
Genomic Profile and Clinical Outcomes in Acute Myeloid Leukemia with Monosomal Karyotype
 , , and
, , and 
Abstract
1. Introduction
2. Results
2.1. Patient Characteristics
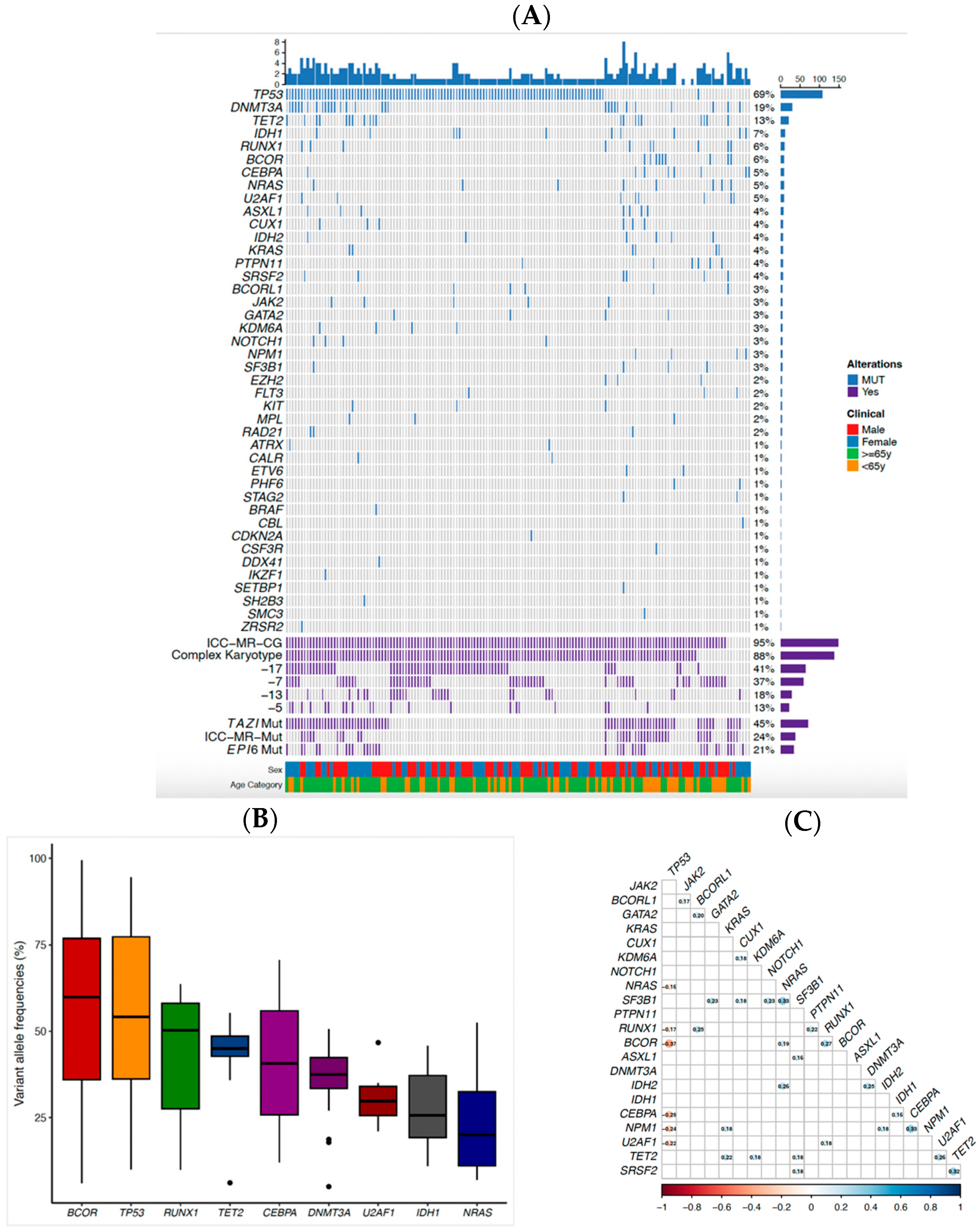
2.2. Genomic Profile
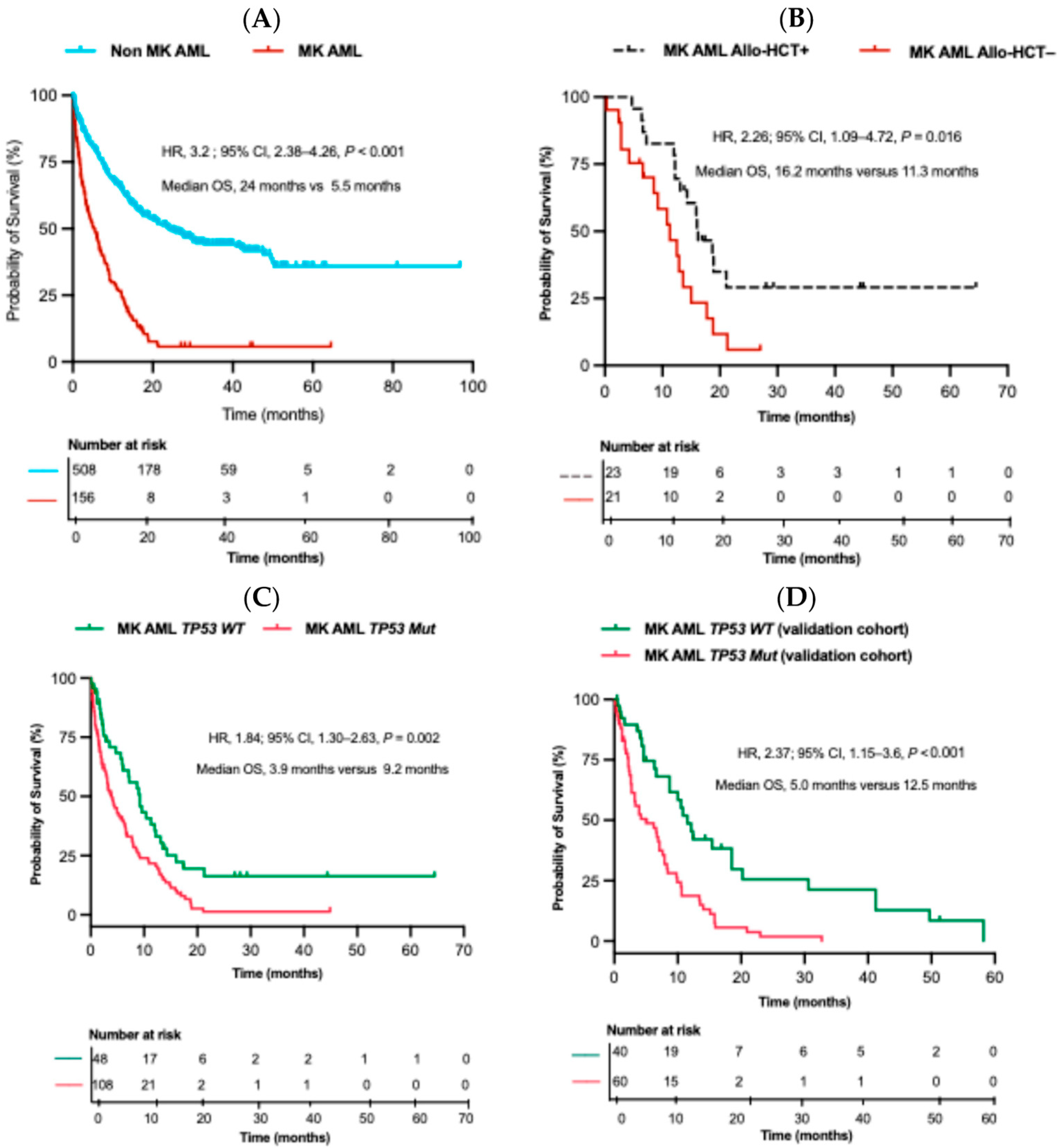
2.3. Overall Survival (OS) and Impact of Allo-HCT on MK AML Patients
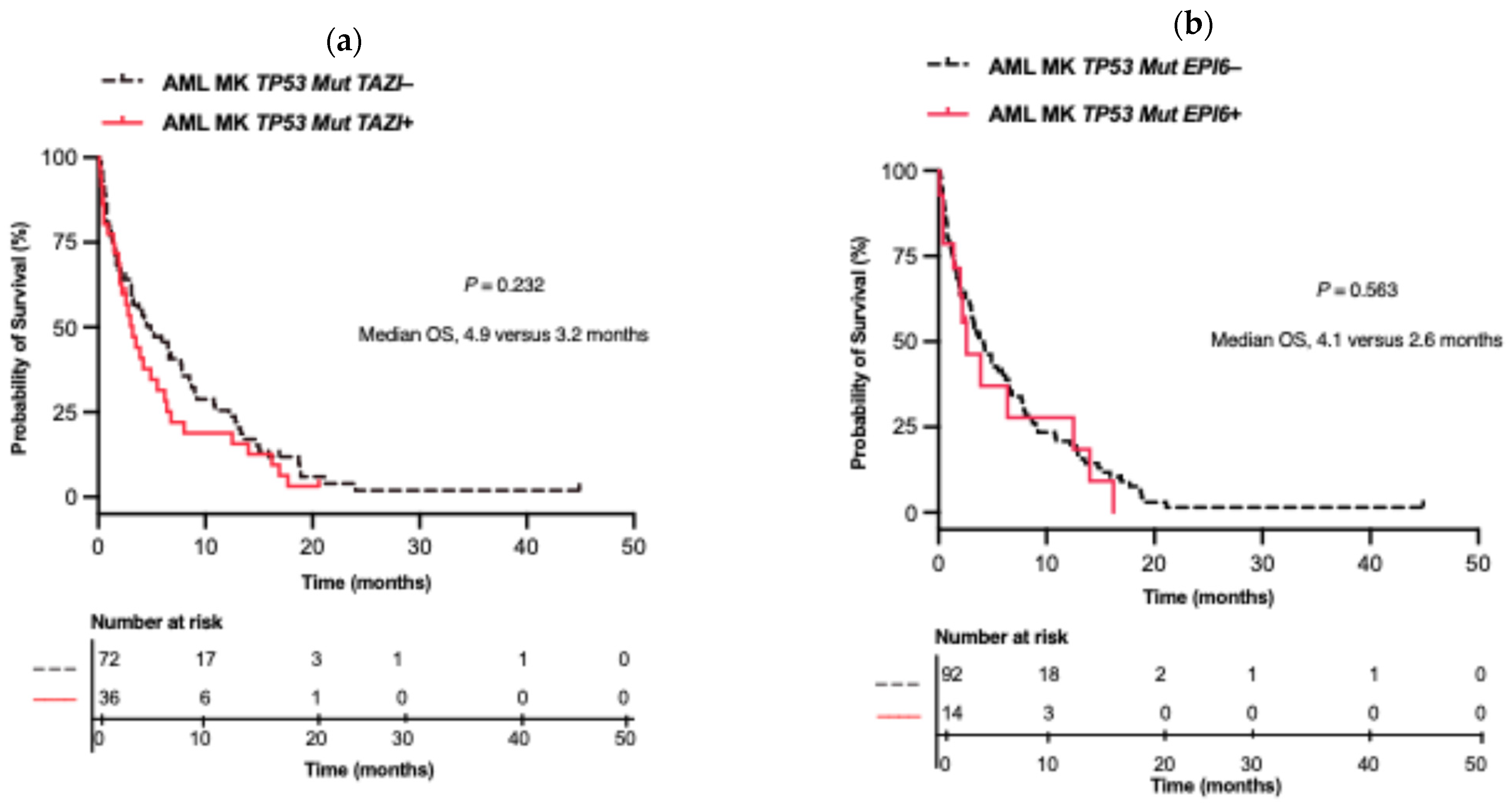
2.4. Impact of TP53 Mutation on OS
2.5. Impact of Co-Alterations on MK AML in TP53 Mut and WT Patients
2.6. Validation Cohort
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Treatment
4.3. Cytogenetic Analysis
4.4. Molecular Testing
4.5. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Allo-HCT | Allogeneic hematopoietic stem cell transplantation |
| AML | Acute Myeloid Leukemia |
| BM | Bone marrow |
| CK | Complex karyotype |
| CR | Complete remission |
| ELN 2022 | European LeukemiaNet 2022 |
| FLAG-IDA | Fludarabine, Cytarabine, Filgrastim, and Idarubicin |
| IC | Induction chemotherapy |
| ICC | International Consensus Classification |
| ISCN | International System for Human Cytogenomic Nomenclature |
| LIT | Low-intensity therapy |
| MK | Monosomal Karyotype |
| MK AML | Acute Myeloid Leukemia with Monosomal Karyotype |
| MN | Myeloid Neoplasia |
| MR | Myelodysplasia-Related |
| NGS | Next-generation sequencing |
| NK | Normal karyotype |
| Nove-HiDAC | Mitoxantrone, Etoposide, and modified high-dose cytarabine) |
| OS | Overall survival |
| SPSS | IBM Statistical Package for the Social Sciences |
| TAR-SEQ | Targeted sequencing |
| TP53 Mut | TP53 mutation |
| UHN | University Health Network |
| VAF | Variant allele frequency |
| WBC | White blood cell |
| WT | Wild-type |
Appendix A

{kind=link}
{kind=link}
{kind=link}
| Variable | MK AML TP53 WT (n = 48) | Median OS (Months) | HR | 95% CI | p Value | |
|---|---|---|---|---|---|---|
| CK | Present | 31 (64.5%) | 7.1 | 1.112 | 0.55–2.25 | 0.771 |
| Absent | 17 (35.4%) | 9.2 | ||||
| MR Cytogenetics | Present | 40 (83.3%) | 7.2 | 0.419 | 0.177–1.99 | 0.135 |
| Absent | 8 (16.7%) | 9.2 | ||||
| MR mutations | Present | 26 (54.1%) | 9.2 | 1.034 | 0.53–2.03 | 0.922 |
| Absent | 22 (45.8%) | 8.7 | ||||
| Variable | MK + (n = 156) | CR | MK + TP53 + (n = 108) | MK + TP53 − (n = 48) | |
|---|---|---|---|---|---|
| Induction Chemotherapy | Supportive | 47 (30.1%) | 33 (30.6%) | 14 (29.2%) | |
| Intensive | 66 (42.3%) | 30 (45.4%) a | 43 (39.8%) | 23 (47.9%) | |
| Less Intensive | 43 (27.6%) | 9 (20.9%) a | 32 (29.6%) | 11 (22.9%) | |
| Allo-HCT | Transplanted | 23 (14.7%) | 23 (14.7%) b | 10 (9.3%) | 13 (27.1%) |
| Not Transplanted | 133 (85.3%) | 21 (13.7%) b | 14 (13%) | 7 (14.6%) | |
| Variable | Study Cohort (n = 156) | Validation Cohort (n = 100) | p-Value | |
|---|---|---|---|---|
| Age (years) | Median (min-max) | 71 (19–94) | 65 (21–83) | 0.002 a |
| Sex | Male Female | 82 (52.6%) 74 (47.4%) | 58(52%) 42 (42%) | 0.394 b |
| TP53 Mut Status | AML MK TP53 Mut | 108 (69%) | 60 (60%) | |
| AML MK TP53 WT | 48 (31%) | 40 (40%) | 0.129 b | |
| Transplant Status | Allo-HCT | 23 (14.7%) | 21 (21%) | 0.196 b |
Appendix B
References
- Anelli, L.; Pasciolla, C.; Zagaria, A.; Specchia, G.; Albano, F. Monosomal Karyotype in Myeloid Neoplasias: A Literature Review. OncoTargets Ther. 2017, 10, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, B.C.; Othus, M.; Fang, M.; Roulston, D.; Appelbaum, F.R. Prognostic Impact of Monosomal Karyotype in Young Adult and Elderly Acute Myeloid Leukemia: The Southwest Oncology Group (SWOG) Experience. Blood 2010, 116, 2224–2228. [Google Scholar] [CrossRef] [PubMed]
- Löwenberg, B.; Ossenkoppele, G.J.; van Putten, W.; Schouten, H.C.; Graux, C.; Ferrant, A.; Sonneveld, P.; Maertens, J.; Jongen-Lavrencic, M.; von Lilienfeld-Toal, M.; et al. High-Dose Daunorubicin in Older Patients with Acute Myeloid Leukemia. N. Engl. J. Med. 2009, 361, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Breems, D.A.; Van Putten, W.L.J.; De Greef, G.E.; Van Zelderen-Bhola, S.L.; Gerssen-Schoorl, K.B.J.; Mellink, C.H.M.; Nieuwint, A.; Jotterand, M.; Hagemeijer, A.; Beverloo, H.B.; et al. Monosomal Karyotype in Acute Myeloid Leukemia: A Better Indicator of Poor Prognosis than a Complex Karyotype. J. Clin. Oncol. 2008, 26, 4791–4797. [Google Scholar] [CrossRef]
- Weinberg, O.K.; Ohgami, R.S.; Ma, L.; Seo, K.; Ren, L.; Gotlib, J.R.; Seetharam, M.; Cherry, A.; Arber, D.A. Acute Myeloid Leukemia with Monosomal Karyotype: Morphologic, Immunophenotypic, and Molecular Findings. Am. J. Clin. Pathol. 2014, 142, 190–195. [Google Scholar] [CrossRef]
- Voutiadou, G.; Papaioannou, G.; Gaitatzi, M.; Lalayanni, C.; Syrigou, A.; Vadikoliou, C.; Saloum, R.; Anagnostopoulos, A.; Athanasiadou, A. Monosomal Karyotype in Acute Myeloid Leukemia Defines a Distinct Subgroup within the Adverse Cytogenetic Risk Category. Cancer Genet. 2013, 206, 32–36. [Google Scholar] [CrossRef]
- Kayser, S.; Zucknick, M.; Döhner, K.; Krauter, J.; Köhne, C.H.; Horst, H.A.; Held, G.; Von Lilienfeld-Toal, M.; Wilhelm, S.; Rummel, M.; et al. Monosomal Karyotype in Adult Acute Myeloid Leukemia: Prognostic Impact and Outcome after Different Treatment Strategies. Blood 2012, 119, 551–558. [Google Scholar] [CrossRef]
- Brands-Nijenhuis, A.V.M.; Labopin, M.; Schouten, H.C.; Volin, L.; Socié, G.; Cornelissen, J.J.; Huynh, A.; Ljungman, P.; Malard, F.; Esteve, J.; et al. Monosomal Karyotype as an Adverse Prognostic Factor in Patients with Acute Myeloid Leukemia Treated with Allogeneic Hematopoietic Stem-Cell Transplantation in First Complete Remission: A Retrospective Survey on Behalf of the Alwp of the Ebmt. Haematologica 2016, 101, 248–255. [Google Scholar] [CrossRef]
- Döhner, H.; Wei, A.H.; Appelbaum, F.R.; Craddock, C.; DiNardo, C.D.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Godley, L.A.; Hasserjian, R.P.; et al. Diagnosis and Management of AML in Adults: 2022 Recommendations from an International Expert Panel on Behalf of the ELN. Blood 2022, 140, 1345–1377. [Google Scholar] [CrossRef]
- Yang, X.F.; Sun, A.N.; Cai, C.S.; Tian, X.P.; Qian, J.; Chen, S.N.; Wu, D.P. Monosomal Karyotypes among 1147 Chinese Patients with Acute Myeloid Leukemia: Prevalence, Features and Prognostic Impact. Asian Pac. J. Cancer Prev. 2012, 13, 5421–5426. [Google Scholar] [CrossRef]
- Gaillard, J.B.; Chiesa, J.; Reboul, D.; Arnaud, A.; Brun, S.; Donadio, D.; Exbrayat, C.; Wickenhauser, S.; Bourquard, P.; Jourdan, E.; et al. Monosomal Karyotype Routinely Defines a Poor Prognosis Subgroup in Acute Myeloid Leukemia and Is Frequently Associated with TP53 Deletion. Leuk. Lymphoma 2012, 53, 336–337. [Google Scholar] [CrossRef] [PubMed]
- Leung, G.M.K.; Zhang, C.; Ng, N.K.L.; Yang, N.; Lam, S.S.Y.; Au, C.H.; Chan, T.L.; Ma, E.S.K.; Tsui, S.P.; Ip, H.W.; et al. Distinct Mutation Spectrum, Clinical Outcome and Therapeutic Responses of Typical Complex/Monosomy Karyotype Acute Myeloid Leukemia Carrying TP53 Mutations. Am. J. Hematol. 2019, 94, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Rücker, F.G.; Schlenk, R.F.; Bullinger, L.; Kayser, S.; Teleanu, V.; Kett, H.; Habdank, M.; Kugler, C.M.; Holzmann, K.; Gaidzik, V.I.; et al. TP53 Alterations in Acute Myeloid Leukemia with Complex Karyotype Correlate with Specific Copy Number Alterations, Monosomal Karyotype, and Dismal Outcome. Blood 2012, 119, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Tyner, J.W.; Tognon, C.E.; Bottomly, D.; Wilmot, B.; Kurtz, S.E.; Savage, S.L.; Long, N.; Schultz, A.R.; Traer, E.; Abel, M.; et al. Functional Genomic Landscape of Acute Myeloid Leukaemia. Nature 2018, 562, 526–531. [Google Scholar] [CrossRef]
- Nazha, A.; Kantarjian, H.M.; Bhatt, V.R.; Nogueras-Gonzalez, G.; Cortes, J.E.; Kadia, T.; Garcia-Manero, G.; Abruzzo, L.; Daver, N.; Pemmaraju, N.; et al. Prognostic Implications of Chromosome 17 Abnormalities in the Context of Monosomal Karyotype in Patients with Acute Myeloid Leukemia and Complex Cytogenetics. Clin. Lymphoma Myeloma Leuk. 2014, 14, 163–171. [Google Scholar] [CrossRef]
- Bowen, D.; Groves, M.J.; Burnett, A.K.; Patel, Y.; Allen, C.; Green, C.; Gale, R.E.; Hills, R.; Linch, D.C. TP53 Gene Mutation Is Frequent in Patients with Acute Myeloid Leukemia and Complex Karyotype, and Is Associated with Very Poor Prognosis. Leukemia 2009, 23, 203–206. [Google Scholar] [CrossRef]
- Qin, G.; Han, X. The Prognostic Value of TP53 Mutations in Adult Acute Myeloid Leukemia: A Meta-Analysis. Transfus. Med. Hemother. 2023, 50, 234–244. [Google Scholar] [CrossRef]
- Shin, D.Y. TP53 Mutation in Acute Myeloid Leukemia: An Old Foe Revisited. Cancers 2023, 15, 4816. [Google Scholar] [CrossRef]
- Kadia, T.M.; Jain, P.; Ravandi, F.; Garcia-Manero, G.; Andreef, M.; Takahashi, K.; Borthakur, G.; Jabbour, E.; Konopleva, M.; Daver, N.G.; et al. TP53 Mutations in Newly Diagnosed Acute Myeloid Leukemia: Clinicomolecular Characteristics, Response to Therapy, and Outcomes. Cancer 2016, 122, 3484–3491. [Google Scholar] [CrossRef]
- Bories, P.; Prade, N.; Lagarde, S.; Cabarrou, B.; Largeaud, L.; Plenecassagnes, J.; Luquet, I.; de Mas, V.; Filleron, T.; Cassou, M.; et al. Impact of TP53 Mutations in Acute Myeloid Leukemia Patients Treated with Azacitidine. PLoS ONE 2020, 15, e0238795. [Google Scholar] [CrossRef]
- Shahzad, M.; Amin, M.K.; Arber, D.A.; Kharfan-dabaja, M.A. What Have We Learned about TP53- Mutated Acute Myeloid Leukemia? Blood Cancer J. 2024, 14, 202. [Google Scholar] [CrossRef]
- Zhao, D.; Zarif, M.; Zhou, Q.; Capo-Chichi, J.M.; Schuh, A.; Minden, M.D.; Atenafu, E.G.; Kumar, R.; Chang, H. TP53 Mutations in AML Patients Are Associated with Dismal Clinical Outcome Irrespective of Frontline Induction Regimen and Allogeneic Hematopoietic Cell Transplantation. Cancers 2023, 15, 3210. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Eladl, E.; Zarif, M.; Capo-Chichi, J.M.; Schuh, A.; Atenafu, E.; Minden, M.; Chang, H. Molecular Characterization of AML-MRC Reveals TP53 Mutation as an Adverse Prognostic Factor Irrespective of MRC-Defining Criteria, TP53 Allelic State, or TP53 Variant Allele Frequency. Cancer Med. 2023, 12, 6511–6522. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Rojek, A.E.; Symes, E.; Nawas, M.T.; Patel, A.A.; Patel, J.L.; Sojitra, P.; Aqil, B.; Sukhanova, M.; McNerney, M.E.; et al. Real World Predictors of Response and 24-Month Survival in High-Grade TP53-Mutated Myeloid Neoplasms. Blood Cancer J. 2024, 14, 99. [Google Scholar] [CrossRef] [PubMed]
- Symes, E.O.; Wang, P.; Sojitra, P.; Menon, M.P.; Patel, A.A.; Hasan, F.; Ghosh, S.; Roloff, G.W.; Zhou, Q.; Findley, A.; et al. Somatic Co-Alteration Signatures Are Prognostic in High-Grade TP53-Mutated Myeloid Neoplasms. Br. J. Haematol. 2024, 206, 1103–1108. [Google Scholar] [CrossRef]
- Tazi, Y.; Arango-Ossa, J.E.; Zhou, Y.; Bernard, E.; Thomas, I.; Gilkes, A.; Freeman, S.; Pradat, Y.; Johnson, S.J.; Hills, R.; et al. Unified Classification and Risk-Stratification in Acute Myeloid Leukemia. Nat. Commun. 2022, 13, 4622. [Google Scholar] [CrossRef]
- Feng, R.; Liu, H.; Chang, N.; Fan, Y.; Li, J.; Zhang, Y.; Chen, W.; Wang, H.; Tian, Y.; Pei, L.; et al. Monosomal Karyotype among Adult Acute Myeloid Leukemia: Clinical Characteristic and Prognostic Analysis. Zhonghua Xue Ye Xue Za Zhi 2014, 35, 393–396. [Google Scholar]
- Weinberg, O.K.; Siddon, A.; Madanat, Y.F.; Gagan, J.; Arber, D.A.; Cin, P.D.; Narayanan, D.; Ouseph, M.M.; Kurzer, J.H.; Hasserjian, R.P. TP53 Mutation Defines a Unique Subgroup within Complex Karyotype de Novo and Therapy-Related MDS/AML. Blood Adv. 2022, 6, 2847–2853. [Google Scholar] [CrossRef]
- Haferlach, C.; Dicker, F.; Herholz, H.; Schnittger, S.; Kern, W.; Haferlach, T. Mutations of the TP53 Gene in Acute Myeloid Leukemia Are Strongly Associated with a Complex Aberrant Karyotype. Leukemia 2008, 22, 1539–1541. [Google Scholar] [CrossRef]
- Bottomly, D.; Long, N.; Schultz, A.R.; Kurtz, S.E.; Tognon, C.E.; Johnson, K.; Abel, M.; Agarwal, A.; Avaylon, S.; Benton, E.; et al. Integrative Analysis of Drug Response and Clinical Outcome in Acute Myeloid Leukemia. Cancer Cell 2022, 40, 850–864.e9. [Google Scholar] [CrossRef]
- Britt, A.; Mohyuddin, G.R.; McClune, B.; Singh, A.; Lin, T.; Ganguly, S.; Abhyankar, S.; Shune, L.; McGuirk, J.; Skikne, B.; et al. Acute Myeloid Leukemia or Myelodysplastic Syndrome with Chromosome 17 Abnormalities and Long-Term Outcomes with or without Hematopoietic Stem Cell Transplantation. Leuk. Res. 2020, 95, 106402. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Jung, J.; Park, S.; Cho, B.S.; Kim, H.J.; Kim, Y.; Lee, J.M.; Kim, H.S.; Ahn, A.; Kim, M.; et al. Genetic Characteristics According to Subgroup of Acute Myeloid Leukemia with Myelodysplasia-Related Changes. J. Clin. Med. 2022, 11, 2378. [Google Scholar] [CrossRef]
- Kadia, T.; Cortes, J.E.; Ravandi, F.; Garcia-Manero, G.; Andreeff, M.; Takahashi, K.; Borthakur, G.; Jabbour, E.J.; Bhalla, K.N.; Konopleva, M.; et al. Clinical and Molecular Characterization of P53-Mutated Acute Myeloid Leukemia. Blood 2015, 126, 564. [Google Scholar] [CrossRef]
- Yanada, M.; Yamamoto, Y.; Iba, S.; Okamoto, A.; Inaguma, Y.; Tokuda, M.; Morishima, S.; Kanie, T.; Mizuta, S.; Akatsuka, Y.; et al. TP53 Mutations in Older Adults with Acute Myeloid Leukemia. Int. J. Hematol. 2016, 103, 429–435. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Simons, A.; Schmid, M. ISCN 2016: An International System for Human Cytogenomic Nomenclature (2016); Karger: Basel, Switzerland, 2016; ISBN 978-3-318-06861-0. [Google Scholar]
- Alduaij, W.; McNamara, C.J.; Schuh, A.; Arruda, A.; Sukhai, M.; Kanwar, N.; Thomas, M.; Spiegel, J.; Kennedy, J.A.; Stockley, T.; et al. Clinical Utility of Next-Generation Sequencing in the Management of Myeloproliferative Neoplasms: A Single-Center Experience. Hemasphere 2018, 2, e44. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Zhao, D.; Zarif, M.; Davidson, M.B.; Minden, M.D.; Tierens, A.; Yeung, Y.W.T.; Wei, C.; Chang, H. A Real-World Analysis of Clinical Outcomes in AML with Myelodysplasia-Related Changes: A Comparison of ICC and WHO-HAEM5 Criteria. Blood Adv. 2024, 8, 1760–1771. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
| Variable | AML (n = 664) | MK − AML (n = 508) | MK + AML (n = 156) | MK − AML vs. MK + AML |
|---|---|---|---|---|
| Male:Female Sex | 377:287 | 295:213 | 82:74 | 0.231 |
| Age (y), median [range] | 69 [18–95] | 69 [18–95] | 71 [19–94] | 0.033 a |
| WBC Count × 109/L, median [range] | 5.9 [0.1–328.7] | 8.0 [0.3–328.7] | 3.4 [0.1–162.3] | <0.001 a |
| Hemoglobin, g/dL, median [range] | 8.6 [3.7–19.7] | 8.6 [3.7–19.7] | 8.4 [5.7–14.2] | 0.598 a |
| Platelet × 109/L, median [range] | 55 [9.0–2726] | 64 [10–2726] | 43 [9–782] | <0.001 a |
| BM Blast percentage, n (%) | 45 (13–97) | 48 (13–97) | 38.5 (20–95) | 0.002 a |
| Complex Karyotype, n (%) | 192 (28.9%) | 54 (10.6%) | 138 (88.5%) | <0.001 b |
| Monosomy 7 | 76 (48.7%) | 18 (3.5%) | 58 (37.2%) | <0.001 b |
| Monosomy 17 | 64 (9.6%) | 0 | 64 (41%) | <0.001 b |
| Monosomy 13 | 28 (4.2%) | 0 | 28 (17.9%) | <0.001 b |
| Monosomy 5 | 21 (3.2%) | 0 | 21 (13.5%) | <0.001 b |
| Transplant, n (%) | 141 (21.2%) | 118 (23.2%) | 23 (14.7%) | 0.023 b |
| Variable | MK + AML TP53 + (n = 108) | MK + AML TP53 − (n = 48) | MK + AML TP53 + vs. MK + AML TP53 − |
|---|---|---|---|
| Male:Female Sex | 53:55 | 29:19 | 0.190 a |
| Age (y), median [range] | 73 [36–94] | 65 [19–91] | 0.008 a |
| WBC Count × 109/L, median [range] | 3.1 [0.1–76.9] | 3.8 [0.1–162.3] | 0.184 a |
| Hemoglobin, g/dL, median [range] | 8.3 [5.7–12.3] | 8.9 [6.4–14.2] | 0.018 a |
| Platelet × 109/L, median [range] | 42 [9–782] | 44 [9–408] | 0.586 a |
| BM Blast percentage, n (%) | 35 (20–91) | 46 (20−95) | 0.070 a |
| Complex Karyotype, n (%) | 107 (99.1%) | 31 (64.6%) | <0.001 b |
| Transplant, n (%) | 10 (9.3%) | 13(27.1%) | 0.004 b |
| Variable | MK AML (n = 156) | Median OS (Months) | HR | 95% CI | p Value | |
|---|---|---|---|---|---|---|
| CK | Present | 138 (88.5%) | 4.9 | 0.64 | 0.39–1.05 | 0.252 |
| Absent | 18 (11.5%) | 9.2 | ||||
| Monosomy 17 | Present | 64 (41.0%) | 3.3 | 1.77 | 1.22–2.57 | 0.009 |
| Absent | 92 (59.0%) | 7.9 | ||||
| Monosomy 7 | Present | 58 (37.2%) | 4.9 | 1.30 | 0.91–1.88 | 0.382 |
| Absent | 98 (62.8%) | 6.6 | ||||
| Monosomy 13 | Present | 29 (18.6%) | 3.3 | 1.20 | 0.73–1.95 | 0.439 |
| Absent | 127 (81.4%) | 5.9 | ||||
| Monosomy 5 | Present | 21 (13.5%) | 3.5 | 1.55 | 0.90−2.69 | 0.183 |
| Absent | 135 (86.5%) | 6.0 | ||||
| ICC MR Mutations | Present | 37 (23.7%) | 7.2 | 1.36 | 0.93–1.99 | 0.252 |
| Absent | 119 (76.3%) | 4.7 | ||||
| DNMT3A | Present | 29 (18.6%) | 3.5 | 0.76 | 0.47–1.22 | 0.264 |
| Absent | 127 (81.4%) | 5.9 | ||||
| TET2 | Present | 20 (12.8%) | 2.3 | 0.72 | 0.41–1.27 | 0.264 |
| Absent | 136 (87.2%) | 5.9 | ||||
| IDH1 | Present | 11 (7.1%) | 8.7 | 1.88 | 1.11−3.18 | 0.183 |
| Absent | 145 (92.9%) | 5.2 | ||||
| Variable | Hazard Ratio | 95% CI | p Value | |
|---|---|---|---|---|
| Age [Years] | 1.01 | 0.66–1.54 | 0.965 | |
| WBC Count | 1.00 | 0.99–1.01 | 0.949 | |
| TP53 Mut | 1.61 | 1.06–2.46 | 0.026 | |
| Induction Chemotherapy | Low-Intensity | 0.25 | 0.16–0.41 | <0.001 |
| Intensive | 0.32 | 0.2–0.52 | <0.001 | |
| Allo-HCT | 0.25 | 0.14–0.47 | <0.001 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wangulu, C.; Bahrami Hezaveh, E.; Zarif, M.; Zhou, Q.; Lo, W.; Wei, C.; Sibai, H.; Chang, H. Genomic Profile and Clinical Outcomes in Acute Myeloid Leukemia with Monosomal Karyotype. Int. J. Mol. Sci. 2025, 26, 5845. https://doi.org/10.3390/ijms26125845
Wangulu C, Bahrami Hezaveh E, Zarif M, Zhou Q, Lo W, Wei C, Sibai H, Chang H. Genomic Profile and Clinical Outcomes in Acute Myeloid Leukemia with Monosomal Karyotype. International Journal of Molecular Sciences. 2025; 26(12):5845. https://doi.org/10.3390/ijms26125845
Chicago/Turabian StyleWangulu, Collins, Ehsan Bahrami Hezaveh, Mojgan Zarif, Qianghua Zhou, Winnie Lo, Cuihong Wei, Hassan Sibai, and Hong Chang. 2025. "Genomic Profile and Clinical Outcomes in Acute Myeloid Leukemia with Monosomal Karyotype" International Journal of Molecular Sciences 26, no. 12: 5845. https://doi.org/10.3390/ijms26125845
APA StyleWangulu, C., Bahrami Hezaveh, E., Zarif, M., Zhou, Q., Lo, W., Wei, C., Sibai, H., & Chang, H. (2025). Genomic Profile and Clinical Outcomes in Acute Myeloid Leukemia with Monosomal Karyotype. International Journal of Molecular Sciences, 26(12), 5845. https://doi.org/10.3390/ijms26125845