
Discovery and Characterization of Novel Non-Hydroxamate HDAC11 Inhibitors


Abstract

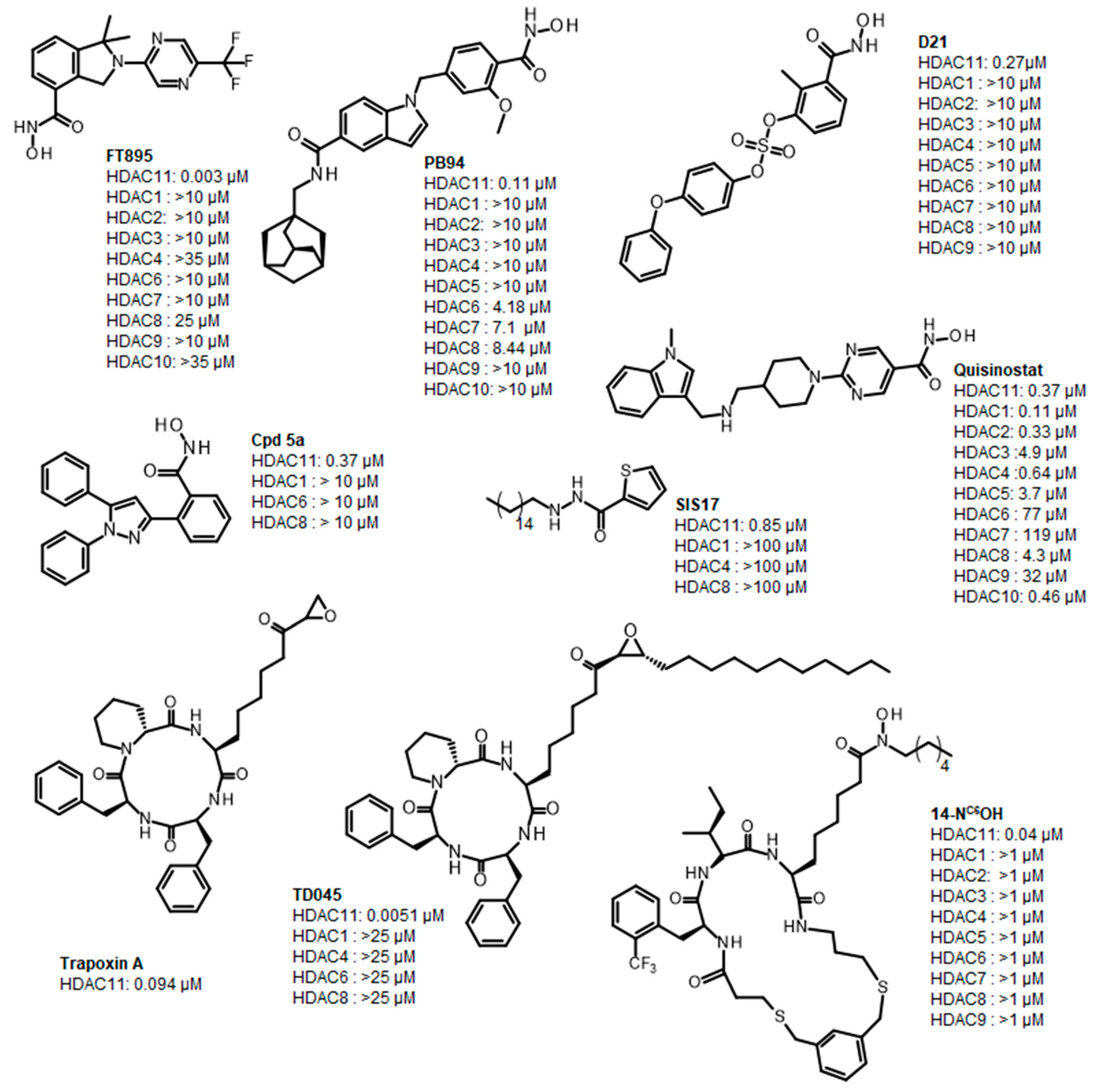
1. Introduction
2. Results
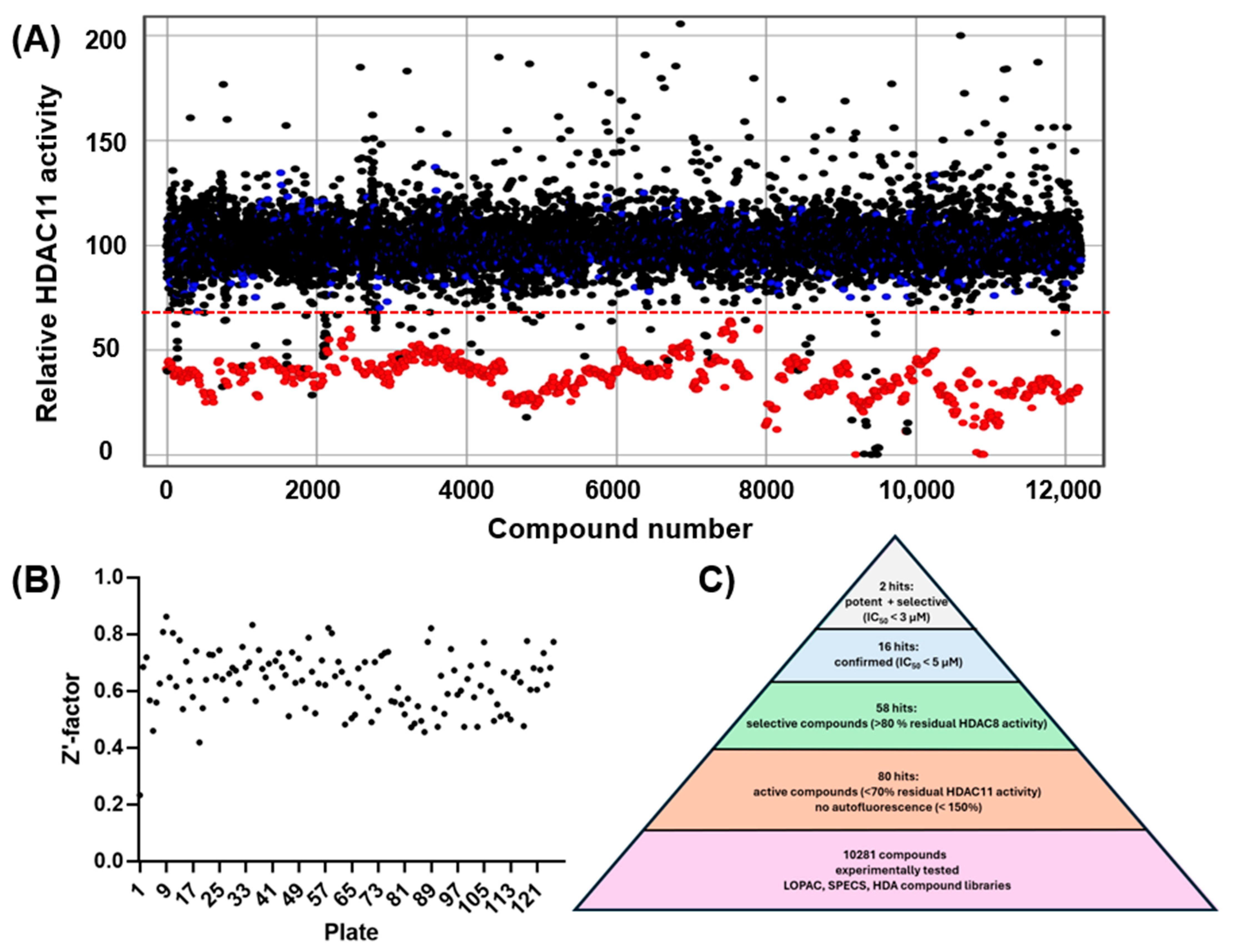
2.1. High-Throughput Screening
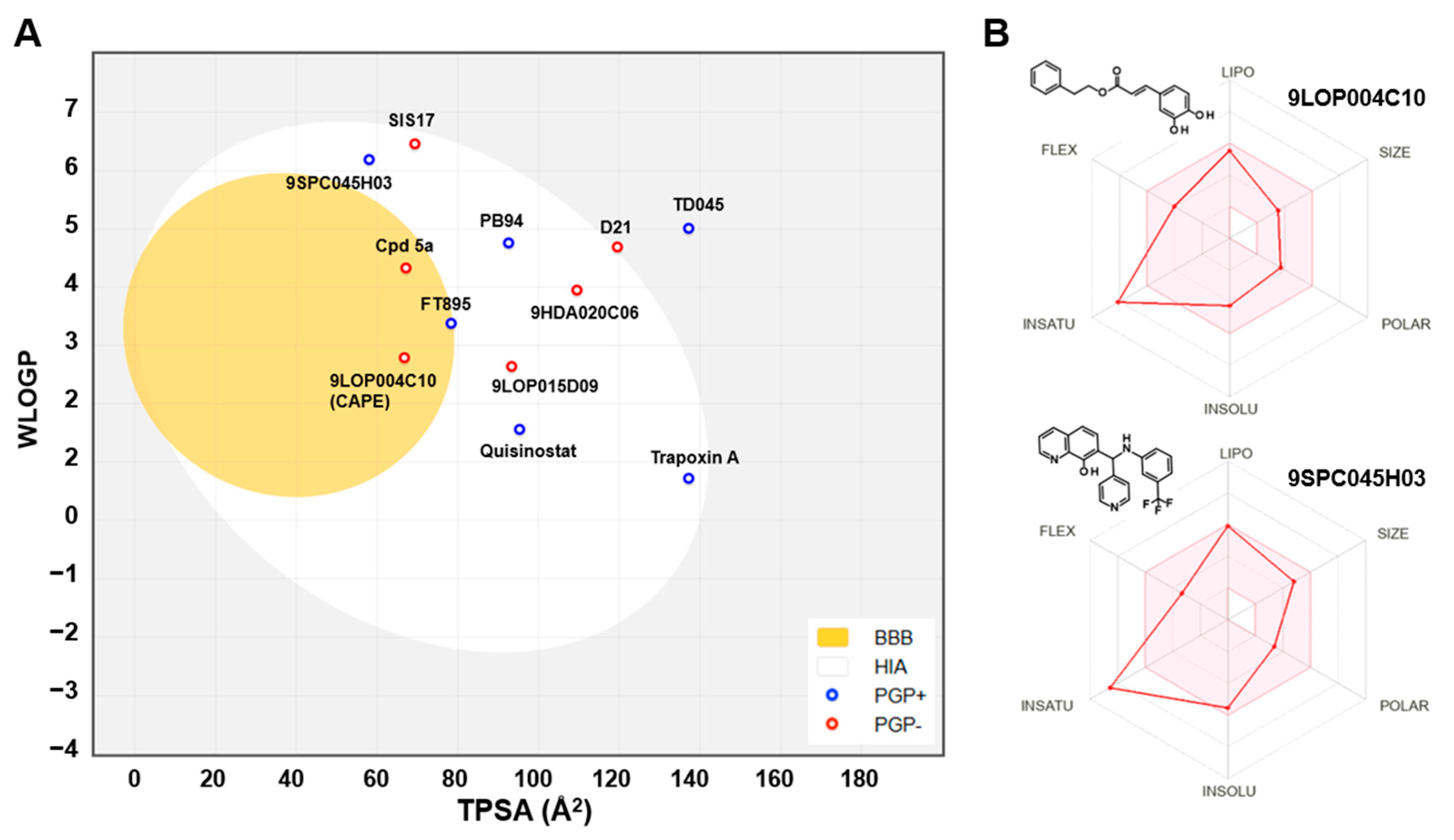
2.2. Pharmacokinetics and Drug-Likeness
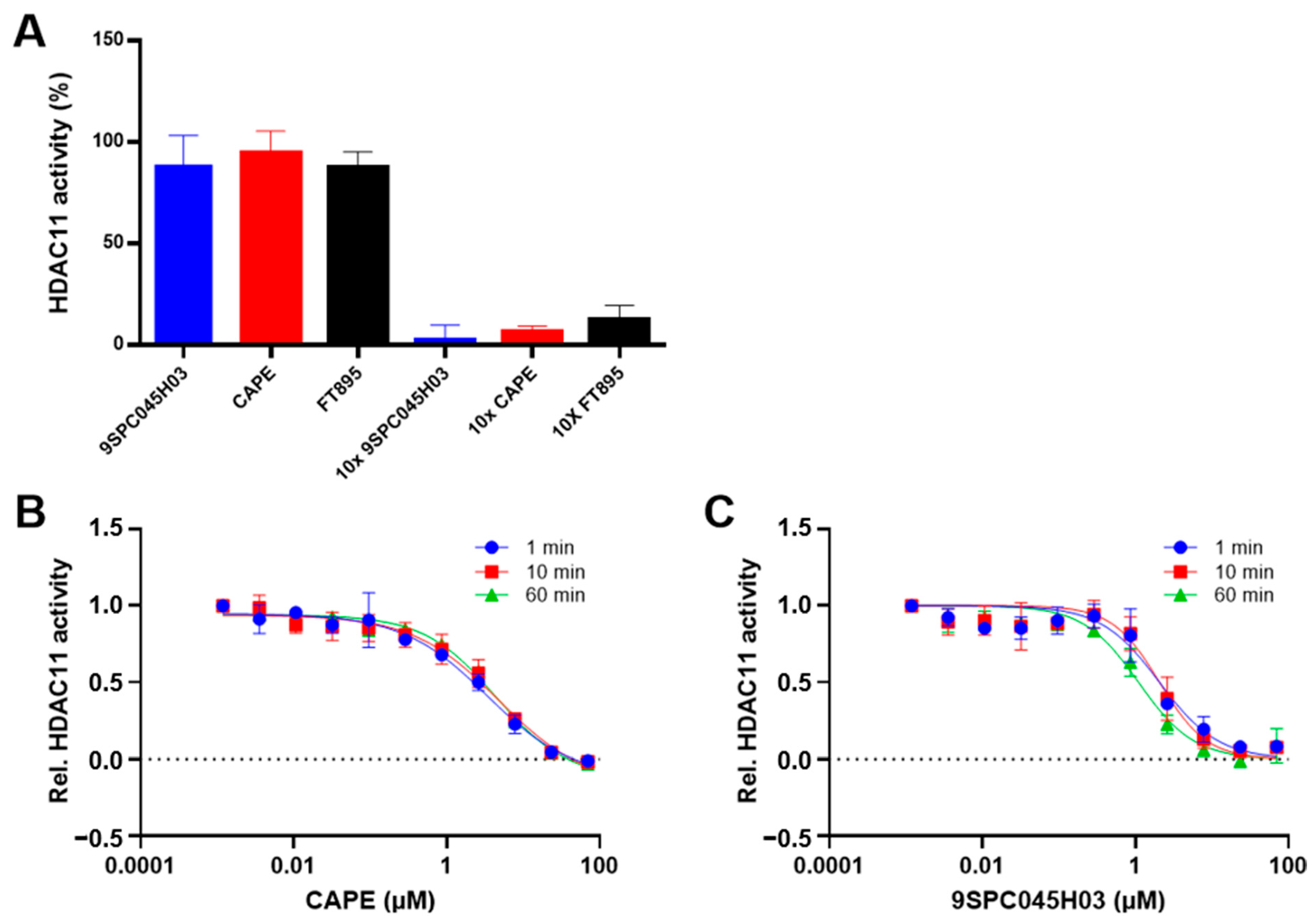
2.3. Reversibility and Time-Dependence of Binding
2.4. Molecular Docking
3. Material and Methods
3.1. High-Throughput Screening
3.2. Production and Purification of HDAC11
3.3. Enzyme Activity Assay
3.4. Time-Dependent IC50 Values
3.5. Rapid Dilution
3.6. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Arts, J.; King, P.; Marin, A.; Floren, W.; Belin, A.; Janssen, L.; Pilatte, I.; Roux, B.; Decrane, L.; Gilissen, R.; et al. JNJ-26481585, a Novel Second-Generation Oral Histone Deacetylase Inhibitor, Shows Broad-Spectrum Preclinical Antitumoral Activity. Clin. Cancer. Res. 2009, 15, 6841–6851. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.N.; Danková, D.; Bæk, M.; Grlaš, L.; Olsen, C.A. Sulfur(VI) Fluoride Exchange Chemistry in Solid-Phase Synthesis of Compound Arrays: Discovery of Histone Deacetylase Inhibitors. JACS Au 2024, 4, 1854–1862. [Google Scholar] [CrossRef]
- Baselious, F.; Hilscher, S.; Hagemann, S.; Tripathee, S.; Robaa, D.; Barinka, C.; Hüttelmaier, S.; Schutkowski, M.; Sippl, W. Utilization of an optimized AlphaFold protein model for structure-based design of a selective HDAC11 inhibitor with anti-neuroblastoma activity. Arch. Pharm. 2024, 357, e2400486. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.W.; Lee, J.Y.; Lancia, D.R., Jr.; Ng, P.Y.; Han, B.; Thomason, J.R.; Lynes, M.S.; Marshall, C.G.; Conti, C.; Collis, A.; et al. Discovery of novel N-hydroxy-2-arylisoindoline-4-carboxamides as potent and selective inhibitors of HDAC11. Bioorg. Med. Chem. Lett. 2018, 28, 2143–2147. [Google Scholar] [CrossRef]
- Son, S.I.; Cao, J.; Zhu, C.L.; Miller, S.P.; Lin, H. Activity-Guided Design of HDAC11-Specific Inhibitors. ACS Chem. Biol. 2019, 14, 1393–1397. [Google Scholar] [CrossRef]
- Ho, T.T.; Peng, C.; Seto, E.; Lin, H. Trapoxin A Analogue as a Selective Nanomolar Inhibitor of HDAC11. ACS Chem. Biol. 2023, 18, 803–809. [Google Scholar] [CrossRef]
- Danková, D.; Nielsen, A.L.; Zarda, A.; Hansen, T.N.; Hesse, M.; Benová, M.; Tsiris, A.; Bartling, C.R.O.; Will, E.J.; Strømgaard, K.; et al. Discovery of De Novo Macrocycle Inhibitors of Histone Deacetylase 11. JACS Au 2025, 5, 1299–1307. [Google Scholar] [CrossRef]
- Marmion, C.J.; Parker, J.P.; Nolan, K.B. 3.23—Hydroxamic Acids: An Important Class of Metalloenzyme Inhibitors. In Comprehensive Inorganic Chemistry II, 2nd ed.; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 683–708. [Google Scholar]
- Verma, R.P. Hydroxamic acids as matrix metalloproteinase inhibitors. Exp. Suppl. 2012, 103, 137–176. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Hydroxamates as Carbonic Anhydrase Inhibitors. In Hydroxamic Acids: A Unique Family of Chemicals with Multiple Biological Activities; Gupta, S.P., Ed.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 55–69. [Google Scholar]
- Mai, A. Hydroxamic Acids: Biological Properties and Potential Uses as Therapeutic Agents. In PATAI’S Chemistry of Functional Groups; Wiley: Hoboken, NJ, USA, 2009. [Google Scholar]
- Day, J.A.; Cohen, S.M. Investigating the selectivity of metalloenzyme inhibitors. J. Med. Chem. 2013, 56, 7997–8007. [Google Scholar] [CrossRef]
- Viana, L.P.S.; Naves, G.M.; Medeiros, I.G.; Guimarães, A.S.; Sousa, E.S.; Santos, J.C.C.; Freire, N.M.L.; de Aquino, T.M.; Modolo, L.V.; de Fátima, Â.; et al. Synergizing structure and function: Cinnamoyl hydroxamic acids as potent urease inhibitors. Bioorg. Chem. 2024, 146, 107247. [Google Scholar] [CrossRef]
- Shen, S.; Kozikowski, A.P. Why Hydroxamates May Not Be the Best Histone Deacetylase Inhibitors—What Some May Have Forgotten or Would Rather Forget? ChemMedChem 2016, 11, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- Baselious, F.; Hilscher, S.; Robaa, D.; Barinka, C.; Schutkowski, M.; Sippl, W. Comparative Structure-Based Virtual Screening Utilizing Optimized AlphaFold Model Identifies Selective HDAC11 Inhibitor. Int. J. Mol. Sci. 2024, 25, 1358. [Google Scholar] [CrossRef] [PubMed]
- Khetmalis, Y.M.; Fathima, A.; Schweipert, M.; Debarnot, C.; Bandaru, N.; Murugesan, S.; Jamma, T.; Meyer-Almes, F.J.; Sekhar, K. Design, Synthesis, and Biological Evaluation of Novel Quinazolin-4(3H)-One-Based Histone Deacetylase 6 (HDAC6) Inhibitors for Anticancer Activity. Int. J. Mol. Sci. 2023, 24, 11044. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, N.; Tilekar, K.; Jansch, N.; Schweipert, M.; Hess, J.D.; Henze Macias, L.; Mrowka, P.; Aguilera, R.J.; Choe, J.Y.; Meyer-Almes, F.J.; et al. Discovery of novel N-substituted thiazolidinediones (TZDs) as HDAC8 inhibitors: In-silico studies, synthesis, and biological evaluation. Bioorg. Chem. 2020, 100, 103934. [Google Scholar] [CrossRef] [PubMed]
- Tilekar, K.; Hess, J.D.; Upadhyay, N.; Schweipert, M.; Flath, F.; Gutierrez, D.A.; Loiodice, F.; Lavecchia, A.; Meyer-Almes, F.-J.; Aguilera, R.J.; et al. HDAC4 Inhibitors with Cyclic Linker and Non-hydroxamate Zinc Binding Group: Design, Synthesis, HDAC Screening and in vitro Cytotoxicity evaluation. ChemistrySelect 2021, 6, 6748–6763. [Google Scholar] [CrossRef]
- Upadhyay, N.; Tilekar, K.; Safuan, S.; Kumar, A.P.; Schweipert, M.; Meyer-Almes, F.-J.; CS, R. Multi-target weapons: Diaryl-pyrazoline thiazolidinediones simultaneously targeting VEGFR-2 and HDAC cancer hallmarks. RSC Med. Chem. 2021, 12, 1540–1554. [Google Scholar] [CrossRef]
- Tilekar, K.; Hess, J.D.; Upadhyay, N.; Bianco, A.L.; Schweipert, M.; Laghezza, A.; Loiodice, F.; Meyer-Almes, F.J.; Aguilera, R.J.; Lavecchia, A.; et al. Thiazolidinedione “Magic Bullets” Simultaneously Targeting PPARgamma and HDACs: Design, Synthesis, and Investigations of their In Vitro and In Vivo Antitumor Effects. J. Med. Chem. 2021, 64, 6949–6971. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings1. Adv. Drug Del. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M., Jr.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I. Pharmacophore features of potential drugs. Chemistry 2002, 8, 1976–1981. [Google Scholar] [CrossRef]
- Cavaliere, V.; Papademetrio, D.L.; Lorenzetti, M.; Valva, P.; Preciado, M.V.; Gargallo, P.; Larripa, I.; Monreal, M.B.; Pardo, M.L.; Hajos, S.E.; et al. Caffeic Acid Phenylethyl Ester and MG-132 Have Apoptotic and Antiproliferative Effects on Leukemic Cells But Not on Normal Mononuclear Cells. Transl. Oncol. 2009, 2, 46–58. [Google Scholar] [CrossRef]
- Jung, W.-K.; Choi, I.; Lee, D.-Y.; Yea, S.S.; Choi, Y.H.; Kim, M.-M.; Park, S.-G.; Seo, S.-K.; Lee, S.-W.; Lee, C.-M.; et al. Caffeic acid phenethyl ester protects mice from lethal endotoxin shock and inhibits lipopolysaccharide-induced cyclooxygenase-2 and inducible nitric oxide synthase expression in RAW 264.7 macrophages via the p38/ERK and NF-κB pathways. Int. J. Biochem. Cell Biol. 2008, 40, 2572–2582. [Google Scholar] [CrossRef]
- Lee, E.S.; Lee, J.-O.; Lee, S.K.; Kim, J.H.; Jung, J.H.; Keum, B.; Park, S.-H.; Kim, H.S. Caffeic acid phenethyl ester accumulates β-catenin through GSK-3β and participates in proliferation through mTOR in C2C12 cells. Life Sci. 2009, 84, 755–759. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Rohman, M.; Harrison-Lavoie, K.J. Separation of copurifying GroEL from glutathione-S-transferase fusion proteins. Protein. Expr. Purif. 2000, 20, 45–47. [Google Scholar] [CrossRef]
- Baselious, F.; Robaa, D.; Sippl, W. Utilization of AlphaFold models for drug discovery: Feasibility and challenges. Histone deacetylase 11 as a case study. Comput. Biol. Med. 2023, 167, 107700. [Google Scholar] [CrossRef]
- Petri, L.; Abranyi-Balogh, P.; Timea, I.; Palfy, G.; Perczel, A.; Knez, D.; Hrast, M.; Gobec, M.; Sosic, I.; Nyiri, K.; et al. Assessment of Tractable Cysteines for Covalent Targeting by Screening Covalent Fragments. ChemBioChem 2021, 22, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Schweipert, M.; Amurthavasan, A.; Meyer-Almes, F.J. Continuous enzyme activity assay for high-throughput classification of histone deacetylase 8 inhibitors. Explor. Target. Anti-Tumor Ther. 2023, 4, 447–459. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster # | Structure | ID | IC50 (µM) |
|---|---|---|---|
| 1 | 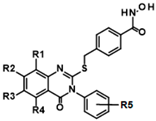 | ||
| R1:H,R2:H,R3H,R4Me,R5:H | 9HDA020C06 | 1.1 ± 0.4 | |
| R1:H,R2:H,R3H,R4:Me,R5:p-Cl | 9HDA020C09 | 1.9 ± 0.4 | |
| R1:H,R2:H,R3H,R4:Me,R5:o-F | 9HDA020C07 | 1.5 ± 0.3 | |
| R1:H,R2:H,R3H,R4:Me,R5:p-Et | 9HDA020B06 | 1.6 ± 0.2 | |
| R1:H,R2:H,R3H,R4:H,R5:m-Me | 9HDA020B09 | 1.6 ± 0.3 | |
| R1:H,R2:H,R3H,R4:H,R5:p-Me | 9HDA020B05 | 2.0 ± 0.2 | |
| R1:H,R2:Cl,R3H,R4:H,R5:o-F | 9HDA020C04 | 2.0 ± 0.5 | |
| R1:H,R2:H,R3H,R4:H,R5:o-Me,p-Me | 9HDA020B08 | 2.0 ± 0.2 | |
| R1:H,R2:H,R3H,R4:H,R5:m-Cl | 9HDA020B10 | 2.0 ± 0.3 | |
| R1:H,R2:H,R3H,R4:H,R5:H | 9HDA020B04 | 2.1 ± 0.2 | |
| R1:H,R2:H,R3H,R4:H,R5:p-Br | 9HDA020B07 | 2.6 ± 0.4 | |
| R1:H,R2:H,R3H,R4:Me,R5:o-Me | 9HDA020C08 | 3.5 ± 0.8 | |
| R1:H,R2:H,R3H,R4:H,R5:o-F | 9HDA020B11 | 4.1 ± 0.5 | |
| 2 |  | 9LOP004C10 (CAPE) | 1.5 ± 0.4 |
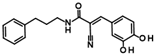 | 9LOP015D09 | 4.3 ± 3 | |
 | 9LOP015E07 | 14 ± 3 | |
| 5 | 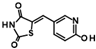 | 9HDA018E08 | 19 ± 2 |
| 6 |  | 9HDA027C02 | >50 |
| Singleton | 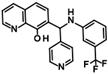 | 9SPC045H03 | 2.3 ± 0.5 |
| Singleton | 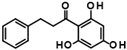 | 9SPC036F04 | >50 |
| Reference | 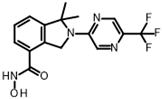 | FT895 | 0.003 ± 0.001 |
| IC50 (µM) | ||||||
|---|---|---|---|---|---|---|
| Cluster # | ID | HDAC1 | HDAC4 | HDAC6 | HDAC8 | HDAC11 |
| 1 | 9HDA020C06 | 1.4 ± 0.4 | 0.4 ± 0.2 | 0.2 ± 0.1 | 2.7 ± 0.6 | 1.1 ± 0.4 |
| 9HDA020C09 | 2.3 ± 0.5 | 0.9 ± 0.4 | 0.9 ± 0.3 | 6 ± 1 | 1.9 ± 0.4 | |
| 2 | CAPE | 22 ± 5 | 68 ± 30 | >70 | 7 ± 2 | 1.5 ± 0.4 |
| Singleton | 9SPC045H03 | >70 | >70 | 67 | 13 ± 5 | 2.3 ± 0.5 |
| Reference | FT895 | >10 * | >10 * | >10 * | 5.6 * | 0.003 ± 0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kopranovic, A.; Meyer-Almes, F.-J. Discovery and Characterization of Novel Non-Hydroxamate HDAC11 Inhibitors. Int. J. Mol. Sci. 2025, 26, 5950. https://doi.org/10.3390/ijms26135950
Kopranovic A, Meyer-Almes F-J. Discovery and Characterization of Novel Non-Hydroxamate HDAC11 Inhibitors. International Journal of Molecular Sciences. 2025; 26(13):5950. https://doi.org/10.3390/ijms26135950
Chicago/Turabian StyleKopranovic, Aleksandra, and Franz-Josef Meyer-Almes. 2025. "Discovery and Characterization of Novel Non-Hydroxamate HDAC11 Inhibitors" International Journal of Molecular Sciences 26, no. 13: 5950. https://doi.org/10.3390/ijms26135950
APA StyleKopranovic, A., & Meyer-Almes, F.-J. (2025). Discovery and Characterization of Novel Non-Hydroxamate HDAC11 Inhibitors. International Journal of Molecular Sciences, 26(13), 5950. https://doi.org/10.3390/ijms26135950