
Influenza a Virus Inhibition: Evaluating Computationally Identified Cyproheptadine Through In Vitro Assessment

 ,
,  ,
,  ,
,  , , , ,
, , , ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
In Vitro Efficacy of Cyproheptadine Against H1N1 Influenza A Viruses
3. Discussion
4. Materials and Methods
4.1. Dataset Preparation
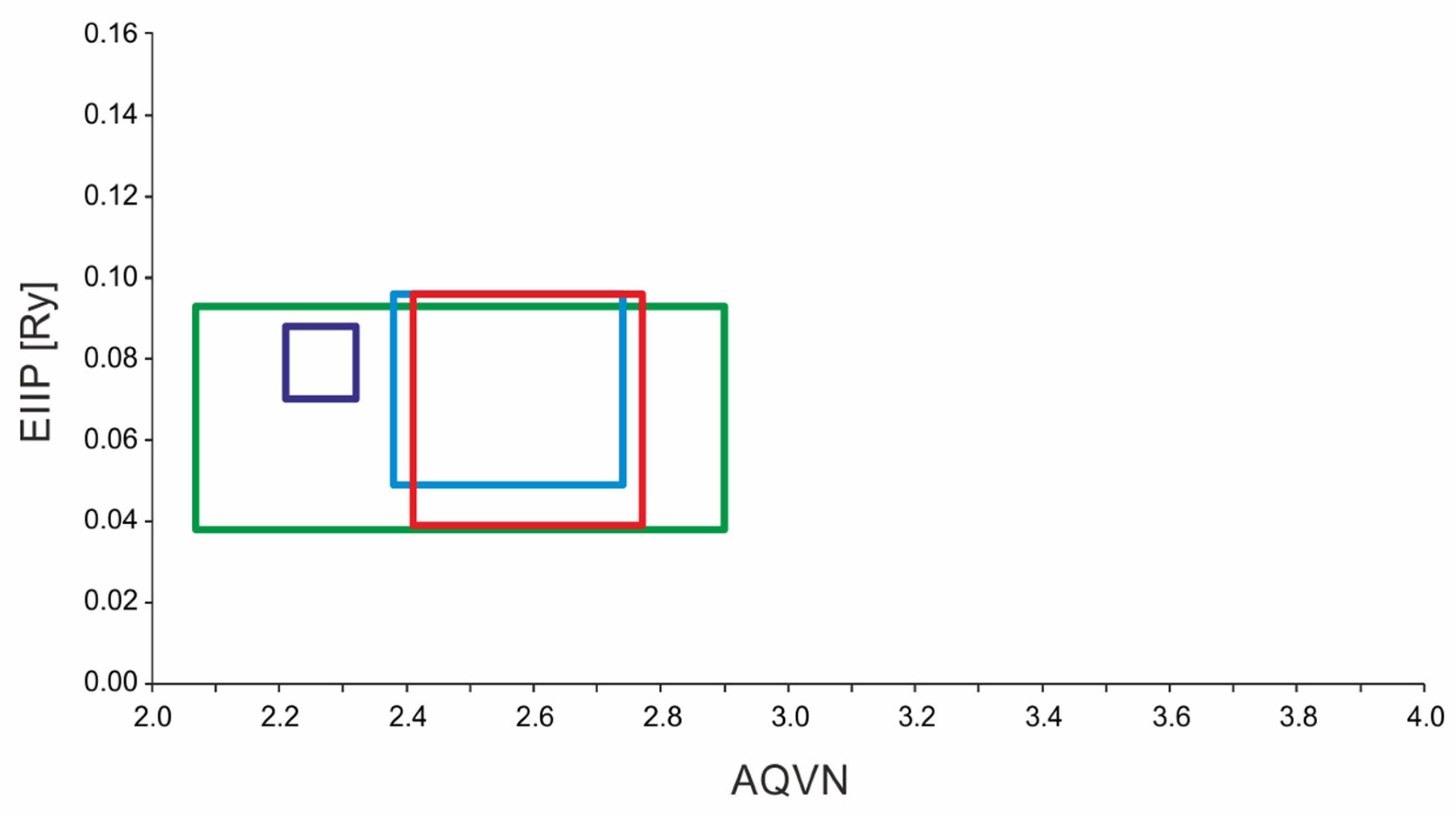
4.2. AQVN/EIIP-Based Virtual Screening
EIIP/AQVN Values
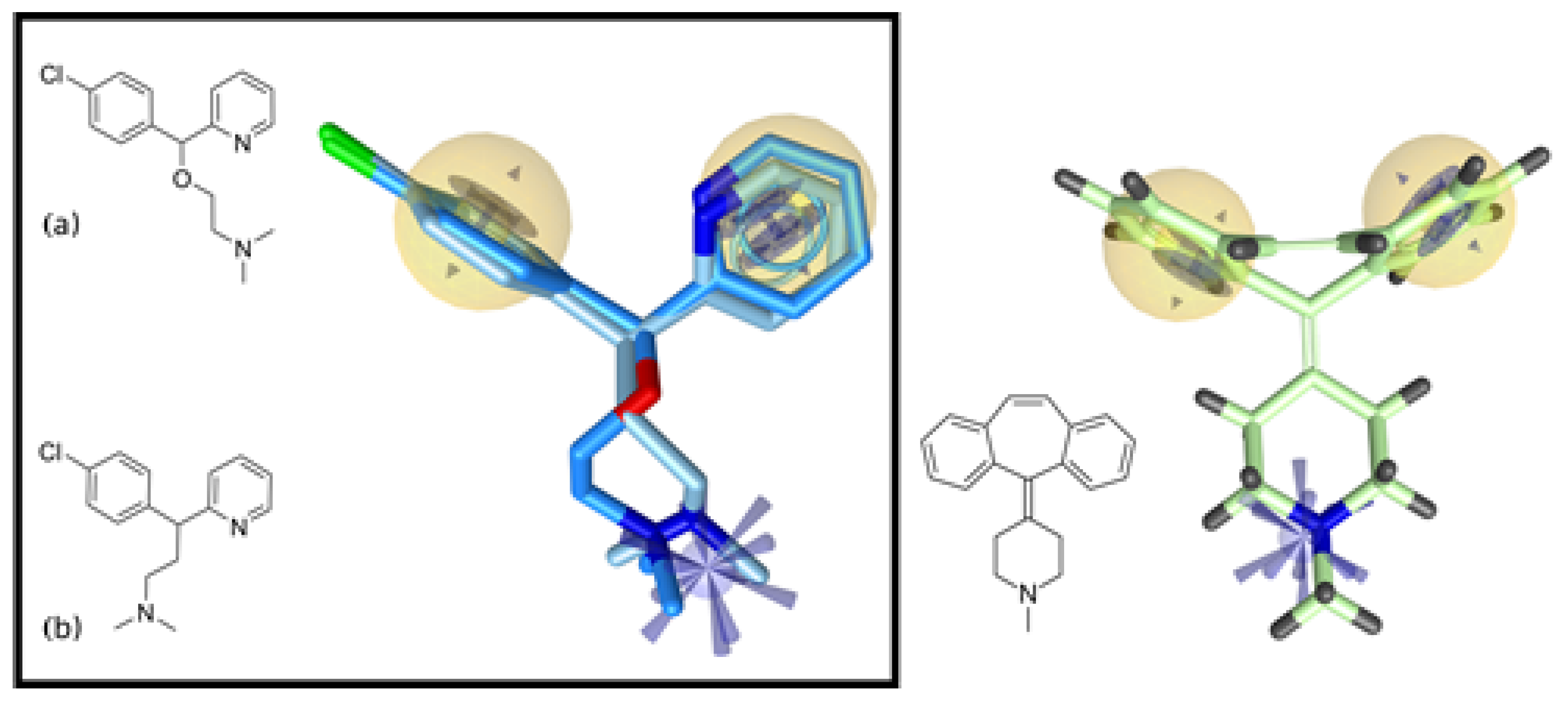
4.3. Pharmacophore Modeling and Screening
4.4. In Vitro Efficacy Testing Against H1N1 Influenza A Viruses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Influenza. Factsheet. March 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/influenza-(seasonal) (accessed on 25 January 2025).
- Bridges, C.; Peasah, S.; Meltzer, M. The control of influenza and cost effectiveness of interventions. In Influenza Textbook; Webster, R.G., Monto, A.S., Braciale, T.J., Lamb, R.A., Eds.; Wiley-Blackwell: Hoboken, NY, USA, 2013; pp. 419–433. [Google Scholar]
- Heo, Y.A. Baloxavir: First global approval. Drugs 2018, 78, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Ison, M.G. Antiviral treatments. Clin. Chest Med. 2017, 38, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Hurt, A.C.; Besselaar, T.G.; Daniels, R.S.; Ermetal, B.; Fry, A.; Gubareva, L.; Huang, W.; Lackenby, A.; Lee, R.T.; Lo, J.; et al. Global update on the susceptibility of human influenza viruses to neuraminidase inhibitors, 2014–2015. Antivir. Res. 2016, 132, 178–185. [Google Scholar] [CrossRef] [PubMed]
- O’Hanlon, R.; Shaw, M.L. Baloxavir marboxil: The new influenza drug on the market. Curr. Opin. Virol. 2019, 35, 14–18. [Google Scholar] [CrossRef]
- Shiraki, K.; Daikoku, T. Favipiravir, an anti-influenza drug against life-threatening RNA virus infections. Pharmacol. Ther. 2020, 209, 107512. [Google Scholar] [CrossRef]
- Bonomini, A.; Mercorelli, B.; Loregian, A. Antiviral strategies against influenza virus: An update on approved and innovative therapeutic approaches. Cell. Mol. Life Sci. 2025, 82, 75. [Google Scholar] [CrossRef]
- Zheng, W.; Sun, W.; Simeonov, A. Drug repurposing screens and synergistic drug-combinations for infectious diseases. Br. J. Pharmacol. 2018, 175, 181–191. [Google Scholar] [CrossRef]
- Strittmatter, S.M. Overcoming drug development bottlenecks with repurposing: Old drugs learn new tricks. Nat. Med. 2014, 20, 590–591. [Google Scholar] [CrossRef]
- Pizzorno, A.; Padey, B.; Terrier, O.; Rosa-Calatrava, M. Drug Repurposing Approaches for the Treatment of Influenza Viral Infection: Reviving Old Drugs to Fight Against a Long-Lived Enemy. Front. Immunol. 2019, 10, 531. [Google Scholar] [CrossRef]
- Tripp, R.A.; Martin, D.E. Repurposing Probenecid to Inhibit SARS-CoV-2, Influenza Virus, and Respiratory Syncytial Virus (RSV) Replication. Viruses 2022, 14, 612. [Google Scholar] [CrossRef]
- Meng, X.; Wang, Y. Drug Repurposing for Influenza Virus Polymerase Acidic (PA) Endonuclease Inhibitor. Molecules 2021, 26, 7326. [Google Scholar] [CrossRef] [PubMed]
- Mercorelli, B.; Palù, G.; Loregian, A. Drug repurposing for viral infectious diseases: How far are we? Trends Microbiol. 2018, 26, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Haffizulla, J.; Hartman, A.; Hoppers, M.; Resnick, H.; Samudrala, S.; Ginocchio, C.; Bardin, M.; Rossignol, J.-F.; US Nitazoxanide Influenza Clinical Study Group. Effect of nitazoxanide in adults and adolescents with acute uncomplicated influenza: A double-blind, randomised, placebo-controlled, phase 2b/3 trial. Lancet Infect. Dis. 2014, 14, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.; Erez, T.; Reynolds, I.J.; Kumar, D.; Ross, J.; Koytiger, G.; Kusko, R.; Zeskind, B.; Risso, S.; Kagan, E.; et al. Drug repurposing from the perspective of pharmaceutical companies. Br. J. Pharmacol. 2018, 175, 168–180. [Google Scholar] [CrossRef]
- McInnes, C. Virtual screening strategies in drug discovery. Curr. Opin. Chem. Biol. 2007, 11, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2013, 66, 334–395. [Google Scholar] [CrossRef]
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful applications of computer aided drug discovery: Moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef]
- Baig, M.H.; Ahmad, K.; Roy, S.; Ashraf, J.M.; Adil, M.; Siddiqui, M.H.; Khan, S.; Kamal, M.A.; Provazník, I.; Choi, I. Computer Aided Drug Design: Success and Limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar] [CrossRef]
- Xu, W.; Xia, S.; Pu, J.; Wang, Q.; Li, P.; Lu, L.; Jiang, S. The Antihistamine Drugs Carbinoxamine Maleate and Chlorpheniramine Maleate Exhibit Potent Antiviral Activity Against a Broad Spectrum of Influenza Viruses. Front. Microbiol. 2018, 9, 2643. [Google Scholar] [CrossRef]
- Radosevic, D.; Sencanski, M.; Perovic, V.; Veljkovic, N.; Prljic, J.; Veljkovic, V.; Mantlo, E.; Bukreyeva, N.; Paessler, S.; Glisic, S. Virtual Screen for Repurposing of Drugs for Candidate Influenza a M2 Ion-Channel Inhibitors. Front. Cell. Infect. Microbiol. 2019, 9, 67. [Google Scholar] [CrossRef]
- Perovic, V.; Stevanovic, K.; Bukreyeva, N.; Paessler, S.; Maruyama, J.; López-Serrano, S.; Darji, A.; Sencanski, M.; Radosevic, D.; Berardozzi, S.; et al. Exploring the Antiviral Potential of Natural Compounds against Influenza: A Combined Computational and Experimental Approach. Int. J. Mol. Sci. 2024, 25, 4911. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, V.; Mouscadet, J.F.; Veljkovic, N.; Glisic, S.; Debyser, Z. Simple criterion for selection of flavonoid compounds with anti-HIV activity. Bioorg. Med. Chem. Lett. 2007, 17, 1226–1232. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, N.; Glisic, S.; Perovic, V.; Veljkovic, V. The role of long-range intermolecular interactions in discovery of new drugs. Exp. Opin. Drug Disc. 2011, 6, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, V.; Loiseau, P.M.; Figadere, B.; Glisic, S.; Veljkovic, N.; Perovic, V.R.; Cavanaugh, D.P.; Branch, D.R. Virtual screen for repurposing approved and experimental drugs for candidate inhibitors of EBOLA virus infection. F1000Research 2015, 4, 34. [Google Scholar] [CrossRef]
- Kirchmair, J.; Markt, P.; Distinto, S.; Wolber, G.; Langer, T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection-what can we learn from earlier mistakes? J. Comput. Aided Mol. Des. 2008, 22, 213–228. [Google Scholar] [CrossRef]
- Mysinger, M.; Carchia, M.; Irwin, J.; Shoichet, B. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Tong, X.; Smith, J.; Bukreyeva, N.; Koma, T.; Manning, J.T.; Kalkeri, R.; Kwong, A.D.; Paessler, S. Merimepodib, an IMPDH inhibitor, suppresses replication of Zika virus and other emerging viral pathogens. Antivir. Res. 2018, 149, 34–40. [Google Scholar] [CrossRef]
- Markland, W.; McQuaid, T.J.; Jain, J.; Kwong, A.D. Broad-spectrum antiviral activity of the IMP dehydrogenase inhibitor VX-497: A comparison with ribavirin and demonstration of antiviral additivity with alpha interferon. Antimicrob. Agents Chemother. 2000, 44, 859–866. [Google Scholar] [CrossRef]
- Hayden, F.G.; de Jong, M.D. Emerging influenza antiviral resistance threats. J. Infect. Dis. 2011, 203, 6–10. [Google Scholar] [CrossRef]
- Sencanski, M.; Radosevic, D.; Perovic, V.; Gemovic, B.; Stanojevic, M.; Veljkovic, N.; Glisic, S. Natural products as promising therapeutics for treatment of influenza disease. Curr. Pharm. Des. 2015, 21, 5573–5588. [Google Scholar] [CrossRef] [PubMed]
- Tintori, C.; Manetti, F.; Veljkovic, N.; Perovic, V.; Vercammen, J.; Hayes, S.; Massa, S.; Witvrouw, M.; Debyser, Z.; Veljkovic, V.; et al. Novel virtual screening protocol based on the combined use of molecular modeling and electron-ion interaction potential techniques to design HIV-1 integrase in-hibitors. J. Chem. Inf. Model. 2007, 47, 1536–1544. [Google Scholar] [CrossRef] [PubMed]
- Veljkovic, N.; Glisic, S.; Prljic, J.; Perovic, V.; Veljkovic, V. Simple and general criterion for “in silico” screening of candidate HIV drugs. Curr. Pharm. Biotechnol. 2013, 14, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Zha, Y.; Re, J.; Harlo, K.; Jone, D.M.; Zeltin, A.; Bowde, T.A.; Padilla-Parra, S.; Fry, E.E.; Stuart, D.I. Toremifene interacts with and destabi-lizes the Ebola virus glycoprotein. Nature 2016, 535, 169–172. [Google Scholar] [CrossRef]
- Paessler, S.; Huang, C.; Sencanski, M.; Veljkovic, N.; Perovic, V.; Glisic, S.; Veljkovic, V. Ibuprofen as a template mole-cule for drug design against Ebola virus. Front. Biosci. 2018, 23, 947–953. [Google Scholar] [CrossRef]
- Todeschini, R.; Consoni, V. Molecular Descriptors for Chemoinformatics; Wiley VCH Verlag: Weinheim, Germany, 2009. [Google Scholar]
- Matejin, S.; Bukreyeva, N.; Radosevic, D.; Sencanski, M.; Mantlo, E.; Veljkovic, V.; Glisic, S.; Paessler, S. In vitro an-ti-influenza activ-ity of in silico repurposed candidate drug cycrimine. Antivir. Ther. 2019, 24, 589–593. [Google Scholar] [CrossRef]
- Luo, G.; Colonno, R.; Krystal, M. Characterization of a hemagglutinin-specific inhibitor of influenza A virus. Virology 1996, 226, 66–76. [Google Scholar] [CrossRef]
- Staschke, K.A.; Hatch, S.D.; Tang, J.C.; Hornback, W.J.; Munroe, J.E.; Colacino, J.M.; Muesing, M.A. Inhibition of influenza virus hemagglutinin-mediated membrane fusion by a compound related to podocarpic acid. Virology 1998, 248, 264–274. [Google Scholar] [CrossRef]
- Shen, X.; Zhang, X.; Liu, S. Novel hemagglutinin-based influenza virus inhibitors. J. Thorac. Dis. 2013, 5 (Suppl. 2), S149–S159. [Google Scholar] [CrossRef]
- Lao, Z.; Li, Y.; Mi, X.; Tang, Q.; Li, J.; Chen, Y.; Yang, Y. Synthetic pentatrideca-valent triazolylsialoside inhibits influenza virus hemagglutinin/neuraminidase and aggregates virion particles. Eur. J. Med. Chem. 2023, 259, 115578. [Google Scholar] [CrossRef]
- Cui, H.; Yang, J.; Yang, B.; Hao, Y.; Shi, X.; Zhang, D.; Yang, X.; Zhang, T.; Zhao, D.; Yuan, X.; et al. Cyproheptadine hydrochloride inhibits African swine fever viral replication in vitro. Microb. Pathog. 2023, 175, 105957. [Google Scholar] [CrossRef] [PubMed]
- Gastaminza, P.; Whitten-Bauer, C.; Chisari, F.V. Unbiased probing of the entire hepatitis C virus life cycle identifies clini-cal compounds that target multiple aspects of the infection. Proc. Natl. Acad. Sci. USA 2010, 107, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Pinzi, L.; Bisi, N.; Rastelli, G. How drug repurposing can advance drug discovery: Challenges and opportuni-ties. Front. Drug Discov. 2024, 4, 1460100. [Google Scholar] [CrossRef]
- Bertrand, V.; Massy, N.; Vegas, N.; Gras, V.; Chalouhi, C.; Tavolacci, M.P.; Abadie, V. Safety of Cyproheptadine, an Orexigenic Drug. Analysis of the French National Pharmacovigilance Data-Base and Systematic Review. Front. Pediatr. 2021, 9, 712413. [Google Scholar] [CrossRef]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2018, 17, 35–56. [Google Scholar] [CrossRef]
- Trimarco, J.D.; Heaton, N.S. From high-throughput to therapeutic: Host-directed interventions against influenza viruses. Curr. Opin. Virol. 2022, 53, 101198. [Google Scholar] [CrossRef]
- Gunja, N.; Collins, M.; Graudins, A. A comparison of the pharmacokinetics of oral and sublingual cyproheptadine. J. Toxicol. Clin. Toxicol. 2004, 42, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Mendes, G.D.; Arruda, A.; Chen, L.S.; de Almeida Magalhães, J.C.; Alkharfy, K.M.; De Nucci, G. Quantification of cyproheptadine in human plasma by high-performance liquid chromatography coupled to electrospray tandem mass spectrometry in a bioequivalence study. Biomed. Chromatogr. 2012, 26, 129–136. [Google Scholar] [CrossRef]
- Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T.N.; Pach, S. Next generation 3D pharmacophore modeling. WIREs Comput. Mol. Sci. 2020, 10, e1468. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glisic, S.; Stevanovic, K.; Perdih, A.; Bukreyeva, N.; Maruyama, J.; Perovic, V.; López-Serrano, S.; Darji, A.; Radosevic, D.; Sencanski, M.; et al. Influenza a Virus Inhibition: Evaluating Computationally Identified Cyproheptadine Through In Vitro Assessment. Int. J. Mol. Sci. 2025, 26, 5962. https://doi.org/10.3390/ijms26135962
Glisic S, Stevanovic K, Perdih A, Bukreyeva N, Maruyama J, Perovic V, López-Serrano S, Darji A, Radosevic D, Sencanski M, et al. Influenza a Virus Inhibition: Evaluating Computationally Identified Cyproheptadine Through In Vitro Assessment. International Journal of Molecular Sciences. 2025; 26(13):5962. https://doi.org/10.3390/ijms26135962
Chicago/Turabian StyleGlisic, Sanja, Kristina Stevanovic, Andrej Perdih, Natalya Bukreyeva, Junki Maruyama, Vladimir Perovic, Sergi López-Serrano, Ayub Darji, Draginja Radosevic, Milan Sencanski, and et al. 2025. "Influenza a Virus Inhibition: Evaluating Computationally Identified Cyproheptadine Through In Vitro Assessment" International Journal of Molecular Sciences 26, no. 13: 5962. https://doi.org/10.3390/ijms26135962
APA StyleGlisic, S., Stevanovic, K., Perdih, A., Bukreyeva, N., Maruyama, J., Perovic, V., López-Serrano, S., Darji, A., Radosevic, D., Sencanski, M., Veljkovic, V., Botta, B., Mori, M., & Paessler, S. (2025). Influenza a Virus Inhibition: Evaluating Computationally Identified Cyproheptadine Through In Vitro Assessment. International Journal of Molecular Sciences, 26(13), 5962. https://doi.org/10.3390/ijms26135962