA1. J. Physiol. Biochem. 2022, 78, 653–666. [Google Scholar] [CrossRef]
- Seldin, M.M.; Peterson, J.M.; Byerly, M.S.; Wei, Z.; Wong, G.W. Myonectin (CTRP15), a Novel Myokine That Links Skeletal Muscle to Systemic Lipid Homeostasis. J. Biol. Chem. 2012, 287, 11968–11980. [Google Scholar] [CrossRef]
- Cai, Y.; Liu, J.; Cai, S.-K.; Miao, E.-Y.; Jia, C.-Q.; Fan, Y.-Z.; Li, Y.-B. Eicosapentaenoic Acid’s Metabolism of 15-LOX-1 Promotes the Expression of miR-101 Thus Inhibits Cox2 Pathway in Colon Cancer. OncoTargets Ther. 2020, 13, 5605–5616. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Ye, F.; Xie, X.; Li, X.; Tang, H.; Li, S.; Huang, X.; Song, C.; Wei, W.; Xie, X. Mir-101-3p Is a Key Regulator of Tumor Metabolism in Triple Negative Breast Cancer Targeting AMPK. Oncotarget 2016, 7, 35188–35198. [Google Scholar] [CrossRef] [PubMed]
- Rincón-Riveros, A.; Morales, D.; Rodríguez, J.A.; Villegas, V.E.; López-Kleine, L. Bioinformatic Tools for the Analysis and Prediction of ncRNA Interactions. Int. J. Mol. Sci. 2021, 22, 11397. [Google Scholar] [CrossRef]
- Wang, H.; Mazzocca, A.; Gao, P. Cadherin Dysregulation in Gastric Cancer: Insights into Gene Expression, Pathways, and Prognosis. J. Gastrointest. Oncol. 2023, 14, 2064–2082. [Google Scholar] [CrossRef] [PubMed]
- Coban, N.; Pirim, D.; Erkan, A.F.; Dogan, B.; Ekici, B. Hsa-miR-584-5p as a Novel Candidate Biomarker in Turkish Men with Severe Coronary Artery Disease. Mol. Biol. Rep. 2020, 47, 1361–1369. [Google Scholar] [CrossRef]
- Lee, H.; Park, C.S.; Deftereos, G.; Morihara, J.; Stern, J.E.; Hawes, S.E.; Swisher, E.; Kiviat, N.B.; Feng, Q. MicroRNA Expression in Ovarian Carcinoma and Its Correlation with Clinicopathological Features. World J. Surg. Oncol. 2012, 10, 174. [Google Scholar] [CrossRef]
- Feng, Q.; Deftereos, G.; Hawes, S.E.; Stern, J.E.; Willner, J.B.; Swisher, E.M.; Xi, L.; Drescher, C.; Urban, N.; Kiviat, N. DNA Hypermethylation, Her-2/Neu Overexpression and P53 Mutations in Ovarian Carcinoma. Gynecol. Oncol. 2008, 111, 320–329. [Google Scholar] [CrossRef]
- Ducoli, L.; Agrawal, S.; Sibler, E.; Kouno, T.; Tacconi, C.; Hon, C.-C.; Berger, S.D.; Müllhaupt, D.; He, Y.; Kim, J.; et al. LETR1 Is a Lymphatic Endothelial-Specific lncRNA Governing Cell Proliferation and Migration through KLF4 and SEMA3C. Nat. Commun. 2021, 12, 925. [Google Scholar] [CrossRef]
- David, C.J.; Massagué, J. Contextual Determinants of TGFβ Action in Development, Immunity and Cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Vanlandewijck, M.; Moustakas, A. Regulation of EMT by TGFβ in Cancer. FEBS Lett. 2012, 586, 1959–1970. [Google Scholar] [CrossRef] [PubMed]
- Gélabert, C.; Papoutsoglou, P.; Golán, I.; Ahlström, E.; Ameur, A.; Heldin, C.-H.; Caja, L.; Moustakas, A. The Long Non-Coding RNA LINC00707 Interacts with Smad Proteins to Regulate TGFβ Signaling and Cancer Cell Invasion. Cell Commun. Signal 2023, 21, 271. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Guo, Y.; Ndandala, C.B.; Chen, H.; Huang, C.; Zhao, G.; Huang, H.; Li, G.; Chen, H. Analysis of circRNA and miRNA Expression Profiles in IGF3-Induced Ovarian Maturation in Spotted Scat (Scatophagus Argus). Front. Endocrinol. 2022, 13, 998207. [Google Scholar] [CrossRef]
- Coen, S.; Keogh, K.; Lonergan, P.; Fair, S.; Kenny, D.A. Early Life Nutrition Affects the Molecular Ontogeny of Testicular Development in the Young Bull Calf. Sci. Rep. 2023, 13, 6748. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).