
Genome-Wide Identification and Expression Profiling of Plasma Membrane-Localized SWEET Gene Family Associated with Sugar Transport During Yam Tuber Development


Abstract
1. Introduction
2. Results
2.1. Identification of Members of the SWEET Gene Family
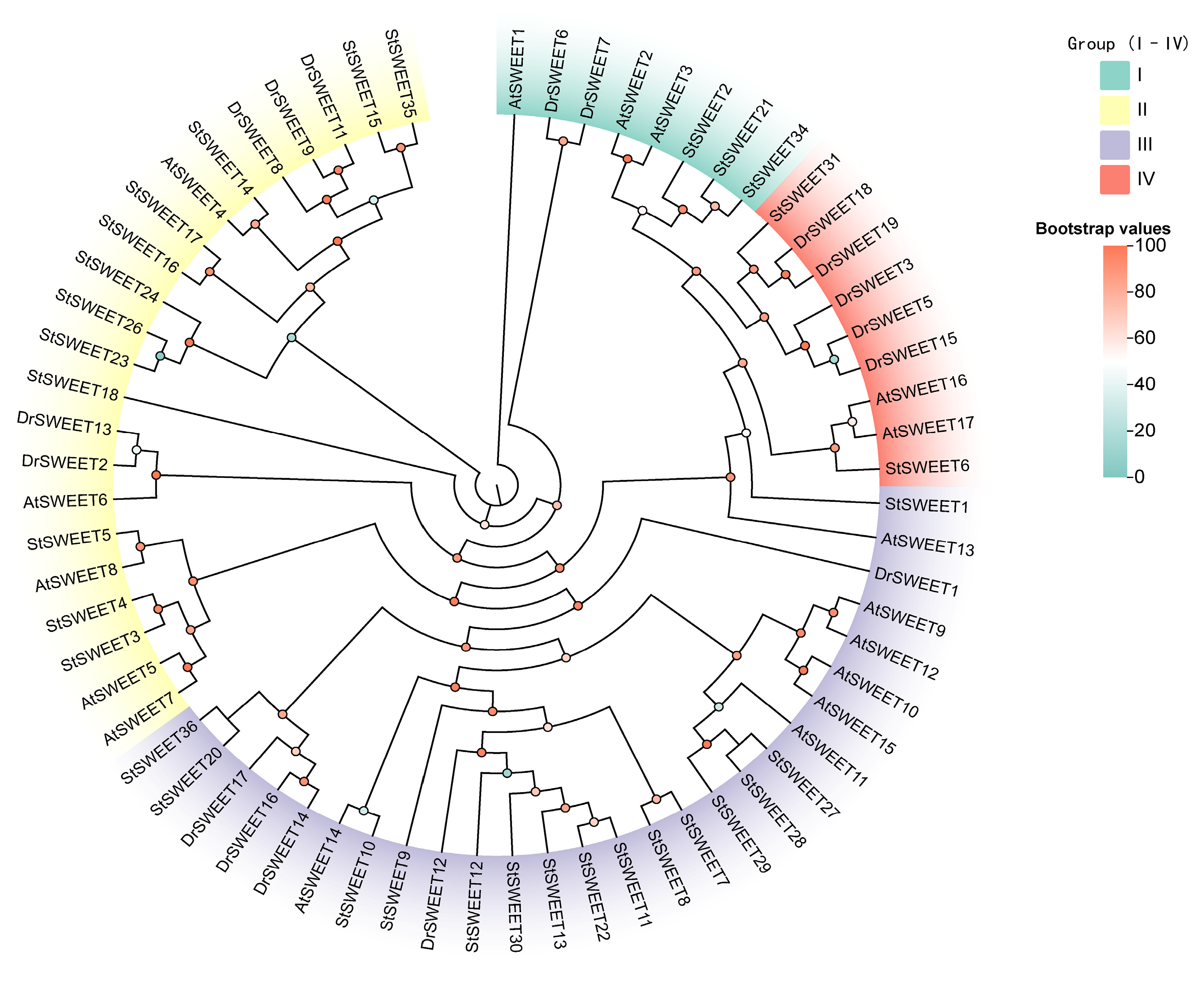
2.2. Phylogenetic Analysis and Classification of DrSWEET Proteins
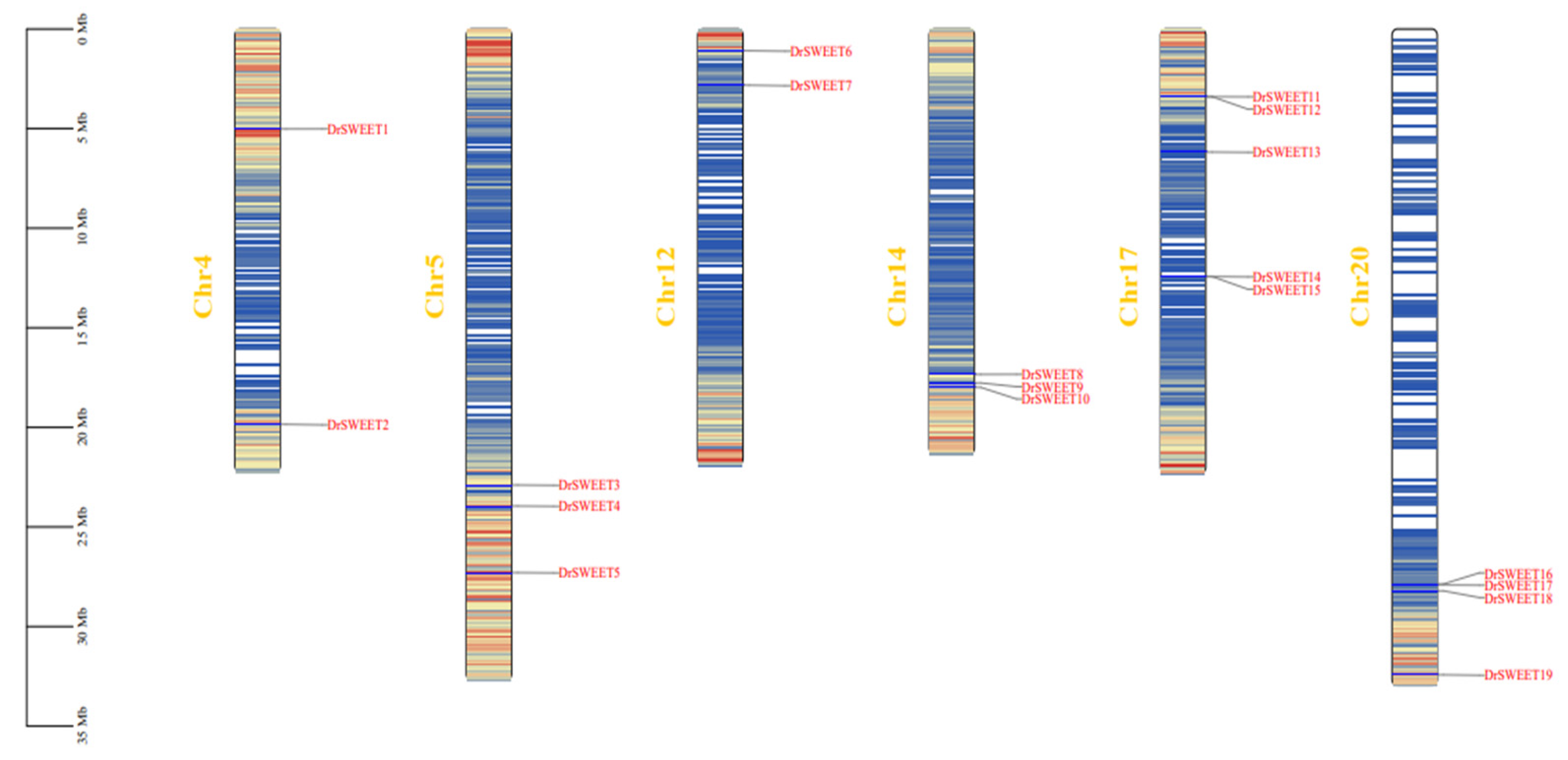
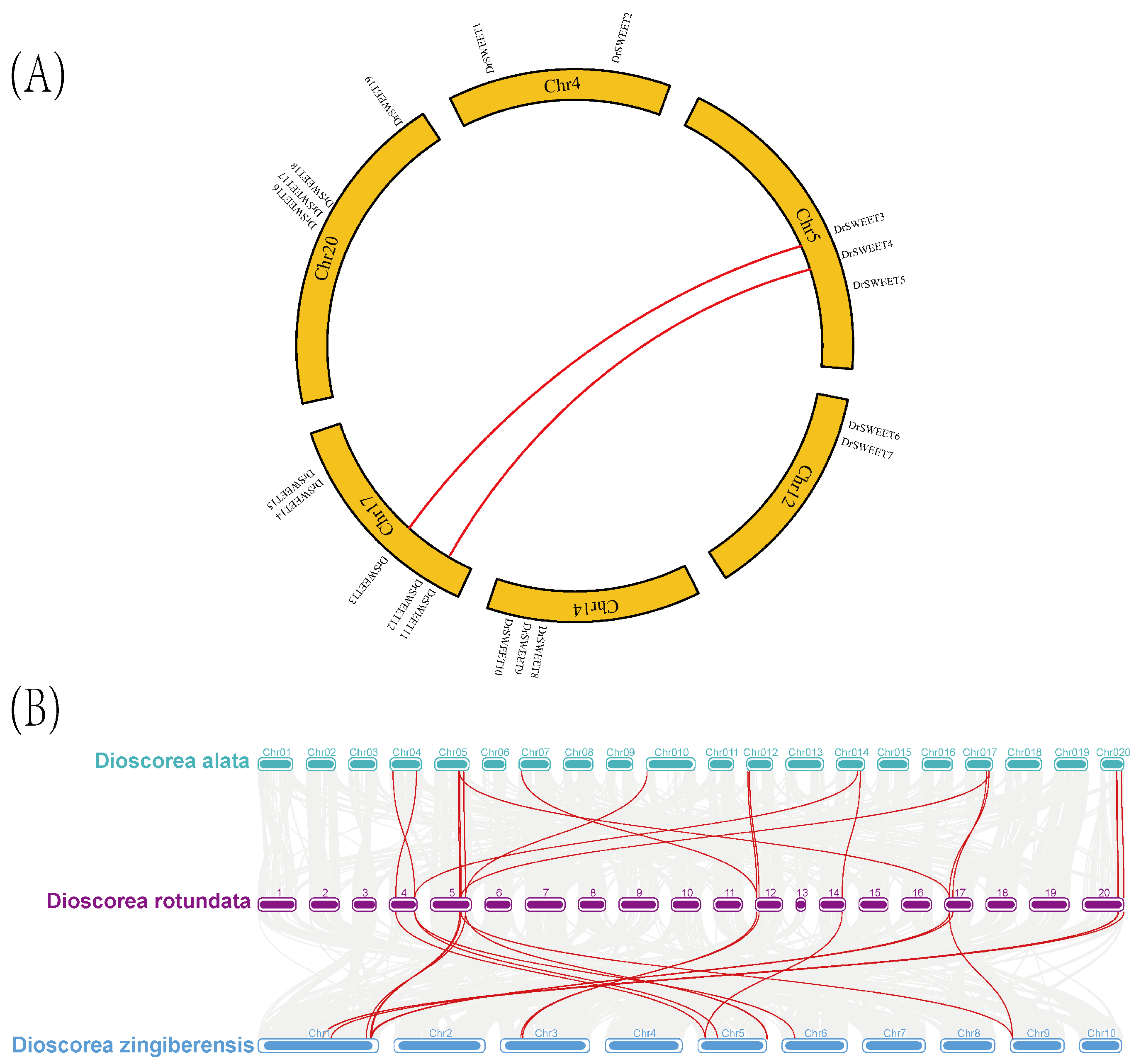
2.3. Chromosome Distribution, Duplication Events, and Collinear Analysis of DrSWEET Genes
2.4. Evolutionary Analysis of DrSWEET Genes and Their Expansion in Several Different Species
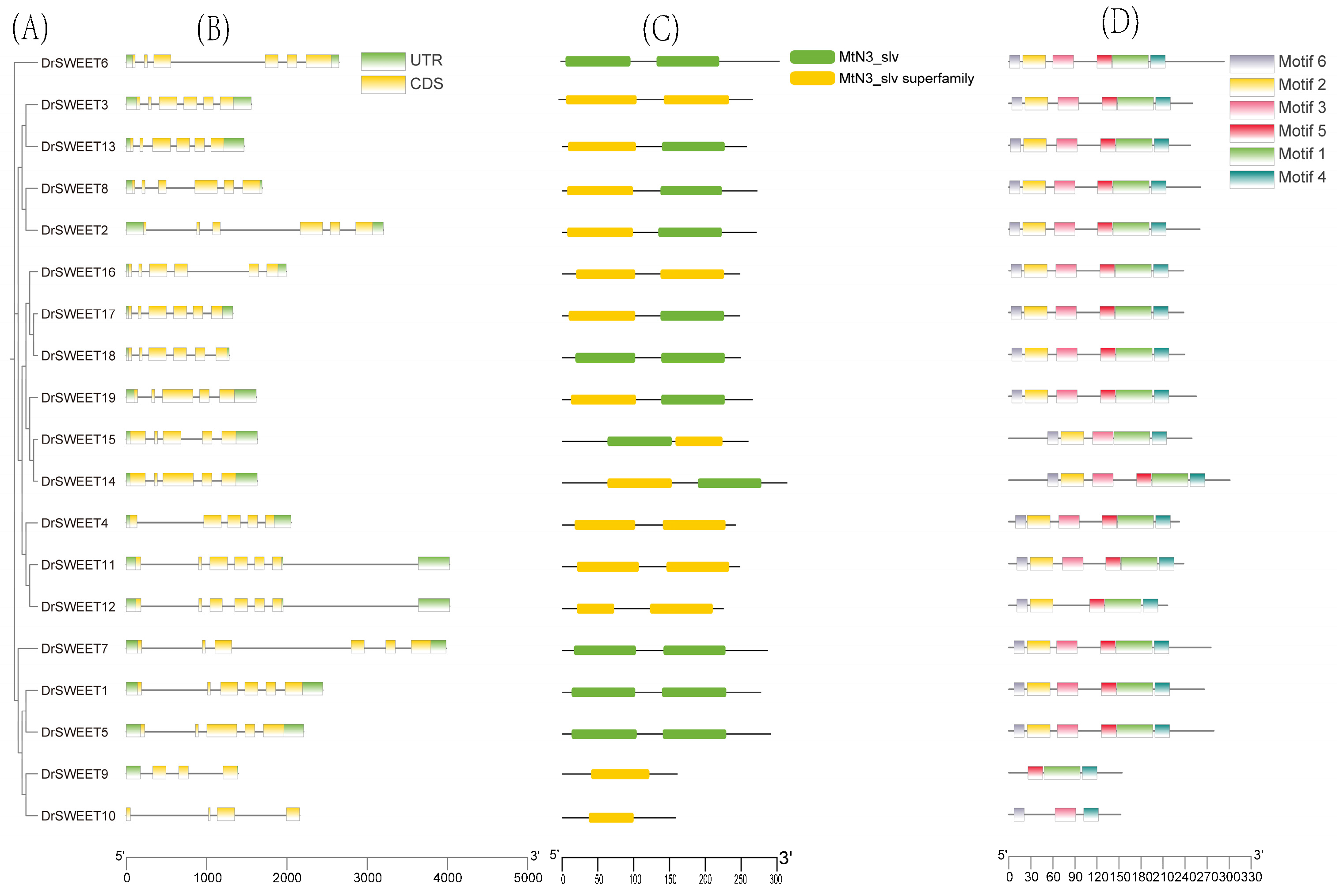
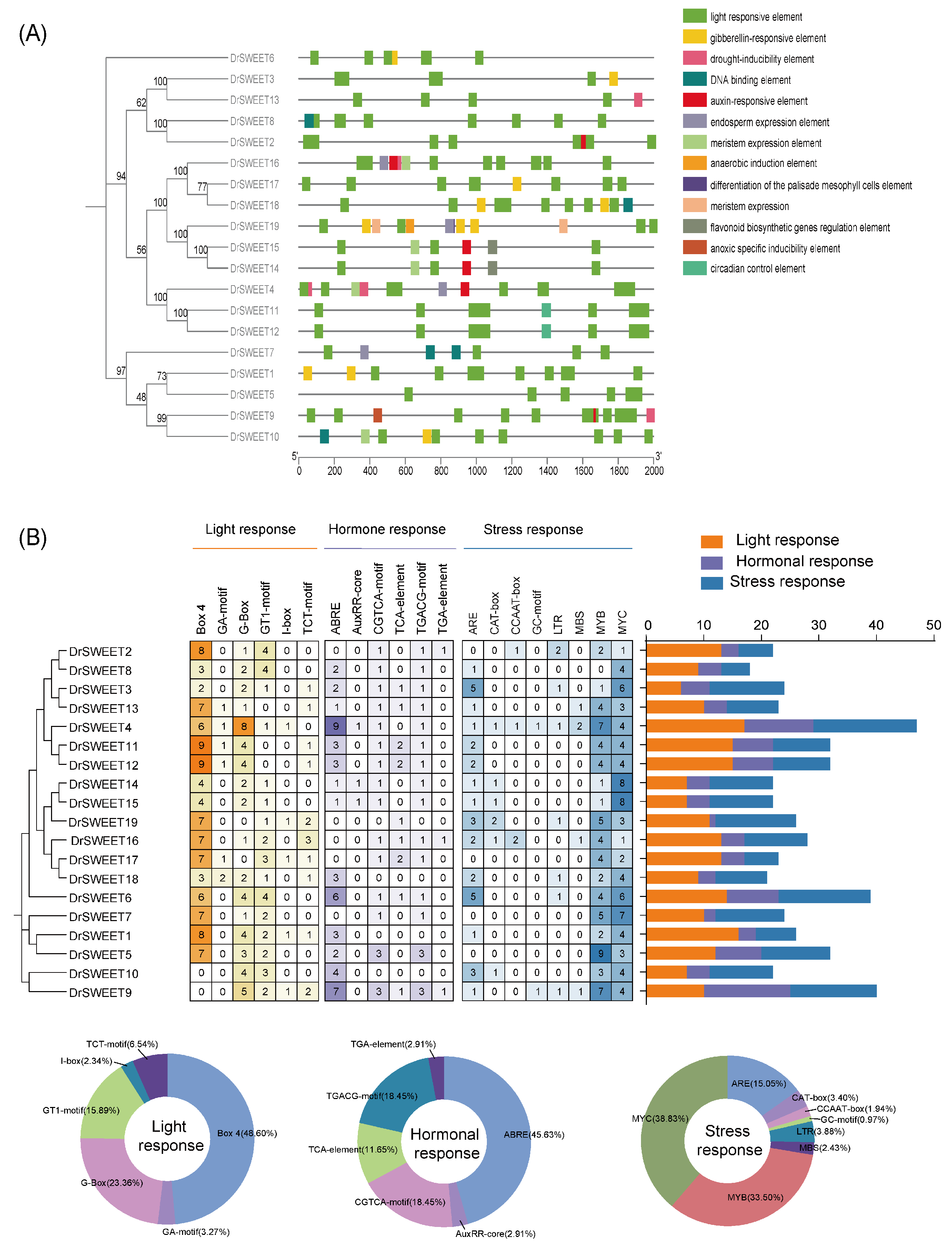
2.5. Gene Structure and Motif Composition of the DrSWEET Genes
2.6. Cis-Regulatory Elements in the Promoters
2.7. Expression Patterns of DrSWEET Genes in Different Tissues
2.8. Expression Patterns of DrSWEET Genes in Tubers at Different Development Stages
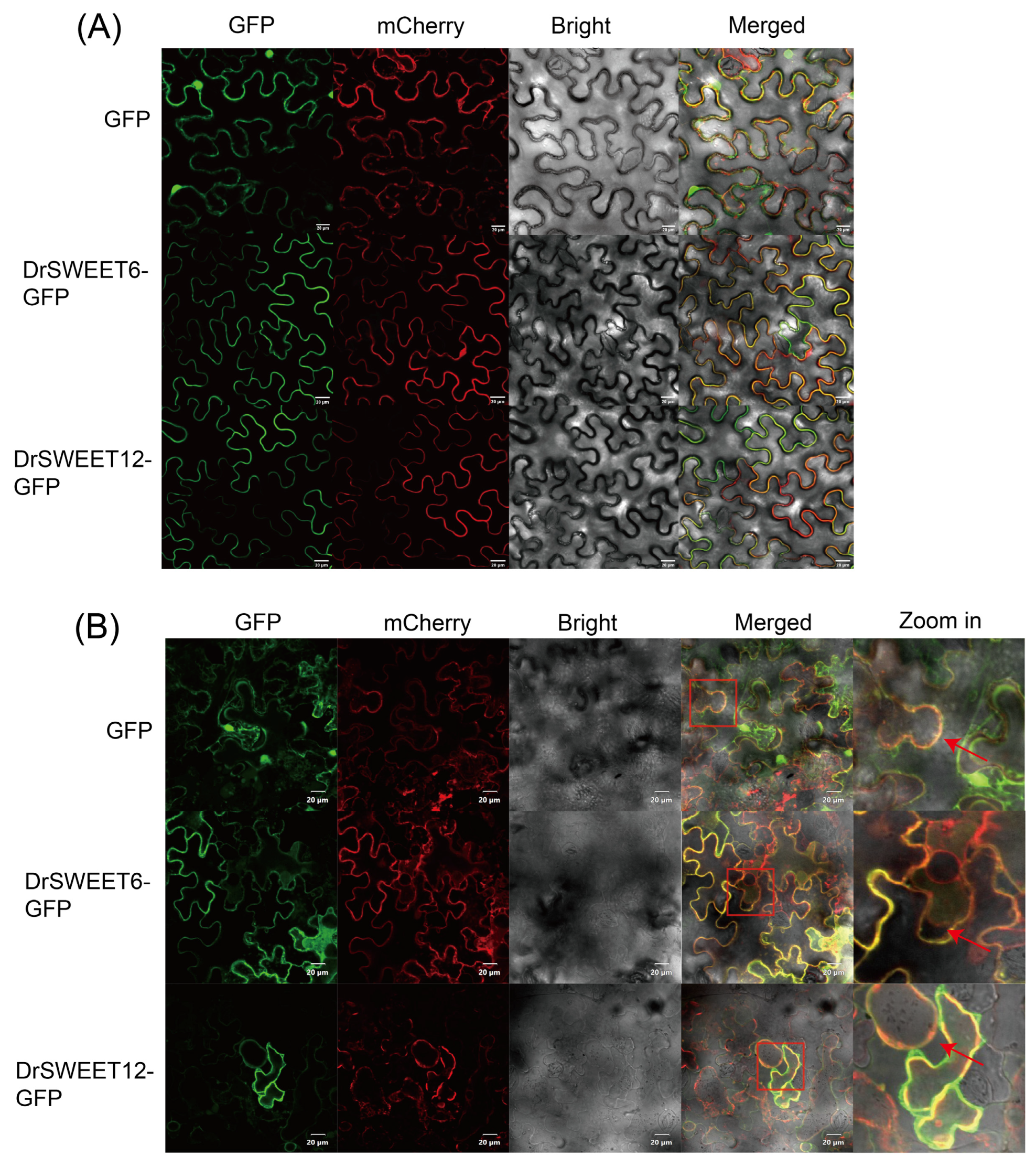
2.9. Subcellular Localization of DrSWEET Proteins
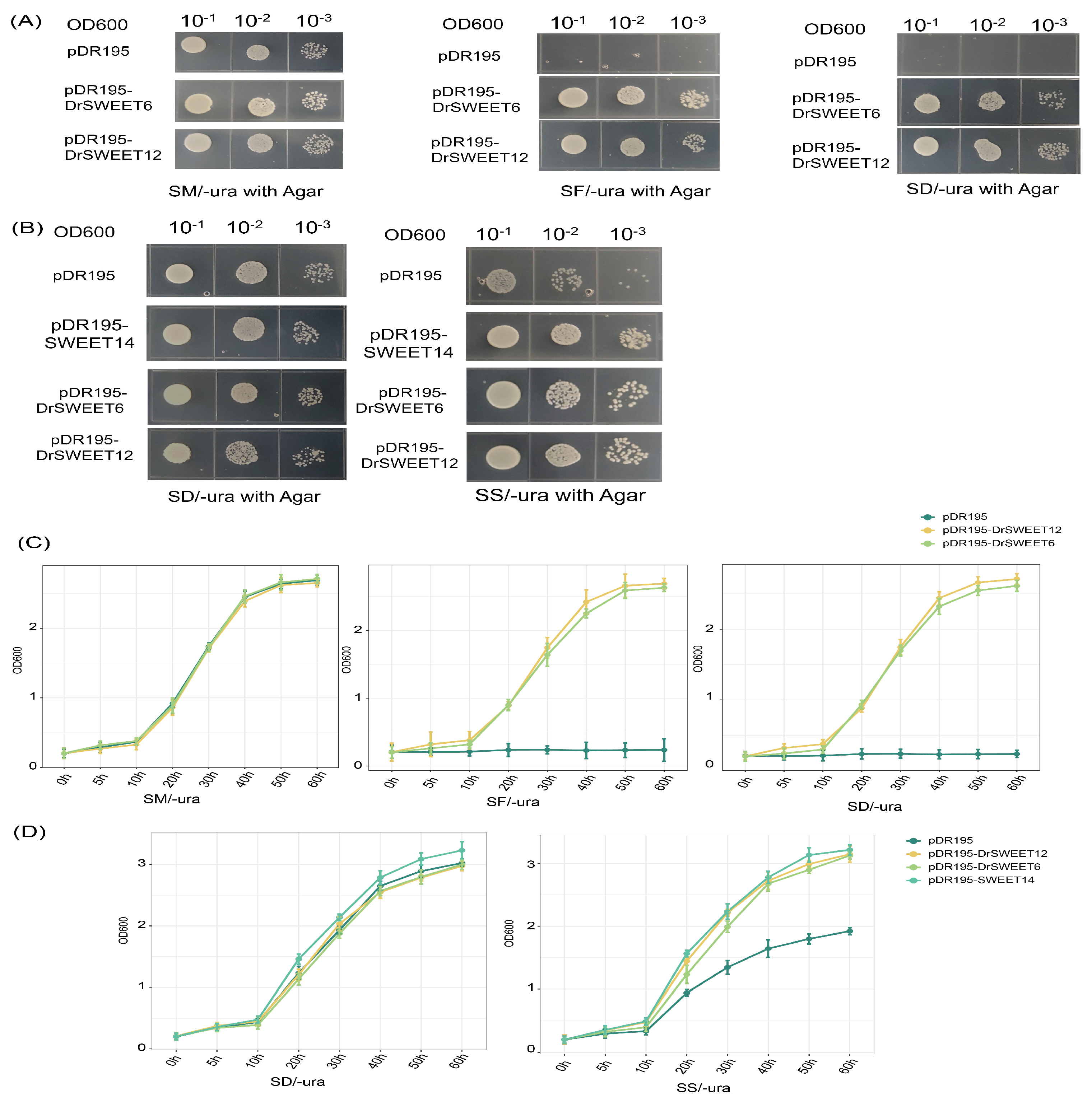
2.10. Transport Substrate Specificity of DrSWEET6 and DrSWEET12 in Yeast
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Identification of the SWEET Genes in Yams
4.3. Multiple Sequence Alignment and Phylogenetic Analysis of SWEET Genes
4.4. Chromosomal Mapping, Gene Replication, and Syntenic Analysis with Other Plant Species
4.5. Identification of Cis-Regulatory Elements in the Promoter Regions of Pear SWEET Genes
4.6. Expression of DrSWEET Genes According to Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.7. Transcriptome Data Analysis
4.8. Yeast Complementation Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, L.-Q.; Qu, X.-Q.; Hou, B.-H.; Sosso, D.; Osorio, S.; Fernie, A.R.; Frommer, W.B. Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 2012, 335, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Comtet, J.; Turgeon, R.; Stroock, A.D. Phloem Loading through Plasmodesmata: A Biophysical Analysis. Plant Physiol. 2017, 175, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Jiang, H.; Zeng, F. The sugar transporter proteins in plants: An elaborate and widespread regulation network—A review. Int. J. Biol. Macromol. 2025, 294, 139252. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wei, X.; Yang, J.; Li, H.; Ma, B.; Zhang, K.; Zhang, Y.; Cheng, L.; Ma, F.; Li, M. Heterologous expression of the apple hexose transporter MdHT2.2 altered sugar concentration with increasing cell wall invertase activity in tomato fruit. Plant Biotechnol. J. 2020, 18, 540–552. [Google Scholar] [CrossRef]
- Gibson, S.I. Control of plant development and gene expression by sugar signaling. Curr. Opin. Plant Biol. 2005, 8, 93–102. [Google Scholar] [CrossRef]
- Chen, L.-Q.; Hou, B.-H.; Lalonde, S.; Takanaga, H.; Hartung, M.L.; Qu, X.-Q.; Guo, W.-J.; Kim, J.-G.; Underwood, W.; Chaudhuri, B.; et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 2010, 468, 527–532. [Google Scholar] [CrossRef]
- Yuan, M.; Wang, S. Rice MtN3/Saliva/SWEET Family Genes and Their Homologs in Cellular Organisms. Mol. Plant 2013, 6, 665–674. [Google Scholar] [CrossRef]
- Gautam, T.; Dutta, M.; Jaiswal, V.; Zinta, G.; Gahlaut, V.; Kumar, S. Emerging Roles of SWEET Sugar Transporters in Plant Development and Abiotic Stress Responses. Cells 2022, 11, 1303. [Google Scholar] [CrossRef]
- Guan, Y.-F.; Huang, X.-Y.; Zhu, J.; Gao, J.-F.; Zhang, H.-X.; Yang, Z.-N. RUPTURED POLLEN GRAIN1, a Member of the MtN3/saliva Gene Family, Is Crucial for Exine Pattern Formation and Cell Integrity of Microspores in Arabidopsis. Plant Physiol. 2008, 147, 852–863. [Google Scholar] [CrossRef]
- Wang, J.; Xue, X.; Zeng, H.; Li, J.; Chen, L.-Q. Sucrose rather than GA transported by AtSWEET13 and AtSWEET14 supports pollen fitness at late anther development stages. New Phytol. 2022, 236, 525–537. [Google Scholar] [CrossRef]
- Eom, J.-S.; Chen, L.-Q.; Sosso, D.; Julius, B.T.; Lin, I.W.; Qu, X.-Q.; Braun, D.M.; Frommer, W.B. SWEETs, transporters for intracellular and intercellular sugar translocation. Curr. Opin. Plant Biol. 2015, 25, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yu, J.; Cai, Q.; Chen, Y.; Li, Y.; Ren, Y.; Miao, Y. Triple-localized WHIRLY2 Influences Leaf Senescence and Silique Development via Carbon Allocation. Plant Physiol. 2020, 184, 1348–1362. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Das, S.; Jagadis Gupta, K.; Ranjan, A.; Foyer, C.H.; Thakur, J.K. Physiological implications of SWEETs in plants and their potential applications in improving source-sink relationships for enhanced yield. Plant Biotechnol. J. 2023, 21, 1528–1541. [Google Scholar] [CrossRef] [PubMed]
- Jeena, G.S.; Kumar, S.; Shukla, R.K. Structure, evolution and diverse physiological roles of SWEET sugar transporters in plants. Plant Mol. Biol. 2019, 100, 351–365. [Google Scholar] [CrossRef]
- Breia, R.; Conde, A.; Badim, H.; Fortes, A.M.; Gerós, H.; Granell, A. Plant SWEETs: From sugar transport to plant-pathogen interaction and more unexpected physiological roles. Plant Physiol. 2021, 186, 836–852. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, H.; Wang, T.; Zhang, J.; Liu, W.; Fang, H.; Zhang, Z.; Peng, F.; Chen, X.; Wang, N. Abscisic acid and regulation of the sugar transporter gene MdSWEET9b promote apple sugar accumulation. Plant Physiol. 2023, 192, 2081–2101. [Google Scholar] [CrossRef]
- Wu, Y.; Di, T.; Wu, Z.; Peng, J.; Wang, J.; Zhang, K.; He, M.; Li, N.; Hao, X.; Fang, W.; et al. CsLHY positively regulates cold tolerance by activating CsSWEET17 in tea plants. Plant Physiol. Biochem. 2024, 207, 108341. [Google Scholar] [CrossRef]
- Fakher, B.; Ashraf, M.A.; Wang, L.; Wang, X.; Zheng, P.; Aslam, M.; Qin, Y. Pineapple SWEET10 is a glucose transporter. Hortic Res 2023, 10, uhad175. [Google Scholar] [CrossRef]
- Chen, Y.; Tariq, H.; Shen, D.; Liu, J.; Dou, D. Omics technologies accelerating research progress in yams. Veg. Res. 2024, 4, e014. [Google Scholar] [CrossRef]
- Yuan, M.; Zhao, J.; Huang, R.; Li, X.; Xiao, J.; Wang, S. Rice MtN3/saliva/SWEET gene family: Evolution, expression profiling, and sugar transport. J. Integr. Plant Biol. 2014, 56, 559–570. [Google Scholar] [CrossRef]
- Qin, J.-X.; Jiang, Y.-J.; Lu, Y.-Z.; Zhao, P.; Wu, B.-J.; Li, H.-X.; Wang, Y.; Xu, S.-B.; Sun, Q.-X.; Liu, Z.-S. Genome-wide identification and transcriptome profiling reveal great expansion of SWEET gene family and their wide-spread responses to abiotic stress in wheat (Triticum aestivum L.). J. Integr. Agric. 2020, 19, 1704–1720. [Google Scholar] [CrossRef]
- Mizuno, H.; Kasuga, S.; Kawahigashi, H. The sorghum SWEET gene family: Stem sucrose accumulation as revealed through transcriptome profiling. Biotechnol. Biofuels 2016, 9, 127. [Google Scholar] [CrossRef]
- Patil, G.; Valliyodan, B.; Deshmukh, R.; Prince, S.; Nicander, B.; Zhao, M.; Sonah, H.; Song, L.; Lin, L.; Chaudhary, J.; et al. Soybean (Glycine max) SWEET gene family: Insights through comparative genomics, transcriptome profiling and whole genome re-sequence analysis. BMC Genom. 2015, 16, 520. [Google Scholar] [CrossRef] [PubMed]
- Heng, S.; He, J.; Zhu, X.; Cai, J.; Fu, M.; Zhang, S.; Zeng, W.; Xing, F.; Mao, G. Genome wide identification of BjSWEET gene family and drought response analysis of BjSWEET12 and BjSWEET17 genes in Brassica juncea. BMC Plant Biol. 2024, 24, 1094. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Osakabe, Y. Sugar compartmentation as an environmental stress adaptation strategy in plants. Semin. Cell Dev. Biol. 2018, 83, 106–114. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, L.; Huang, W.; Yuan, M.; Zhou, F.; Li, X.; Lin, Y. Overexpression of OsSWEET5 in Rice Causes Growth Retardation and Precocious Senescence. PLoS ONE 2014, 9, e94210. [Google Scholar] [CrossRef]
- Zhang, Z.; Zou, L.; Ren, C.; Ren, F.; Wang, Y.; Fan, P.; Li, S.; Liang, Z. VvSWEET10 Mediates Sugar Accumulation in Grapes. Genes 2019, 10, 255. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Z.; Tang, R.; Wang, L.; Chen, C.; Ren, Z. Genome-Wide Identification and Expression Analysis of Hsf and Hsp Gene Families in Cucumber (Cucumis sativus L.). Plant Growth Regul. 2021, 95, 223–239. [Google Scholar] [CrossRef]
- Ceylan, Y.; Altunoglu, Y.C.; Horuz, E. HSF and Hsp Gene Families in sunflower: A comprehensive genome-wide determination survey and expression patterns under abiotic stress conditions. Protoplasma 2023, 260, 1473–1491. [Google Scholar] [CrossRef]
- Wu, D.; Luo, J.; Chen, J.; Zhang, L.; Lim, K.J.; Wang, Z. Selection pressure causes differentiation of the SPL gene family in the Juglandaceae. Mol. Genet. Genom. 2019, 294, 1037–1048. [Google Scholar] [CrossRef]
- Han, X.; Han, S.; Zhu, Y.; Liu, Y.; Gao, S.; Yin, J.; Wang, F.; Yao, M. Genome-Wide Identification and Expression Analysis of the SWEET Gene Family in Capsicum annuum L. Int. J. Mol. Sci. 2023, 24, 17408. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Oikawa, T.; Chiba, Y.; Ishimaru, Y.; Shimizu, T.; Sano, N.; Koshiba, T.; Kamiya, Y.; Ueda, M.; Seo, M. AtSWEET13 and AtSWEET14 regulate gibberellin-mediated physiological processes. Nat. Commun. 2016, 7, 13245. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Wang, J.; Wang, L.; Liu, H.; Wu, W.; Hou, F.; Liu, Y.; Gao, Y.; Cheng, X.; Li, S.; et al. Genome-wide analysis of the SWEET gene family in Hemerocallis citrina and functional characterization of HcSWEET4a in response to salt stress. BMC Plant Biol. 2024, 24, 661. [Google Scholar] [CrossRef] [PubMed]
- Klemens, P.A.W.; Patzke, K.; Deitmer, J.; Spinner, L.; Le Hir, R.; Bellini, C.; Bedu, M.; Chardon, F.; Krapp, A.; Neuhaus, H.E. Overexpression of the Vacuolar Sugar Carrier AtSWEET16 Modifies Germination, Growth, and Stress Tolerance in Arabidopsis. Plant Physiol. 2013, 163, 1338–1352. [Google Scholar] [CrossRef]
- Kaur, H.; Manna, M.; Thakur, T.; Gautam, V.; Salvi, P. Imperative role of sugar signaling and transport during drought stress responses in plants. Physiol. Plant. 2021, 171, 833–848. [Google Scholar] [CrossRef]
- Durand, M.; Porcheron, B.; Hennion, N.; Maurousset, L.; Lemoine, R.; Pourtau, N. Water Deficit Enhances C Export to the Roots in Arabidopsis thaliana Plants with Contribution of Sucrose Transporters in Both Shoot and Roots. Plant Physiol. 2016, 170, 1460–1479. [Google Scholar] [CrossRef]
- Feng, C.-Y.; Han, J.-X.; Han, X.-X.; Jiang, J. Genome-wide identification, phylogeny, and expression analysis of the SWEET gene family in tomato. Gene 2015, 573, 261–272. [Google Scholar] [CrossRef]
- Hu, Z.; Tang, Z.; Zhang, Y.; Niu, L.; Yang, F.; Zhang, D.; Hu, Y. Rice SUT and SWEET Transporters. Int. J. Mol. Sci. 2021, 22, 11198. [Google Scholar] [CrossRef]
- Anjali, A.; Fatima, U.; Manu, M.S.; Ramasamy, S.; Senthil-Kumar, M. Structure and regulation of SWEET transporters in plants: An update. Plant Physiol. Biochem. 2020, 156, 1–6. [Google Scholar] [CrossRef]
- Chen, L.; Cai, M.; Liu, J.; Jiang, X.; Liu, J.; Zhenxing, W.; Wang, Y.; Li, Y. Genome-wide identification and expression analyses of SWEET gene family reveal potential roles in plant development, fruit ripening and abiotic stress responses in cranberry (Vaccinium macrocarpon Ait). PeerJ 2024, 12, e17974. [Google Scholar] [CrossRef]
- Reiser, L.; Bakker, E.; Subramaniam, S.; Chen, X.; Sawant, S.; Khosa, K.; Prithvi, T.; Berardini, T.Z. The Arabidopsis Information Resource in 2024. Genetics 2024, 227, iyae027. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, D.; Xiao, Q.; Wang, H.; Wen, J.; Tu, J.; Shen, J.; Fu, T.; Yi, B. An in planta haploid induction system in Brassica napus. J. Integr. Plant Biol. 2022, 64, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids Res. 2021, 49, D458–D460. [Google Scholar] [CrossRef]
- Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Gonzales, N.R.; Gwadz, M.; Lu, S.; Marchler, G.H.; Song, J.S.; Thanki, N.; Yamashita, R.A.; et al. The conserved domain database in 2023. Nucleic Acids Res. 2023, 51, D384–D388. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Xing, L.; Zhang, Y.; Ge, M.; Zhao, L.; Huo, X. Identification of WRKY gene family in Dioscorea opposita Thunb. reveals that DoWRKY71 enhanced the tolerance to cold and ABA stress. PeerJ 2024, 12, e17016. [Google Scholar] [CrossRef]
- Maness, N. Extraction and Analysis of Soluble Carbohydrates. In Plant Stress Tolerance: Methods and Protocols; Sunkar, R., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 341–370. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Number of Amino Acids | Molecular Weight | Theoretical pI | Instability Index | Aliphatic Index | Grand Average of Hydropathicity | Subcellular Localization |
|---|---|---|---|---|---|---|---|
| DrSWEET1 | 265 | 30,100.93 | 8.84 | 34.82 | 116.15 | 0.728 | PlasmaMembrane |
| DrSWEET2 | 259 | 28,879.07 | 8.87 | 36.92 | 107.61 | 0.55 | PlasmaMembrane |
| DrSWEET3 | 249 | 27,792.97 | 8.59 | 45.54 | 116.67 | 0.646 | PlasmaMembrane |
| DrSWEET4 | 231 | 25,941.74 | 9.05 | 36.57 | 123.55 | 0.824 | PlasmaMembrane |
| DrSWEET5 | 278 | 31,401.43 | 8.71 | 30.48 | 116.01 | 0.655 | PlasmaMembrane |
| DrSWEET6 | 292 | 32,414.34 | 9.63 | 39.47 | 109.76 | 0.401 | PlasmaMembrane |
| DrSWEET7 | 274 | 30,542.61 | 8.78 | 33.73 | 126.28 | 0.726 | PlasmaMembrane |
| DrSWEET8 | 260 | 28,541.97 | 9.01 | 27.58 | 116.88 | 0.754 | PlasmaMembrane |
| DrSWEET9 | 153 | 17,072.41 | 6.39 | 49.59 | 130 | 0.666 | PlasmaMembrane |
| DrSWEET10 | 151 | 16,772.87 | 5.58 | 37.26 | 109.67 | 0.695 | PlasmaMembrane |
| DrSWEET11 | 237 | 26,533.57 | 9.49 | 39.49 | 121.31 | 0.916 | PlasmaMembrane |
| DrSWEET12 | 215 | 23,990.57 | 9.46 | 41.9 | 120.14 | 0.927 | PlasmaMembrane |
| DrSWEET13 | 246 | 27,264.68 | 9.3 | 38.95 | 122.4 | 0.652 | PlasmaMembrane |
| DrSWEET14 | 300 | 33,682.69 | 9.56 | 27.54 | 131.2 | 0.859 | PlasmaMembrane |
| DrSWEET15 | 248 | 27,944.52 | 9.61 | 28.89 | 126.9 | 0.667 | PlasmaMembrane |
| DrSWEET16 | 237 | 26,257.78 | 9.25 | 40.75 | 128.61 | 0.84 | PlasmaMembrane |
| DrSWEET17 | 237 | 26,821.32 | 9.04 | 38.6 | 115.11 | 0.757 | PlasmaMembrane |
| DrSWEET18 | 238 | 26,763.48 | 9.04 | 43.35 | 128.53 | 0.83 | PlasmaMembrane |
| DrSWEET19 | 254 | 28,296.95 | 9 | 30.54 | 119.29 | 0.771 | PlasmaMembrane |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, N.; Zhang, Y.; Huo, X.; Xing, L.; Ge, M.; Suo, N. Genome-Wide Identification and Expression Profiling of Plasma Membrane-Localized SWEET Gene Family Associated with Sugar Transport During Yam Tuber Development. Int. J. Mol. Sci. 2025, 26, 5847. https://doi.org/10.3390/ijms26125847
Li N, Zhang Y, Huo X, Xing L, Ge M, Suo N. Genome-Wide Identification and Expression Profiling of Plasma Membrane-Localized SWEET Gene Family Associated with Sugar Transport During Yam Tuber Development. International Journal of Molecular Sciences. 2025; 26(12):5847. https://doi.org/10.3390/ijms26125847
Chicago/Turabian StyleLi, Na, Yanfang Zhang, Xiuwen Huo, Linan Xing, Mingran Ge, and Ningning Suo. 2025. "Genome-Wide Identification and Expression Profiling of Plasma Membrane-Localized SWEET Gene Family Associated with Sugar Transport During Yam Tuber Development" International Journal of Molecular Sciences 26, no. 12: 5847. https://doi.org/10.3390/ijms26125847
APA StyleLi, N., Zhang, Y., Huo, X., Xing, L., Ge, M., & Suo, N. (2025). Genome-Wide Identification and Expression Profiling of Plasma Membrane-Localized SWEET Gene Family Associated with Sugar Transport During Yam Tuber Development. International Journal of Molecular Sciences, 26(12), 5847. https://doi.org/10.3390/ijms26125847