

Long-Range Allosteric Communication Modulated by Active Site Mn(II) Coordination Drives Catalysis in Xanthobacter autotrophicus Acetone Carboxylase

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
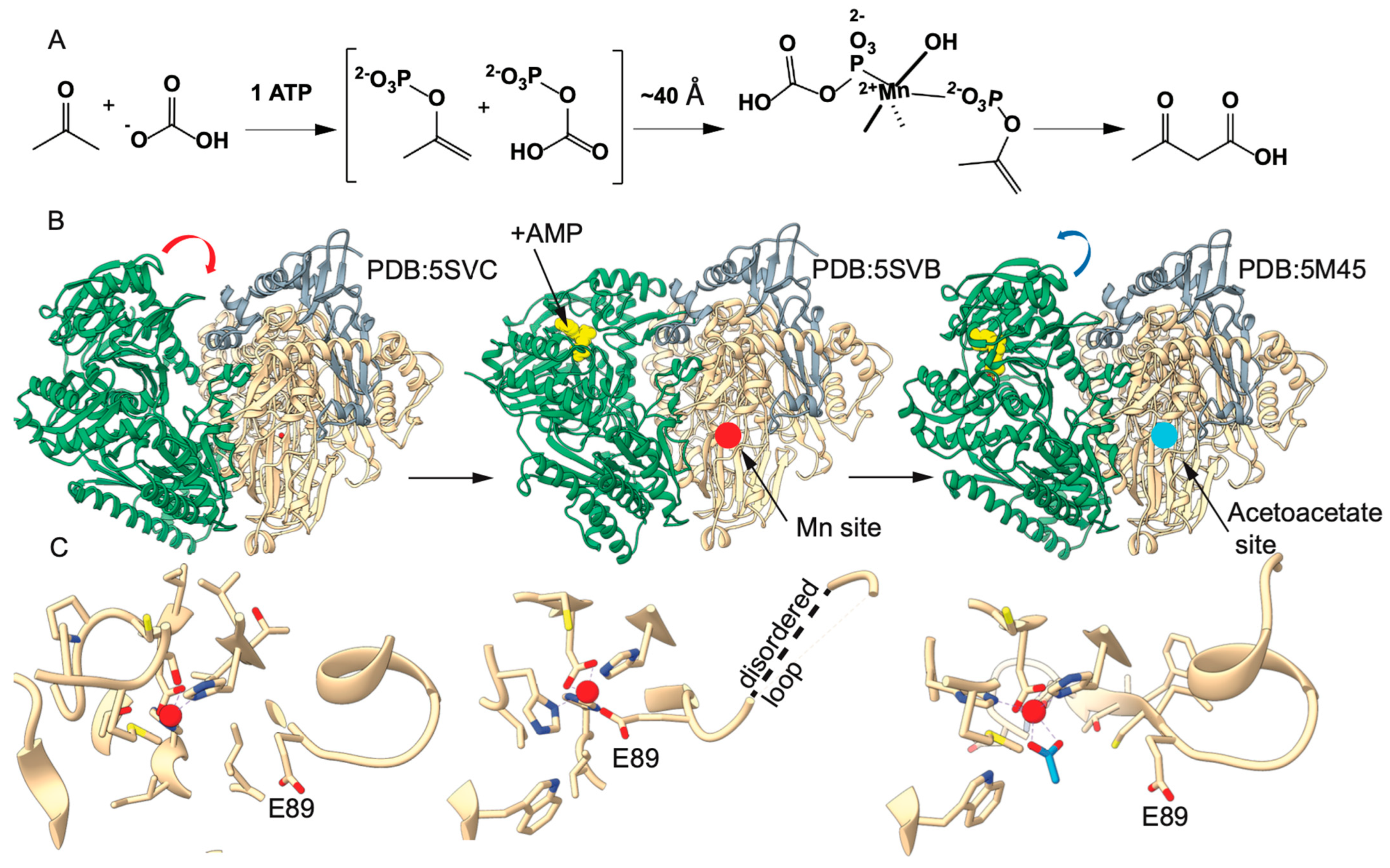
2.1. Substitution of αE89 Confirms an Essential Role in Catalysis
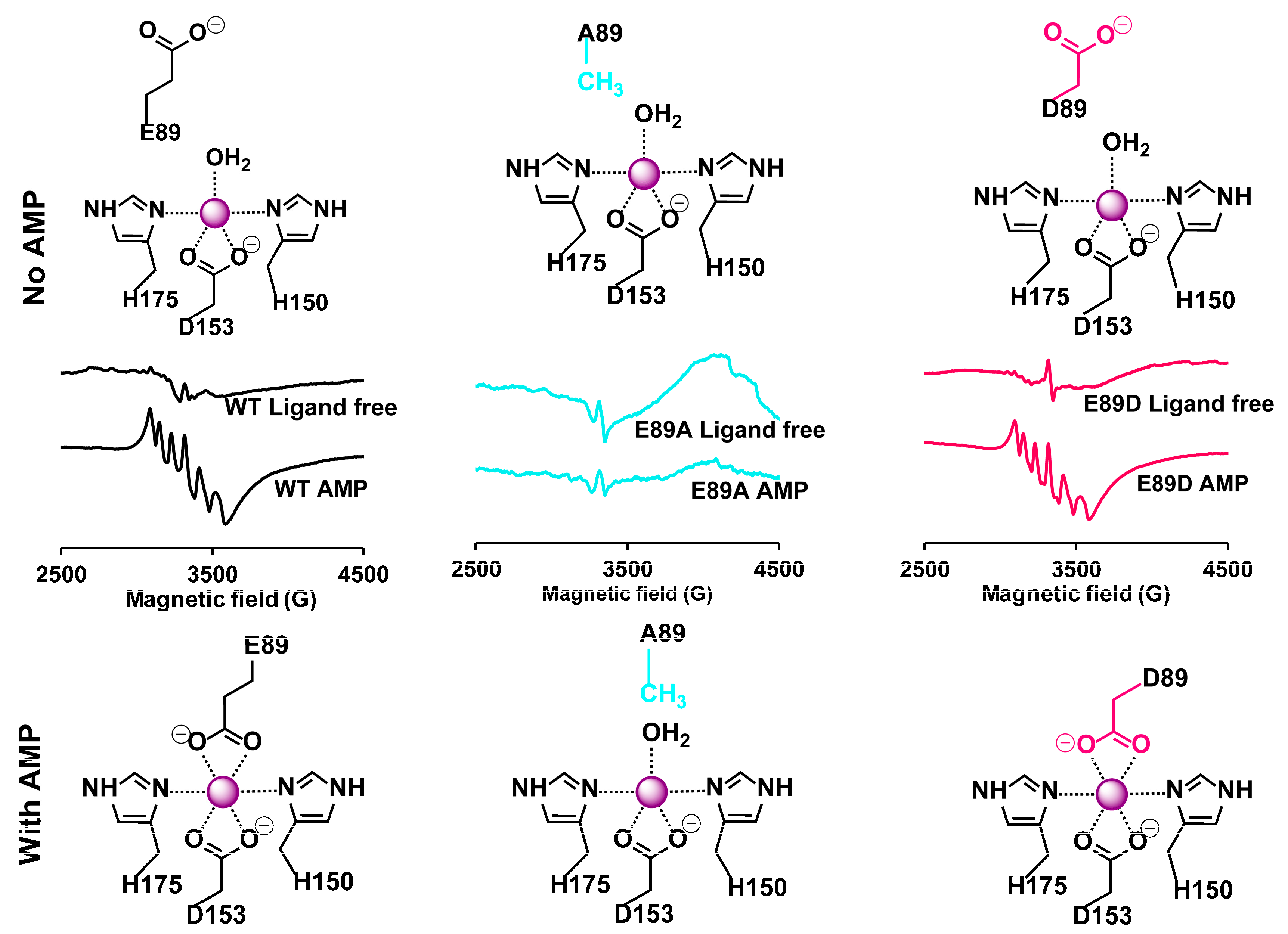
2.2. EPR Studies Confirm the Role of αE89 in Mn(II) Coordination
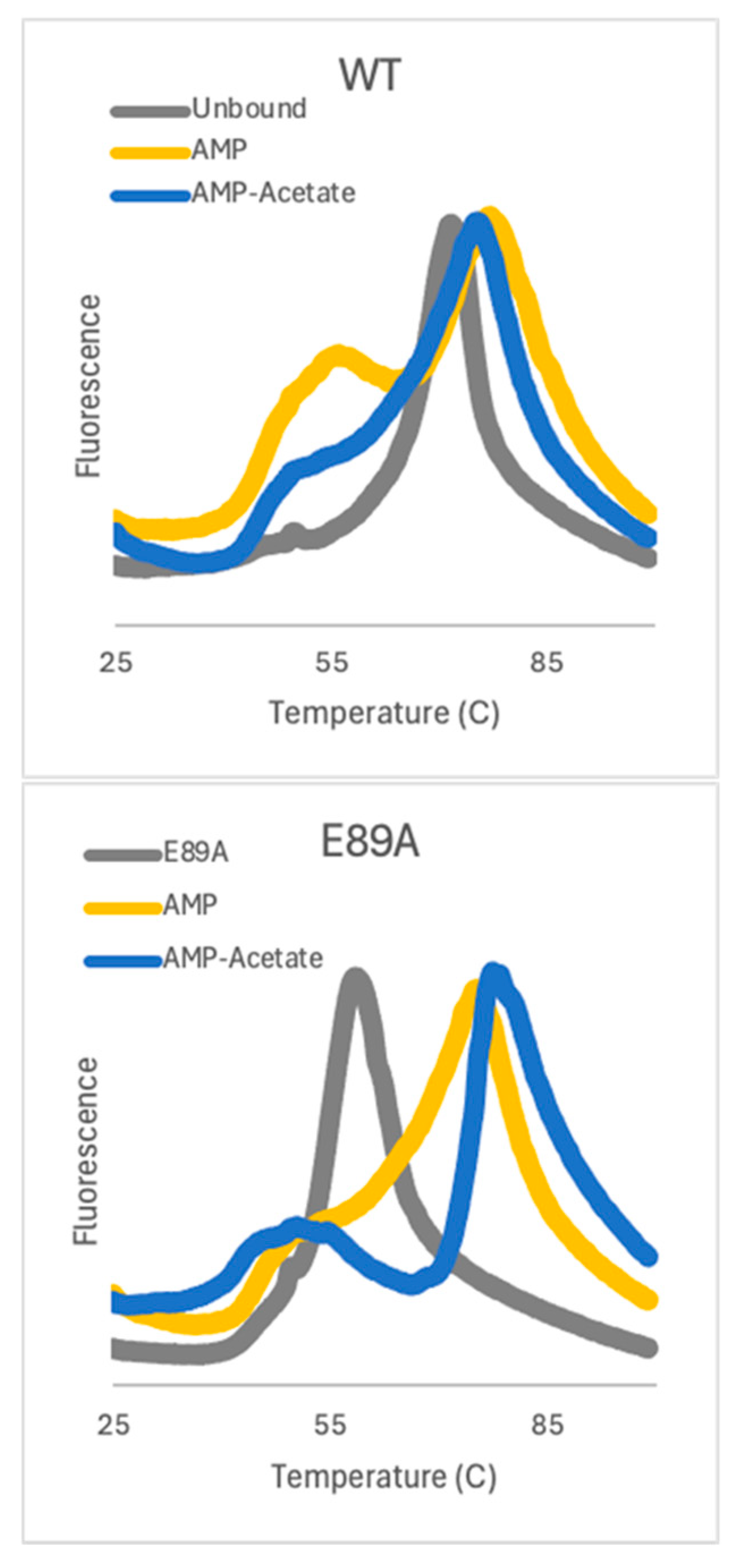
2.3. Thermal Stability Shifts with Substrates Bound to both WT and αE89A
2.4. Ligand Binding Alters the Hydrogen Bonding Network of AC
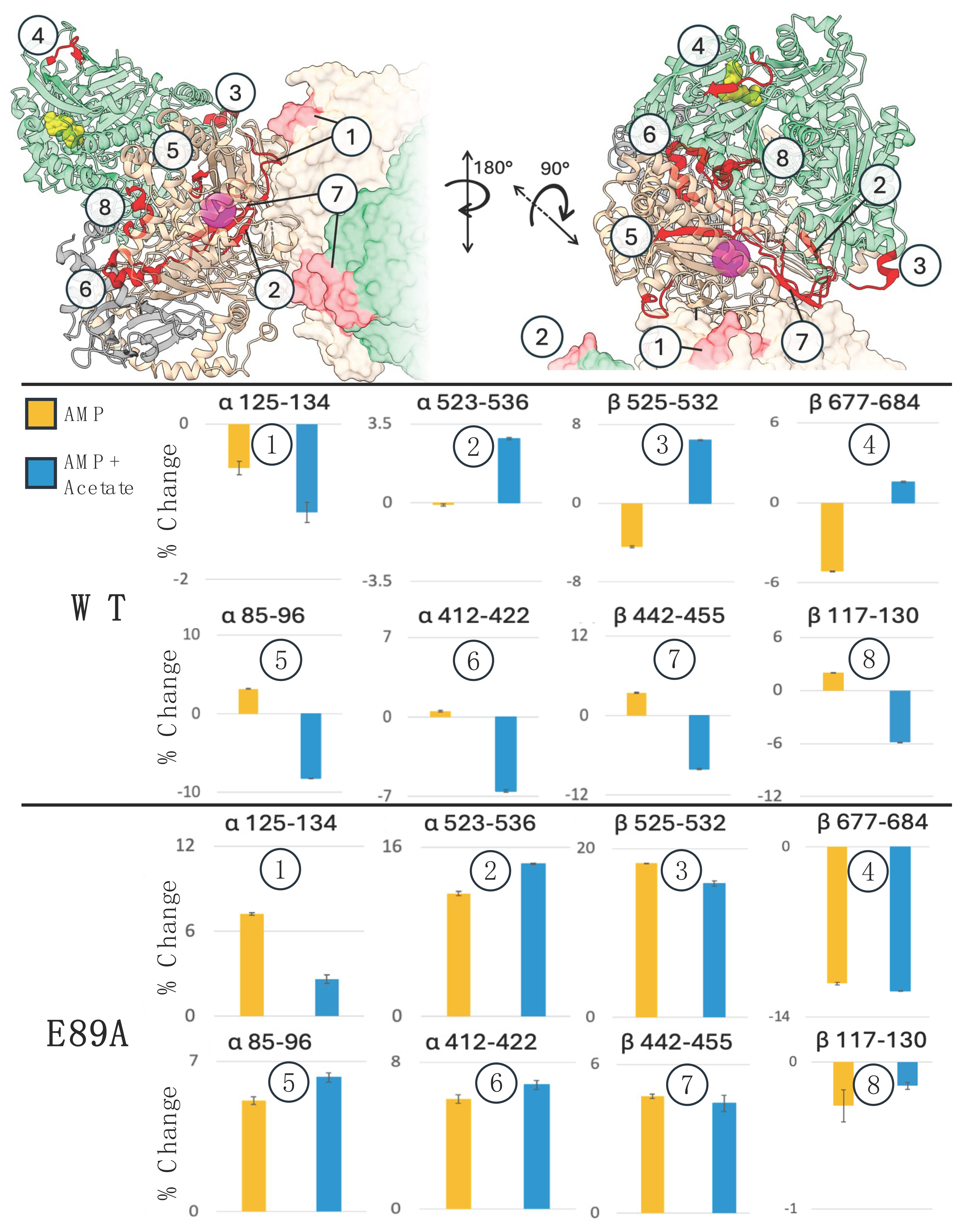
2.5. Mechanical Connectivity Between the Substrate Binding and Mn(II) Sites
2.6. αE89A Variant Has Local and Distal Changes in HDX Pattern
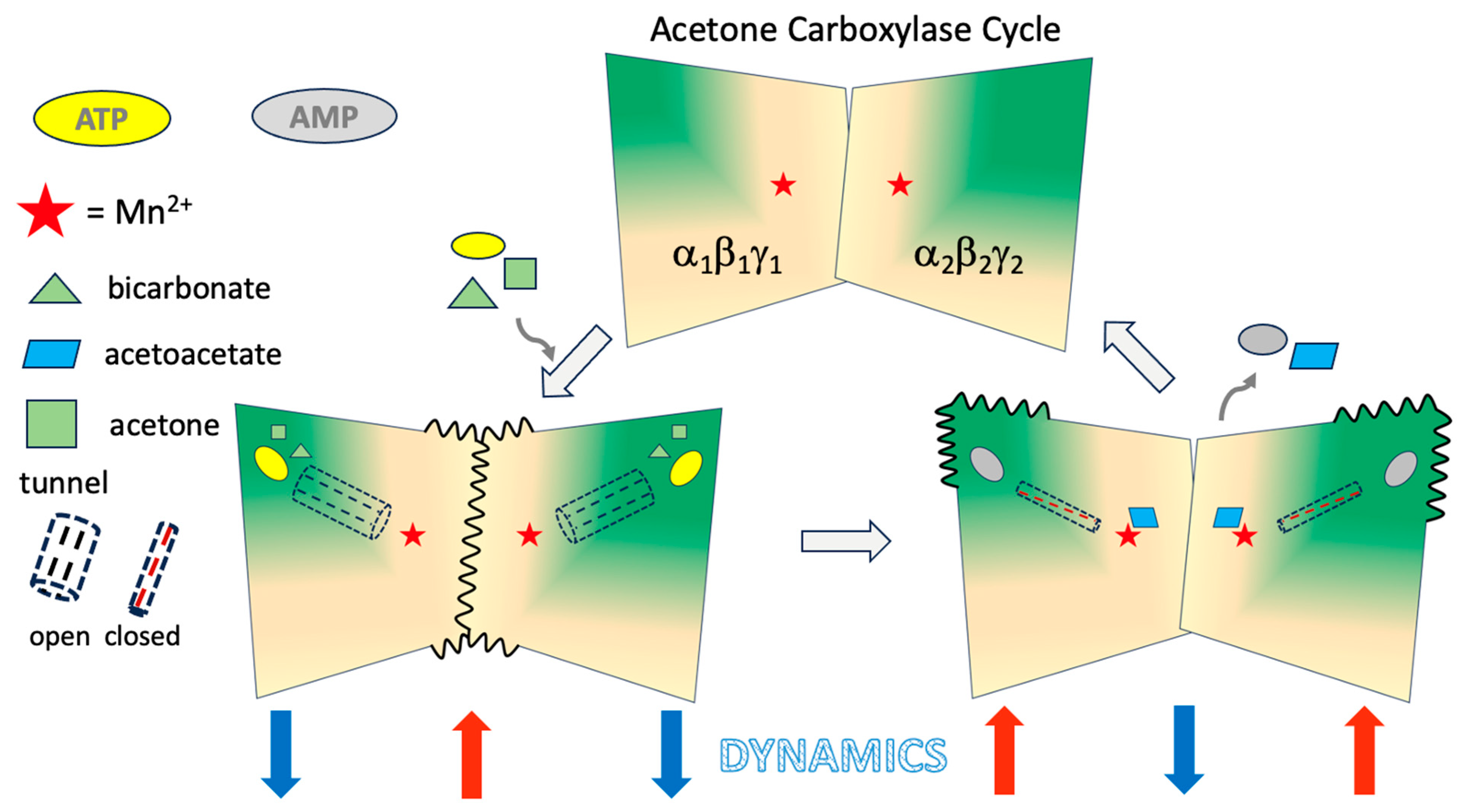
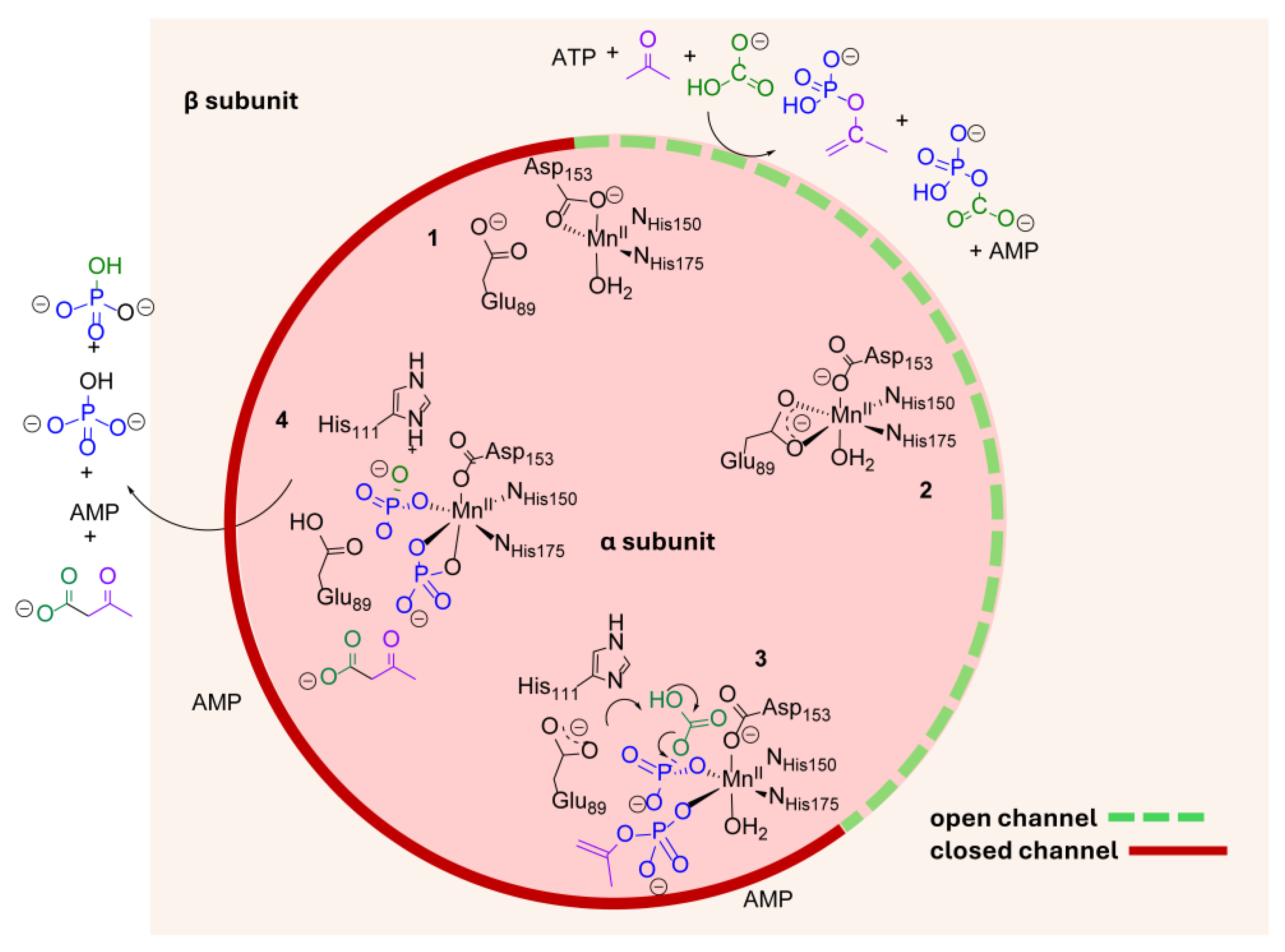
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of Recombinant Acetone Carboxylase
4.2. EPR Spectroscopy of WT and Variant AC
4.3. Enzymatic Assays of WT and Variant AC
4.4. Intact Protein HDX of WT and Variant AC
4.5. Peptide-Level HDX of WT and Variant AC
4.6. Differential Scanning Fluorimetry
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Erb, T.J. Carboxylases in natural and synthetic microbial pathways. Appl. Environ. Microbiol. 2011, 77, 8466–8477. [Google Scholar] [CrossRef] [PubMed]
- Mus, F.; Eilers, B.J.; Alleman, A.B.; Kabasakal, B.V.; Wells, J.N.; Murray, J.W.; Nocek, B.P.; DuBois, J.L.; Peters, J.W. Structural Basis for the Mechanism of ATP-Dependent Acetone Carboxylation. Sci. Rep. 2017, 7, 7234. [Google Scholar] [CrossRef]
- Sluis, M.K.; Ensign, S.A. Purification and characterization of acetone carboxylase from Xanthobacter strain Py2. Biochemistry 1997, 94, 8456–8461. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.M.; Ellsworth, H.; Ensign, S.A. Bacterial acetone carboxylase is a manganese-dependent metalloenzyme. J. Biol. Chem. 2004, 279, 46644–46651. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.M.; Ensign, S.A. ATP-dependent enolization of acetone by acetone carboxylase from Rhodobacter capsulatus. Biochemistry 2005, 44, 8543–8553. [Google Scholar] [CrossRef]
- Stich, T.A.; Lahiri, S.; Yeagle, G.; Dicus, M.; Brynda, M.; Gunn, A.; Aznar, C.; Derose, V.J.; Britt, R.D. Multifrequency Pulsed EPR Studies of Biologically Relevant Manganese (II) Complexes. Appl. Magn. Reson. 2007, 31, 321. [Google Scholar] [CrossRef]
- Rayaprolu, V.; Kruse, S.; Kant, R.; Movahed, N.; Brooke, D.; Bothner, B. Fluorometric Estimation of Viral Thermal Stability. Bio-protocol 2014, 4, e1199. [Google Scholar] [CrossRef]
- Tokmina-Lukaszewska, M.; Patterson, A.; Berry, L.; Scott, L.; Balasubramanian, N.; Bothner, B. The Role of Mass Spectrometry in Structural Studies of Flavin-Based Electron Bifurcating Enzymes. Front. Microbiol. 2018, 9, 1397. [Google Scholar] [CrossRef]
- Konermann, L.; Pan, J.; Liu, Y.H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem. Soc. Rev. 2011, 40, 1224–1234. [Google Scholar] [CrossRef]
- Mus, F.; Wu, H.H.; Alleman, A.B.; Shisler, K.A.; Zadvornyy, O.A.; Bothner, B.; DuBois, J.L.; Peters, J.W. Insights into the unique carboxylation reactions in the metabolism of propylene and acetone. Biochem. J. 2020, 477, 2027–2038. [Google Scholar] [CrossRef]
- Radin, R.; Tolman, W.; Lippard, S. Monodentate carboxylate complexes and the carboxylate shift: Implications for polymetalloprotein structure and function. New J. Chem. 1991, 15, 417–430. [Google Scholar]
- Nordlund, P.; Eklund, H. Structure and function of the Escherichia coli ribonucleotide reductase protein R2. J. Mol. Biol. 1993, 232, 123–164. [Google Scholar] [CrossRef] [PubMed]
- Whittington, D.A.; Lippard, S.J. Crystal Structures of the Soluble Methane Monooxygenase Hydroxylase from Methylococcus capsulatus (Bath) Demonstrating Geometrical Variability at the Dinuclear Iron Active Site. J. Am. Chem. Soc. 2001, 123, 827–838. [Google Scholar] [CrossRef]
- Torrent, M.; Musaev, D.G.; Morokuma, K. The Flexibility of Carboxylate Ligands in Methane Monooxygenase and Ribonucleotide Reductase: A Density Functional Study. J. Phys. Chem. B 2001, 105, 322–327. [Google Scholar] [CrossRef]
- Zimmermann, T.P.; Limpke, T.; Stammler, A.; Bogge, H.; Walleck, S.; Glaser, T. Reversible Carboxylate Shift in a μ-Oxo Diferric Complex in Solution by Acid-/Base-Addition. Inorg. Chem. 2018, 57, 5400–5405. [Google Scholar] [CrossRef]
- Costas, M.; Cady, C.W.; Kryatov, S.V.; Ray, M.; Ryan, M.J.; Rybak-Akimova, E.V.; Que, L. Role of Carboxylate Bridges in Modulating Nonheme Diiron(II)/O2 Reactivity. Inorg. Chem. 2003, 42, 7519–7530. [Google Scholar] [CrossRef]
- DeLucia, A.A.; Olshansky, L. Carboxylate Shift Dynamics in Biomimetic CO2(μ−OH)2 Complexes. Inorg. Chem. 2024, 63, 1109–1118. [Google Scholar] [CrossRef]
- Jeoung, J.H.; Dobbek, H. Carbon dioxide activation at the Ni,Fe-cluster of anaerobic carbon monoxide dehydrogenase. Science 2007, 318, 1461–1464. [Google Scholar] [CrossRef]
- Ragsdale, S.; Pierce, E. Acetogenesis and the Wood-Ljungdahl pathway of CO2 fixation. Biochim. Biophys. Acta 2008, 1784, 1873–1898. [Google Scholar] [CrossRef]
- Kung, Y.; Drennan, C. A role for nickel-iron cofactors in biological carbon monoxide and carbon dioxide utilization. Curr. Opin. Chem. Biol. 2010, 15, 276–283. [Google Scholar] [CrossRef]
- Doukov, T.I.; Blasiak, L.C.; Seravalli, J.; Ragsdale, S.W.; Drennan, C.L. Xenon in and at the end of the tunnel of bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Biochemistry 2008, 47, 3474–3483. [Google Scholar] [CrossRef] [PubMed]
- Rae, B.D.; Long, B.M.; Badger, M.R.; Price, G.D. Functions, Compositions, and Evolution of the Two Types of Carboxysomes: Polyhedral Microcompartments That Facilitate CO2 Fixation in Cyanobacteria and Some Proteobacteria. Microbiol. Mol. Biol. Rev. 2013, 77, 357–379. [Google Scholar] [CrossRef]
- Fleming, J.R.; Schupfner, M.; Busch, F.; Baslé, A.; Ehrmann, A.; Sterner, R.; Mayans, O. Evolutionary Morphing of Tryptophan Synthase: Functional Mechanisms for the Enzymatic channeling of Indole. J. Mol. Biol. 2018, 430, 5066–5079. [Google Scholar] [CrossRef]
- Bhat, J.Y.; Venkatachala, R.; Singh, K.; Gupta, K.; Sarma, S.P.; Balaram, H. Ammonia channeling in Plasmodium falciparum GMP synthetase: Investigation by NMR spectroscopy and biochemical assays. Biochemistry 2011, 50, 3346–3356. [Google Scholar] [CrossRef] [PubMed]
- Patterson, A.; Zhao, Z.; Waymire, E.; Zlotnick, A.; Bothner, B. Dynamics of Hepatitis B Virus Capsid Protein Dimer Regulate Assembly through an Allosteric Network. ACS Chem. Biol. 2020, 15, 2273–2280. [Google Scholar] [CrossRef] [PubMed]
- Berry, L.; Patterson, A.; Pence, N.; Peters, J.W.; Bothner, B. Hydrogen Deuterium Exchange Mass Spectrometry of Oxygen Sensitive Proteins. Bioprotocols 2018, 8, e2769. [Google Scholar] [CrossRef]
- Vaudel, M.; Burkhart, J.M.; Zahedi, R.P.; Oveland, E.; Berven, F.S.; Sickmann, A.; Martens, L.; Barsnes, H. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 2015, 33, 22–24. [Google Scholar] [CrossRef]
- Kavan, D.; Man, P. MSTools—Web based application for visualization and presentation of HXMS data. Int. J. Mass. Spectrom. 2011, 302, 53–58. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mattice, J.R.; Shisler, K.A.; Malone, J.R.; Murray, N.A.; Tokmina-Lukaszewska, M.; Nath, A.K.; Flusche, T.; Mus, F.; DuBois, J.L.; Peters, J.W.; et al. Long-Range Allosteric Communication Modulated by Active Site Mn(II) Coordination Drives Catalysis in Xanthobacter autotrophicus Acetone Carboxylase. Int. J. Mol. Sci. 2025, 26, 5945. https://doi.org/10.3390/ijms26135945
Mattice JR, Shisler KA, Malone JR, Murray NA, Tokmina-Lukaszewska M, Nath AK, Flusche T, Mus F, DuBois JL, Peters JW, et al. Long-Range Allosteric Communication Modulated by Active Site Mn(II) Coordination Drives Catalysis in Xanthobacter autotrophicus Acetone Carboxylase. International Journal of Molecular Sciences. 2025; 26(13):5945. https://doi.org/10.3390/ijms26135945
Chicago/Turabian StyleMattice, Jenna R., Krista A. Shisler, Jadyn R. Malone, Nic A. Murray, Monika Tokmina-Lukaszewska, Arnab K. Nath, Tamara Flusche, Florence Mus, Jennifer L. DuBois, John W. Peters, and et al. 2025. "Long-Range Allosteric Communication Modulated by Active Site Mn(II) Coordination Drives Catalysis in Xanthobacter autotrophicus Acetone Carboxylase" International Journal of Molecular Sciences 26, no. 13: 5945. https://doi.org/10.3390/ijms26135945
APA StyleMattice, J. R., Shisler, K. A., Malone, J. R., Murray, N. A., Tokmina-Lukaszewska, M., Nath, A. K., Flusche, T., Mus, F., DuBois, J. L., Peters, J. W., & Bothner, B. (2025). Long-Range Allosteric Communication Modulated by Active Site Mn(II) Coordination Drives Catalysis in Xanthobacter autotrophicus Acetone Carboxylase. International Journal of Molecular Sciences, 26(13), 5945. https://doi.org/10.3390/ijms26135945