
Coagulation Factor XII Is an Antibacterial Protein That Acts Against Bacterial Infection via Its Heavy Chain

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. FXII Exhibits Antibacterial Activity In Vitro
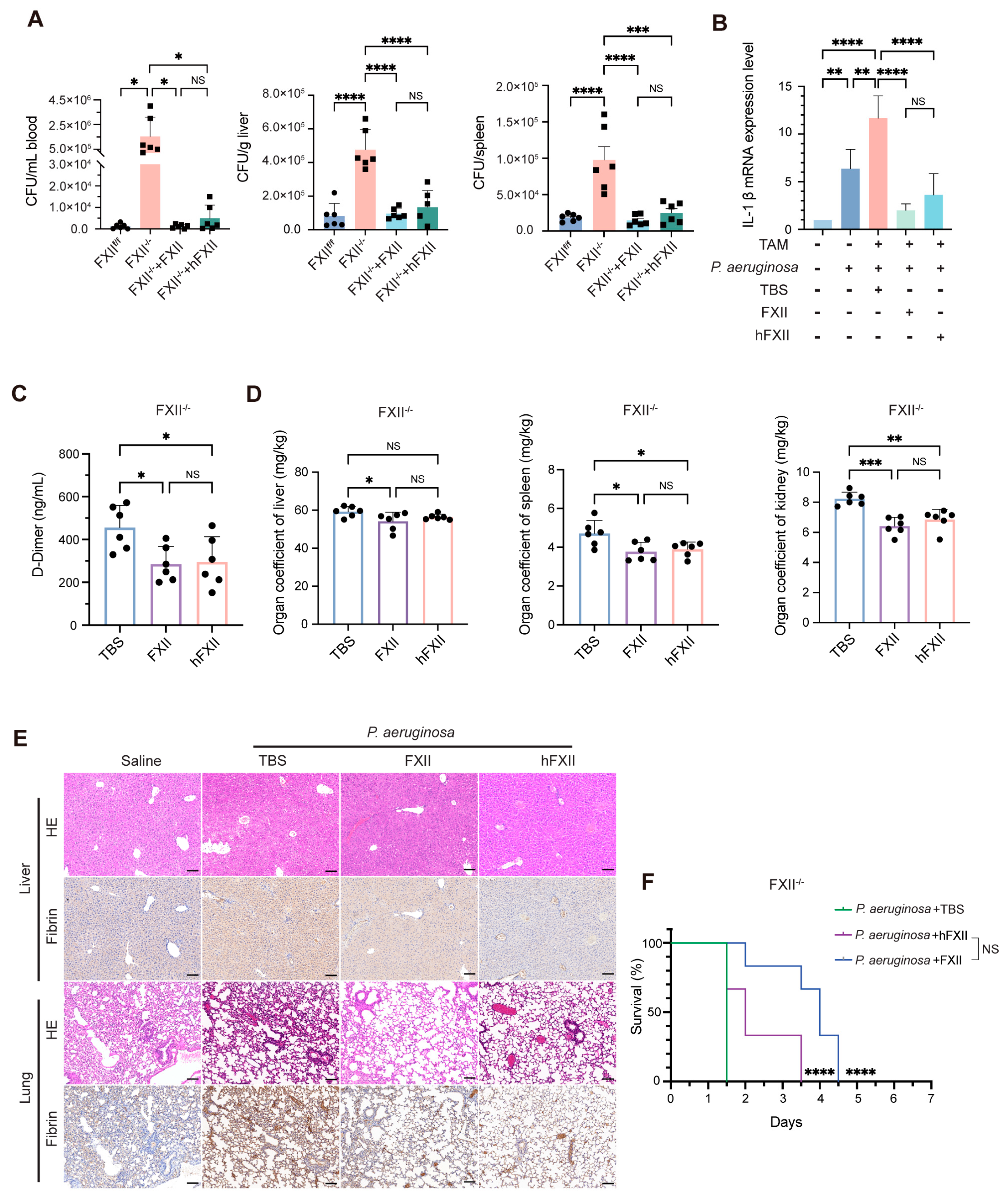
2.2. FXII Deficiency Impairs Antibacterial Capacity
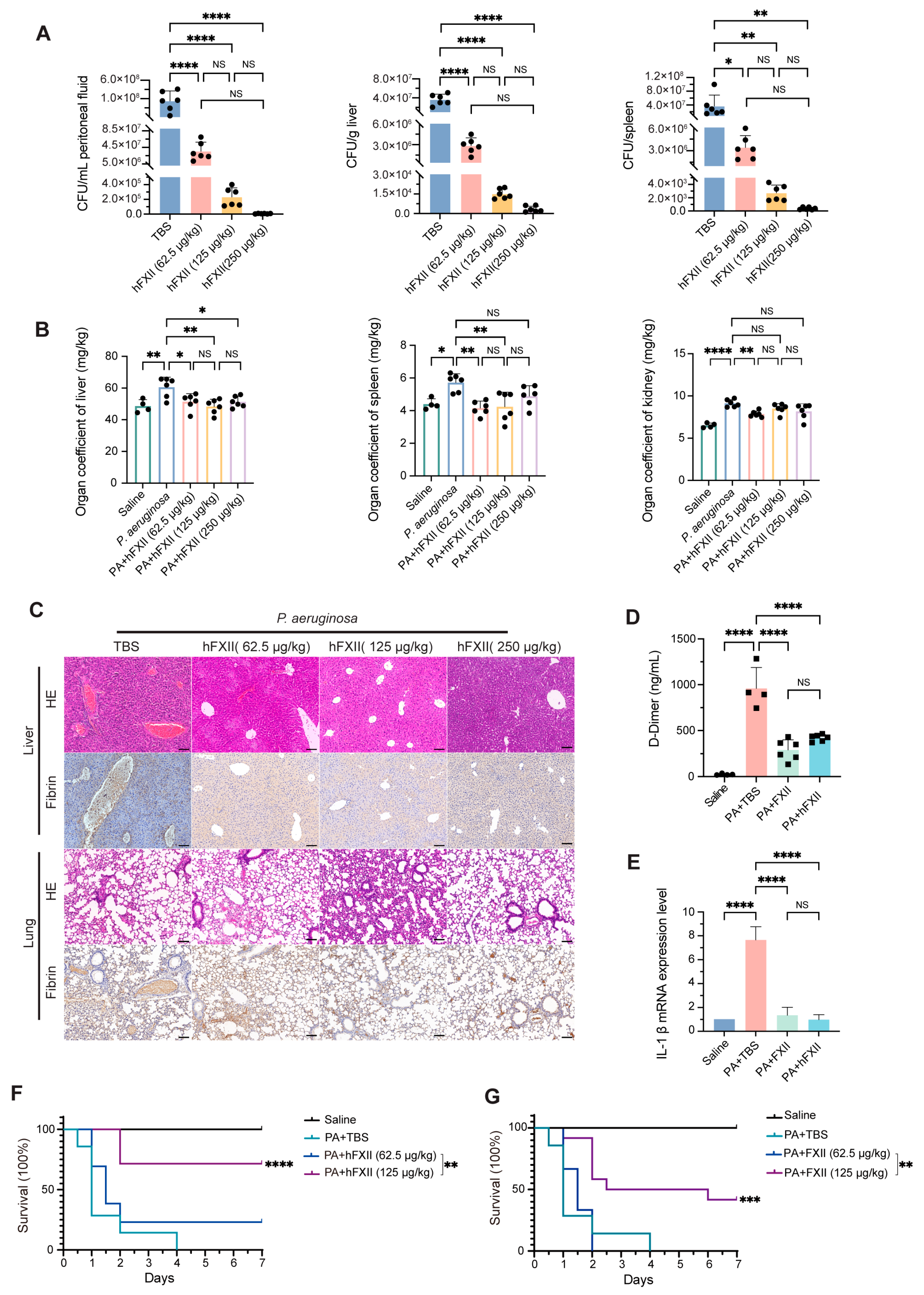
2.3. Recombinant Proteins Restore Antibacterial Capacity Impaired by FXII Deficiency
2.4. Antibacterial Role of FXII in a DIC Model
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Bacterial Strain
4.2. Mice
- FXII-P3: 5′-ACTGTTGGGCGCTGGTTGG-3′;
- FXII-P4: 5′-GCTGGATTTGGTCATGGTGTTT-3′;
- Cre-Mut-F: 5′-TGCGGGCTCTACTTCATCG-3′;
- Cre-Mut-R: 5′-GAGGGGGAAAGCGAAAGTCC-3′;
- Cre-Wt-F: 5′-AGTCGCTCTGAGTTGTTATCAG-3′;
- Cre-Wt-R: 5′-TGAGCATGTCTTTAATCTACCTCGATG-3′.
4.3. Protein Production
4.4. Growth Kinetics Analysis
4.5. Analysis of LPS Degradation by Electrophoresis
4.6. Western Blot and ELISA
4.7. Quantitative Real-Time Polymerase Chain Reaction
- FXII-F: 5′-AATCCGTGCCTTAATGGGGG-3′,
- FXII-R: 5′-TCATAGCAGGTCGCCCAAAG-3′;
- GAPDH-F: 5′-AGGTCGGTGTGAACGGATTTG-3′,
- GAPDH-R: 5′-TGTAGACCATGTAGTTGAGGTCA-3′;
- IL-1β-F: 5′-TCAAATCTCGCAGCAGCACATC-3′,
- IL-1β-R: 5′-CGTCACACACCAGCAGGTTATC-3′.
4.8. Bacterial Load in Blood, Peritoneal Fluid, Liver, and Spleen
4.9. Measurement of Coagulation Parameters
4.10. Liver and Lung Histology and IHC
4.11. Antibodies and Reagents
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Palta, S.; Saroa, R.; Palta, A. Overview of the coagulation system. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef]
- Dahlback, B. Blood coagulation. Lancet 2000, 355, 1627–1632. [Google Scholar] [CrossRef] [PubMed]
- Renne, T.; Schmaier, A.H.; Nickel, K.F.; Blomback, M.; Maas, C. In Vivo roles of factor XII. Blood 2012, 120, 4296–4303. [Google Scholar] [CrossRef]
- Shamanaev, A.; Ivanov, I.; Sun, M.F.; Litvak, M.; Srivastava, P.; Mohammed, B.M.; Shaban, R.; Maddur, A.; Verhamme, I.M.; McCarty, O.J.T.; et al. Model for surface-dependent factor XII activation: The roles of factor XII heavy chain domains. Blood Adv. 2022, 6, 3142–3154. [Google Scholar] [CrossRef]
- de Maat, S.; Maas, C. Factor XII: Form determines function. J. Thromb. Haemost. 2016, 14, 1498–1506. [Google Scholar] [CrossRef] [PubMed]
- Renne, T. The procoagulant and proinflammatory plasma contact system. Semin. Immunopathol. 2012, 34, 31–41. [Google Scholar] [CrossRef]
- Xu, P.; Zhang, Y.; Guo, J.; Li, H.; Konrath, S.; Zhou, P.; Cai, L.; Rao, H.; Chen, H.; Lin, J.; et al. A single-domain antibody targeting factor XII inhibits both thrombosis and inflammation. Nat. Commun. 2024, 15, 7898. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y. The plasma contact system as a modulator of innate immunity. Curr. Opin. Hematol. 2018, 25, 389–394. [Google Scholar] [CrossRef]
- Revenko, A.S.; Gao, D.; Crosby, J.R.; Bhattacharjee, G.; Zhao, C.; May, C.; Gailani, D.; Monia, B.P.; MacLeod, A.R. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood 2011, 118, 5302–5311. [Google Scholar] [CrossRef]
- Yau, J.W.; Liao, P.; Fredenburgh, J.C.; Stafford, A.R.; Revenko, A.S.; Monia, B.P.; Weitz, J.I. Selective depletion of factor XI or factor XII with antisense oligonucleotides attenuates catheter thrombosis in rabbits. Blood 2014, 123, 2102–2107. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Y.; Kang, H.; Wu, J.; Wang, Z.; Liu, Y.; Chen, F.; et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity 2019, 51, 983–996.e986. [Google Scholar] [CrossRef]
- Renne, T.; Pozgajova, M.; Gruner, S.; Schuh, K.; Pauer, H.U.; Burfeind, P.; Gailani, D.; Nieswandt, B. Defective thrombus formation in mice lacking coagulation factor XII. J. Exp. Med. 2005, 202, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Gruber, A. The role of the contact pathway in thrombus propagation. Thromb. Res. 2014, 133 (Suppl. S1), S45–S47. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. Brit. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar] [CrossRef]
- Brown, E.D.; Wright, G.D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. [Google Scholar] [CrossRef]
- Global Leaders Group on Antimicrobial Resistance. Towards Specific Commitments and Action in the Response to Antimicrobial Resistance: Recommendations for Consideration by UN Member States in the Outcome Document of the High-Level Meeting on AMR in September 2024; Global Leaders Group on Antimicrobial Resistance: Bridgetown, Barbados, 2024. [Google Scholar]
- Ferreira, A.R.; Ferreira, M.; Nunes, C.; Reis, S.; Teixeira, C.; Gomes, P.; Gameiro, P. The unusual aggregation and fusion activity of the antimicrobial peptide W-BP100 in anionic vesicles. Membranes 2023, 13, 138. [Google Scholar] [CrossRef]
- World Health Organization. Global Research Agenda for Antimicrobial Resistance in Human Health; World Health Organization: Geneva, Switzerland, 2024; ISBN 978-92-4-010230-9.
- Oehmcke, S.; Herwald, H. Contact system activation in severe infectious diseases. J. Mol. Med. 2010, 88, 121–126. [Google Scholar] [CrossRef]
- Renne, T.; Stavrou, E.X. Roles of factor XII in innate immunity. Front. Immunol. 2019, 10, 2011. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, X.; Tang, Y.; Qiu, X.; Wang, Z.; Fu, G.; Wu, J.; Kang, H.; Wang, J.; Wang, H.; et al. The role of type 1 interferons in coagulation induced by gram-negative bacteria. Blood 2020, 135, 1087–1100. [Google Scholar] [CrossRef]
- Popescu, N.I.; Lupu, C.; Lupu, F. Disseminated intravascular coagulation and its immune mechanisms. Blood 2022, 139, 1973–1986. [Google Scholar] [CrossRef]
- Mazuski, J.E.; Solomkin, J.S. Intra-abdominal infections. Surg. Clin. N. Am. 2009, 89, 421–437. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.L.; Albin, O.R.; Mobley, H.L.T.; Bachman, M.A. Bloodstream infections: Mechanisms of pathogenesis and opportunities for intervention. Nat. Rev. Microbiol. 2025, 23, 210–224. [Google Scholar] [CrossRef]
- Long, A.T.; Kenne, E.; Jung, R.; Fuchs, T.A.; Renne, T. Contact system revisited: An interface between inflammation, coagulation, and innate immunity. J. Thromb. Haemost. 2016, 14, 427–437. [Google Scholar] [CrossRef]
- Jiang, H.; Guo, Y.; Wang, Q.; Wang, Y.; Peng, D.; Fang, Y.; Yan, L.; Ruan, Z.; Zhang, S.; Zhao, Y.; et al. The dysfunction of complement and coagulation in diseases: The implications for the therapeutic interventions. MedComm 2024, 5, e785–e810. [Google Scholar] [CrossRef] [PubMed]
- Lavranou, G.A.; Mentzelopoulos, S.; Katsaounou, P.; Siempos, I.; Kalomenidis, I.; Geranaki, A.; Routsi, C.; Zakynthinos, S. Can coagulation cystem disorders and cytokine and inflammatory marker levels predict the temporary clinical deterioration or improvement of septic patients on ICU admission? J. Clin. Med. 2021, 10, 1548. [Google Scholar] [CrossRef]
- Sala-Cunill, A.; Björkqvist, J.; Senter, R.; Guilarte, M.; Cardona, V.; Labrador, M.; Nickel, K.F.; Butler, L.; Luengo, O.; Kumar, P.; et al. Plasma contact system activation drives anaphylaxis in severe mast cell–mediated allergic reactions. J. Allergy Clin. Immunol. 2015, 135, 1031–1043.e6. [Google Scholar] [CrossRef] [PubMed]
- Berends, E.T.; Kuipers, A.; Ravesloot, M.M.; Urbanus, R.T.; Rooijakkers, S.H. Bacteria under stress by complement and coagulation. FEMS Microbiol. Rev. 2014, 38, 1146–1171. [Google Scholar] [CrossRef]
- Chen, J.; Li, X.; Li, L.; Zhang, T.; Zhang, Q.; Wu, F.; Wang, D.; Hu, H.; Tian, C.; Liao, D.; et al. Coagulation factors VII, IX and X are effective antibacterial proteins against drug-resistant Gram-negative bacteria. Cell Res. 2019, 29, 711–724. [Google Scholar] [CrossRef]
- Maas, C.; Renné, T. Coagulation factor XII in thrombosis and inflammation. Blood 2018, 131, 1903. [Google Scholar] [CrossRef]
- Schmaier, A.H. The elusive physiologic role of Factor XII. J. Clin. Investig. 2008, 118, 3006–3009. [Google Scholar] [CrossRef]
- Feil, S.; Valtcheva, N.; Feil, R. Inducible Cre mice. Methods Mol. Biol. 2009, 530, 343–363. [Google Scholar] [CrossRef] [PubMed]
- Bachler, M.; Niederwanger, C.; Hell, T.; Hofer, J.; Gerstmeyr, D.; Schenk, B.; Treml, B.; Fries, D. Influence of factor XII deficiency on activated partial thromboplastin time (aPTT) in critically ill patients. J. Thromb. Thrombolysis 2019, 48, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I.; Fredenburgh, J.C.; Eikelboom, J.W. A Test in Context: D-Dimer. J. Am. Coll. Cardiol. 2017, 70, 2411–2420. [Google Scholar] [CrossRef]
- Tsantes, A.G.; Parastatidou, S.; Tsantes, E.A.; Bonova, E.; Tsante, K.A.; Mantzios, P.G.; Vaiopoulos, A.G.; Tsalas, S.; Konstantinidi, A.; Houhoula, D.; et al. Sepsis-induced coagulopathy: An update on pathophysiology, biomarkers, and current guidelines. Life 2023, 13, 350. [Google Scholar] [CrossRef] [PubMed]
- Shet, A.S. FXII and sickle cell: The clot thickens. Blood 2023, 141, 1787–1789. [Google Scholar] [CrossRef]
- Sparkenbaugh, E.M.; Henderson, M.W.; Miller-Awe, M.; Abrams, C.; Ilich, A.; Trebak, F.; Ramadas, N.; Vital, S.; Bohinc, D.; Bane, K.L.; et al. Factor XII contributes to thrombotic complications and vaso-occlusion in sickle cell disease. Blood 2023, 141, 1871–1883. [Google Scholar] [CrossRef]
- Sargis, T.; Youn, S.W.; Thakkar, K.; Naiche, L.A.; Paik, N.Y.; Pajcini, K.V.; Kitajewski, J.K. Notch1 and Notch4 core binding domain peptibodies exhibit distinct ligand-binding and anti-angiogenic properties. Angiogenesis 2023, 26, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Tombling, B.J.; Wang, C.K.; Craik, D.J. EGF-like and other disulfide-rich microdomains as therapeutic scaffolds. Angew Chem Int. Ed. Engl. 2020, 59, 11218–11232. [Google Scholar] [CrossRef]
- Nickel, K.F.; Jamsa, A.; Konrath, S.; Papareddy, P.; Butler, L.M.; Stavrou, E.X.; Frye, M.; Gelderblom, M.; Nieswandt, B.; Hammerschmidt, S.; et al. Factor XII-driven coagulation traps bacterial infections. J. Exp. Med. 2025, 222, e20250049. [Google Scholar] [CrossRef]
- Tirsoaga, A.; Novikov, A.; Adib-Conquy, M.; Werts, C.; Fitting, C.; Cavaillon, J.M.; Caroff, M. Simple method for repurification of endotoxins for biological use. Appl. Environ. Microbiol. 2007, 73, 1803–1808. [Google Scholar] [CrossRef]
- Westphal, O.; Jann, J.K. Bacterial lipopolysaccharides: Extraction with phenol-water and further appplications of the procedure. Methods Biochem. Anal. 1964, 43, 83–91. [Google Scholar]
- Hood, D.W.; Makepeace, K.; Deadman, M.E.; Rest, R.F.; Moxon, E.R. Sialic acid in the lipopolysaccharide of Haemophilus influenzae: Strain distribution, influence on serum resistance and structural characterization. Mol. Microbiol. 2010, 33, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Fomsgaard, A.; Freudenberg, M.A.; Galanos, C. Modification of the silver staining technique to detect lipopolysaccharide in polyacrylamide gels. J. Clin. Microbiol. 1990, 28, 2627–2631. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.J.; O’Brien-Simpson, N.M.; Pantarat, N.; Sulistio, A.; Wong, E.H.H.; Chen, Y.-Y.; Lenzo, J.C.; Holden, J.A.; Blencowe, A.; Reynolds, E.C.; et al. Combating multidrug-resistant Gram-negative bacteria with structurally nanoengineered antimicrobial peptide polymers. Nat. Microbiol. 2016, 1, 16162. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Takahashi, K.; Dundee, J.; Shahroor-Karni, S.; Thiel, S.; Jensenius, J.C.; Gad, F.; Hamblin, M.R.; Sastry, K.N.; Ezekowitz, R.A.B. Mannose-binding lectin-deficient mice are susceptible to infection with Staphylococcus aureus. J. Exp. Med. 2004, 199, 1379–1390. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Wang, D.; Pan, S.; Song, X. Coagulation Factor XII Is an Antibacterial Protein That Acts Against Bacterial Infection via Its Heavy Chain. Int. J. Mol. Sci. 2025, 26, 6009. https://doi.org/10.3390/ijms26136009
Liu J, Wang D, Pan S, Song X. Coagulation Factor XII Is an Antibacterial Protein That Acts Against Bacterial Infection via Its Heavy Chain. International Journal of Molecular Sciences. 2025; 26(13):6009. https://doi.org/10.3390/ijms26136009
Chicago/Turabian StyleLiu, Junnan, Diyue Wang, Sirui Pan, and Xu Song. 2025. "Coagulation Factor XII Is an Antibacterial Protein That Acts Against Bacterial Infection via Its Heavy Chain" International Journal of Molecular Sciences 26, no. 13: 6009. https://doi.org/10.3390/ijms26136009
APA StyleLiu, J., Wang, D., Pan, S., & Song, X. (2025). Coagulation Factor XII Is an Antibacterial Protein That Acts Against Bacterial Infection via Its Heavy Chain. International Journal of Molecular Sciences, 26(13), 6009. https://doi.org/10.3390/ijms26136009