
Molecular Determinants of the Human Retinal Pigment Epithelium Cell Fate and Potential Pharmacogenomic Targets for Precision Medicine


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
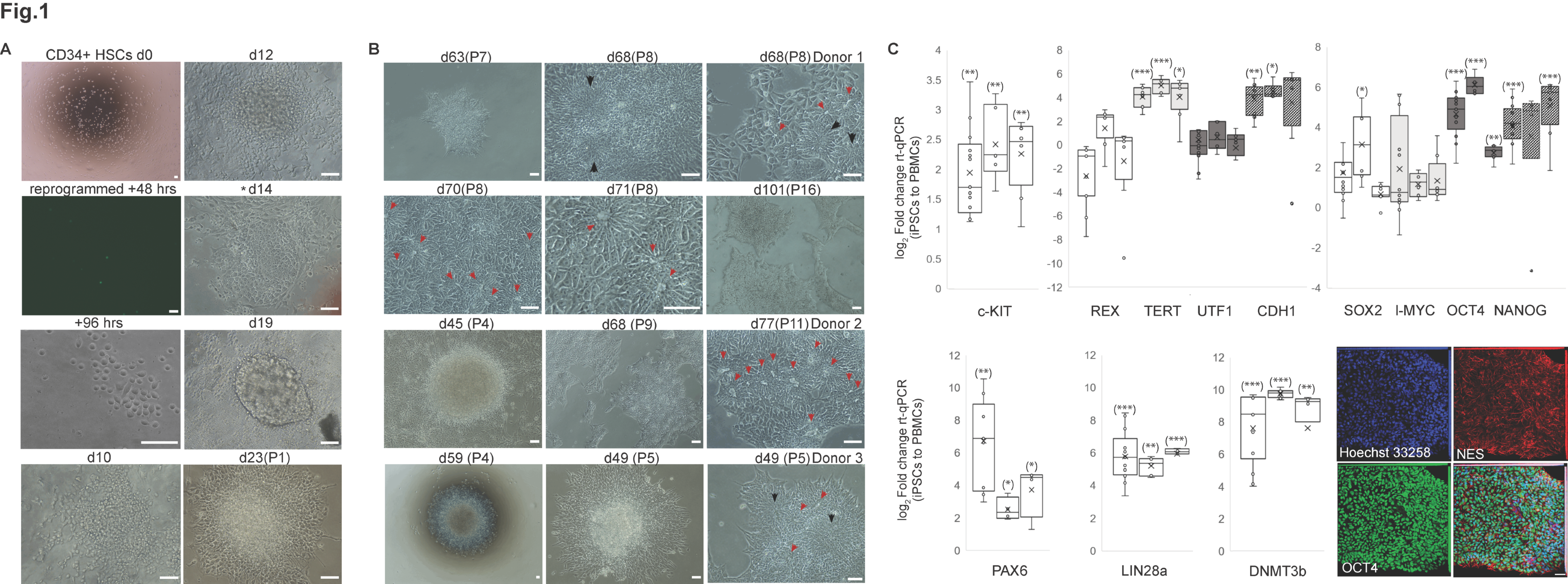
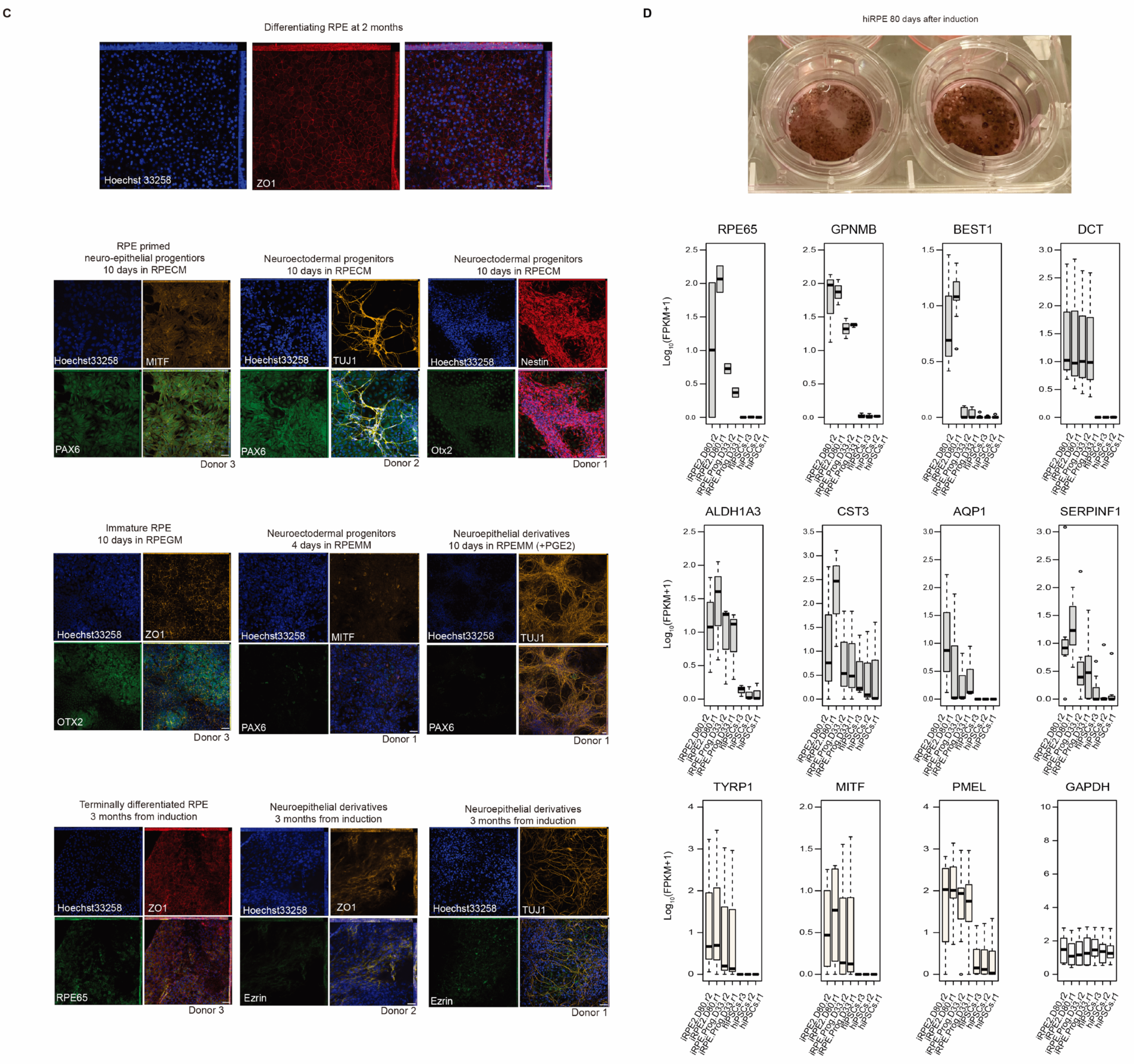
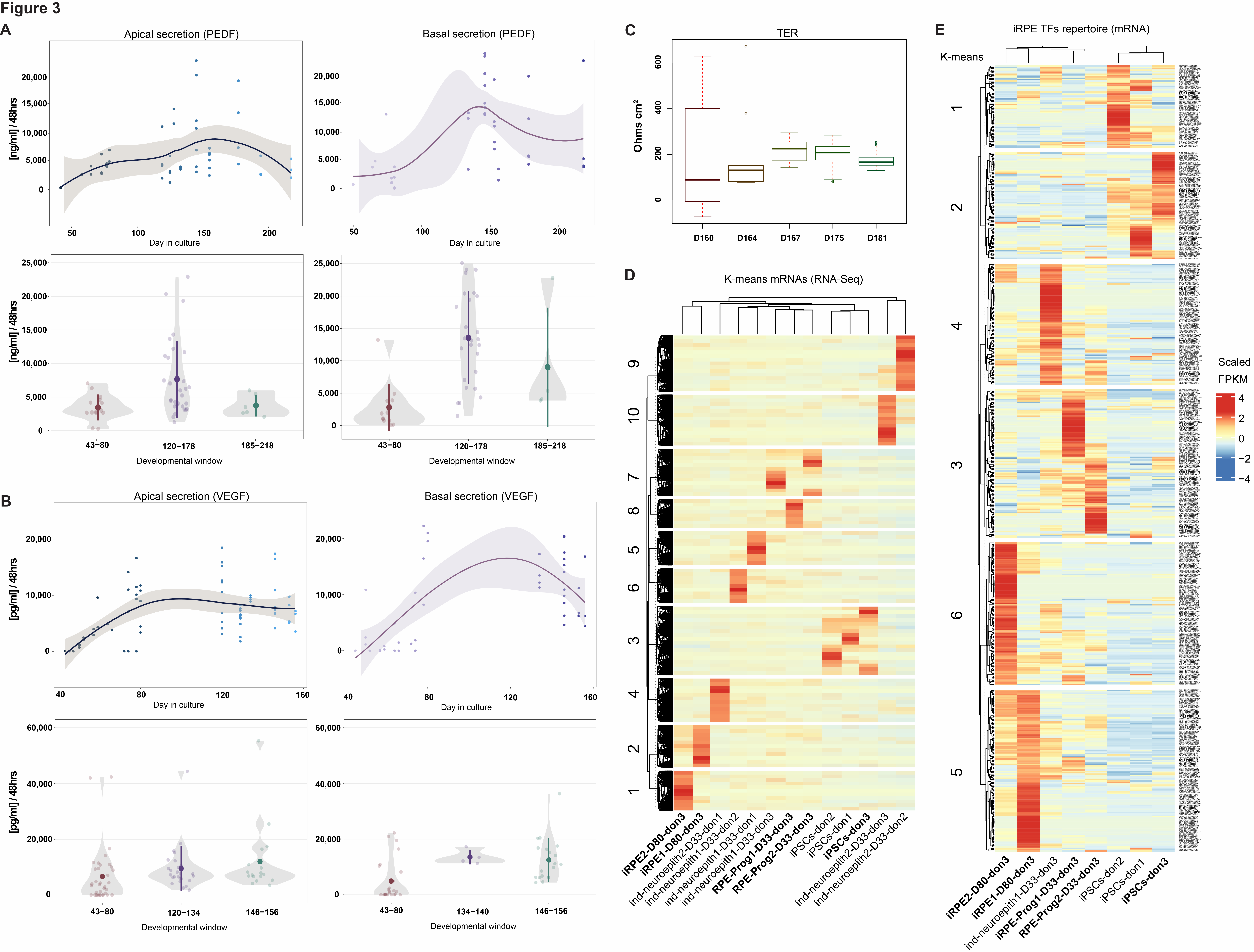
2.1. Bipotent, Neuroepithelial Progenitors Give Rise to Presumptive RPE Progenitors, Following hiPSCs Mesenchymal-Epithelial Transition to a Primed State, and Neurons
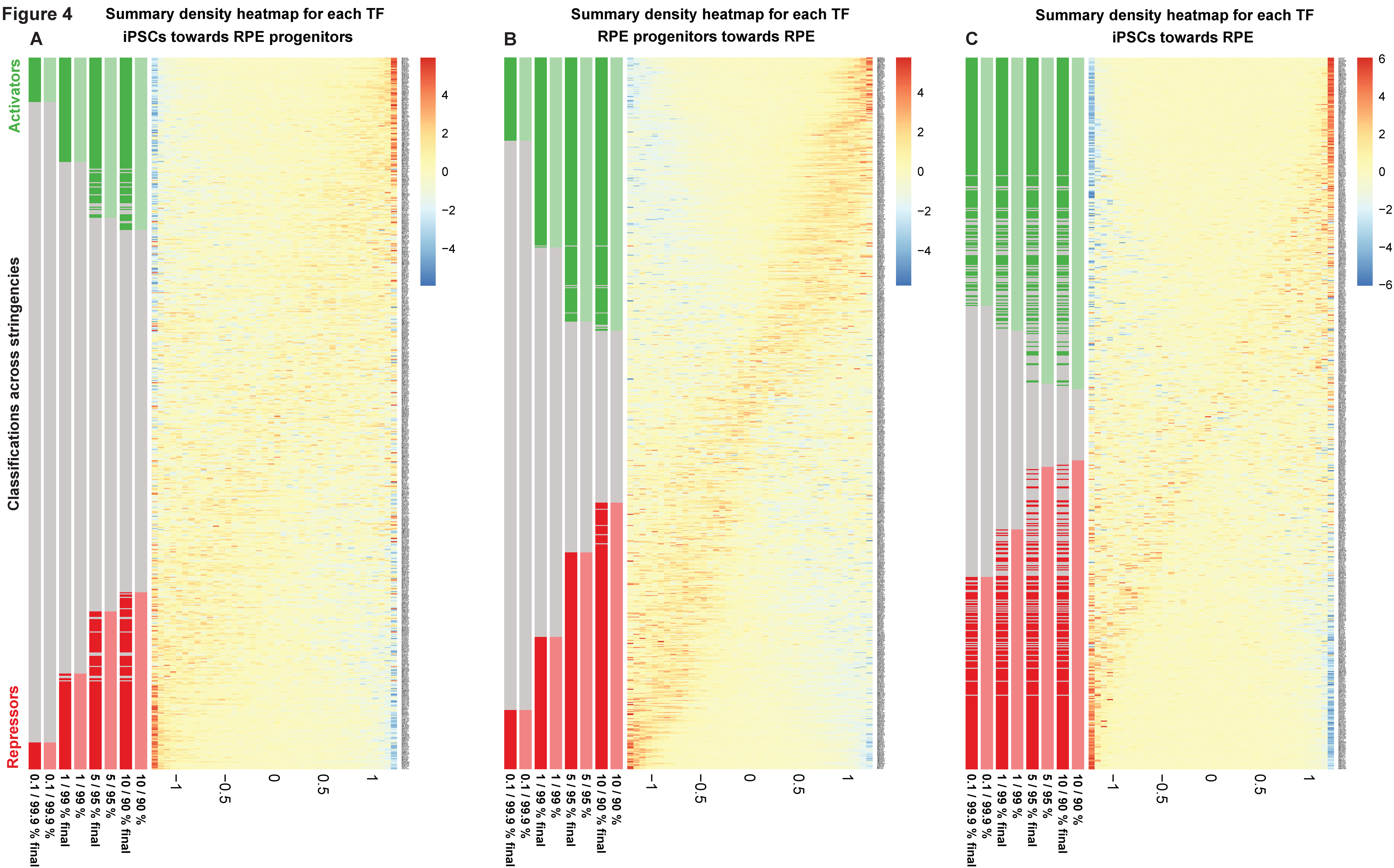
2.2. Transcription Factors Footprinting for the Agnostic Identification of Human RPE Master Regulators
2.3. Predicted Pioneer Factors in the Transition from Human-Induced Pluripotent Stem Cells Towards a Retinal Pigment Epithelium Cell Fate
2.4. TFs Regulatory Targets and New Avenues for Pharmacological Intervention on AMD
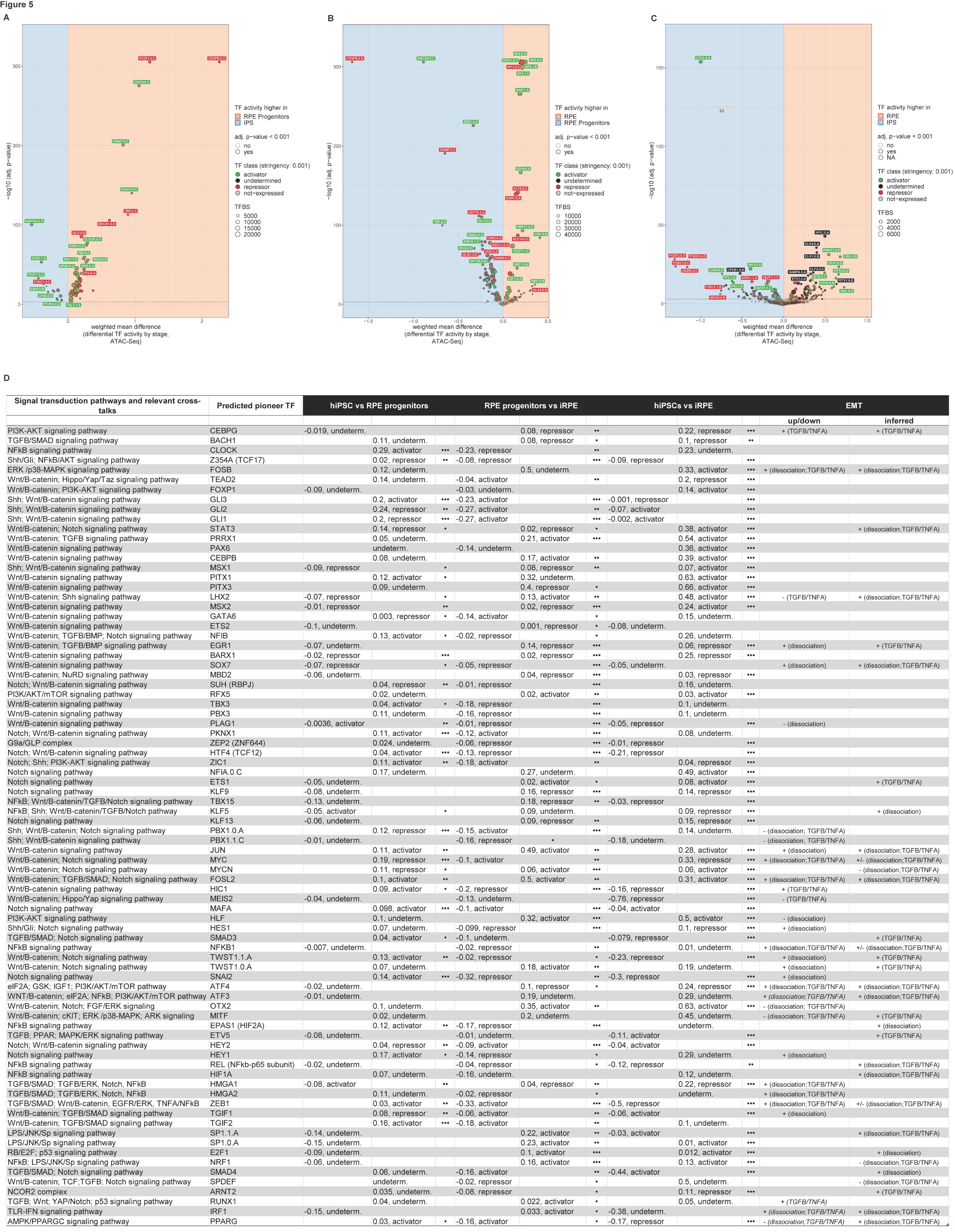
2.5. Predicted Pioneer Factors and Their Regulatory Targets Are Associated with Latent Developmental Pathways or Alternate Neuro-Mesodermal Cell Fates and the Epithelial-Mesenchymal Transition
3. Discussion
3.1. Dual Regulators and Preventive Developmental Gatekeepers Suppress Latent, Alternate Neuro-Mesodermal Cell Fates in the Consolidation of the Human RPE Cell Fate Identity
3.2. Pharmacogenomics to Restore the RPE and Pleiotropic Cis-Regulatory Elements in Complex Diseases
4. Materials and Methods
4.1. Human iPSCs Derivation, Phenotyping, and Reprogramming to Retinal Pigment Epithelium
4.2. RPE Phenotyping
4.3. Next-Generation Sequencing
4.4. De Novo and Known Motifs Enrichment
4.5. Gene Ontology Enrichment on Open Chromatin
Limitations of the Study
4.6. Detection of Differential Transcription Factor Activity in Human iPSC-Derived RPE Cell Fate Determination and RPE Differentiation
Limitations of the Study
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef] [PubMed]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N. Engl. J. Med. 2008, 358, 2606–2617, Erratum in N. Engl. J. Med. 2008, 359, 1736. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.C.; Grassmann, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, J.; Zibetti, C.; Shang, P.; Sripathi, S.R.; Zhang, P.; Cano, M.; Hoang, T.; Xia, S.; Ji, H.; Merbs, S.L.; et al. ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration. Nat. Commun. 2018, 9, 1364. [Google Scholar] [CrossRef]
- Porter, L.F.; Saptarshi, N.; Fang, Y.; Rathi, S.; den Hollander, A.I.; e Jong, E.K.; Clark, S.J.; Bishop, P.N.; Olsen, T.W.; Liloglou, T.; et al. Whole-genome methylation profiling of the retinal pigment epithelium of individuals with age-related macular degeneration reveals differential methylation of the SKI, GTF2H4, and TNXB genes. Clin. Epigenetics 2019, 11, 6. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, C.; Qin, B.; Liu, G.; Sun, X.; Shi, H.; Ding, S.; Liu, Y.; Zhu, M.; Chen, X.; Zhao, C. MicroRNA-184 promotes differentiation of the retinal pigment epithelium by targeting the AKT2/mTOR signaling pathway. Oncotarget 2016, 7, 52340–52353. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dalvi, S.; Galloway, C.A.; Winschel, L.; Hashim, A.; Soto, C.; Tang, C.; MacDonald, L.A.; Singh, R. Environmental stress impairs photoreceptor outer segment (POS) phagocytosis and degradation and induces autofluorescent material accumulation in hiPSC-RPE cells. Cell Death Discov. 2019, 5, 96. [Google Scholar] [CrossRef]
- Zhang, M.; Jiang, N.; Chu, Y.; Postnikova, O.; Varghese, R.; Horvath, A.; Cheema, A.K.; Golestaneh, N. Dysregulated metabolic pathways in age-related macular degeneration. Sci. Rep. 2020, 10, 2464. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- La Cunza, N.; Tan, L.X.; Thamban, T.; Germer, C.J.; Rathnasamy, G.; Toops, K.A.; Lakkaraju, A. Mitochondria-dependent phase separation of disease-relevant proteins drives pathological features of age-related macular degeneration. JCI Insight 2021, 6, e142254. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sharma, R.; Khristov, V.; Rising, A.; Jha, B.S.; Dejene, R.; Hotaling, N.; Li, Y.; Stoddard, J.; Stankewicz, C.; Wan, Q.; et al. Clinical-grade stem cell-derived retinal pigment epithelium patch rescues retinal degeneration in rodents and pigs. Sci. Transl. Med. 2019, 11, eaat5580, Erratum in Sci. Transl. Med. 2019, 11, eaaw7624. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tian, L.; Al-Nusaif, M.; Chen, X.; Li, S.; Le, W. Roles of Transcription Factors in the Development and Reprogramming of the Dopaminergic Neurons. Int. J. Mol. Sci. 2022, 23, 845. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zibetti, C. Deciphering the Retinal Epigenome during Development, Disease and Reprogramming: Advancements, Challenges and Perspectives. Cells 2022, 11, 806. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cohen-Gulkar, M.; David, A.; Messika-Gold, N.; Eshel, M.; Ovadia, S.; Zuk-Bar, N.; Idelson, M.; Cohen-Tayar, Y.; Reubinoff, B.; Ziv, T.; et al. The LHX2-OTX2 transcriptional regulatory module controls retinal pigmented epithelium differentiation and underlies genetic risk for age-related macular degeneration. PLoS Biol. 2023, 21, e3001924. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- D’Alessio, A.C.; Fan, Z.P.; Wert, K.J.; Baranov, P.; Cohen, M.A.; Saini, J.S.; Cohick, E.; Charniga, C.; Dadon, D.; Hannett, N.M.; et al. A Systematic Approach to Identify Candidate Transcription Factors that Control Cell Identity. Stem Cell Rep. 2015, 5, 763–775. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Berest, I.; Arnold, C.; Reyes-Palomares, A.; Palla, G.; Rasmussen, K.D.; Giles, H.; Bruch, P.M.; Huber, W.; Dietrich, S.; Helin, K.; et al. Quantification of Differential Transcription Factor Activity and Multiomics-Based Classification into Activators and Repressors: DiffTF. Cell Rep. 2019, 29, 3147–3159.E12. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.J.; Yun, S.; Clark, A.M.; Monuki, E.S.; Murtaugh, L.C.; Levine, E.M. Lhx2 balances progenitor maintenance with neurogenic output and promotes competence state progression in the developing retina. J. Neurosci. 2013, 33, 12197–12207. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- de Melo, J.; Zibetti, C.; Clark, B.S.; Hwang, W.; Miranda-Angulo, A.L.; Qian, J.; Blackshaw, S. Lhx2 Is an Essential Factor for Retinal Gliogenesis and Notch Signaling. J. Neurosci. 2016, 36, 2391–2405. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zibetti, C.; Liu, S.; Wan, J.; Qian, J.; Blackshaw, S. Epigenomic profiling of retinal progenitors reveals LHX2 is required for developmental regulation of open chromatin. Commun. Biol. 2019, 2, 142. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hu, H.; Li, B.; Zhou, C.; Ying, X.; Chen, M.; Huang, T.; Chen, Y.; Ji, H.; Pan, R.; Wang, T.; et al. Diagnostic value of WIF1 methylation for colorectal cancer: A meta-analysis. Oncotarget 2018, 9, 5378–5386. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, X.; Shan, W.; Li, T.; Gao, X.; Kong, F.; You, H.; Kong, D.; Qiao, S.; Tang, R. Cellular retinol binding protein-1 inhibits cancer stemness via upregulating WIF1 to suppress Wnt/β-catenin pathway in hepatocellular carcinoma. BMC Cancer 2021, 21, 1224. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zeitz, C.; Robson, A.G.; Audo, I. Congenital stationary night blindness: An analysis and update of genotype–phenotype correlations and pathogenic mechanisms. Prog. Retin. Eye Res. 2015, 45, 58–110. [Google Scholar] [CrossRef] [PubMed]
- Ratnapriya, R.; Sosina, O.A.; Starostik, M.R.; Kwicklis, M.; Kapphahn, R.J.; Fritsche, L.G.; Walton, A.; Arvanitis, M.; Gieser, L.; Pietraszkiewicz, A.; et al. Retinal transcriptome and eQTL analyses identify genes associated with age-related macular degeneration. Nat. Genet. 2019, 51, 606–610, Erratum in Nat. Genet. 2019, 51, 1067. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Advani, J.; Mehta, P.A.; Hamel, A.R.; Mehrotra, S.; Kiel, C.; Strunz, T.; Corso-Díaz, X.; Kwicklis, M.; van Asten, F.; Ratnapriya, R.; et al. QTL mapping of human retina DNA methylation identifies 87 gene-epigenome interactions in age-related macular degeneration. Nat. Commun. 2024, 15, 1972. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- DeVera, C.; Dixon, J.; Chrenek, M.A.; Baba, K.; Le, Y.Z.; Iuvone, P.M.; Tosini, G. The Circadian Clock in the Retinal Pigment Epithelium Controls the Diurnal Rhythm of Phagocytic Activity. Int. J. Mol. Sci. 2022, 23, 5302. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blasiak, J.; Reiter, R.J.; Kaarniranta, K. Melatonin in Retinal Physiology and Pathology: The Case of Age-Related Macular Degeneration. Oxidative Med. Cell. Longev. 2016, 2016, 6819736. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mihaylov, S.R.; Castelli, L.M.; Lin, Y.-H.; Gül, A.; Soni, N.; Hastings, C.; Flynn, H.R.; Păun, O.; Dickman, M.J.; Snijders, A.P.; et al. The master energy homeostasis regulator PGC-1α exhibits an mRNA nuclear export function. Nat. Commun. 2023, 14, 5496. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ghosh, S.; Sharma, R.; Bammidi, S.; Koontz, V.; Nemani, M.; Yazdankhah, M.; Kedziora, K.M.; Stolz, D.B.; Wallace, C.T.; Yu-Wei, C.; et al. The AKT2/SIRT5/TFEB pathway as a potential therapeutic target in non-neovascular AMD. Nat. Commun. 2024, 15, 6150. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pastore, N.; Brady, O.A.; Diab, H.I.; Martina, J.A.; Sun, L.; Huynh, T.; Lim, J.-A.; Zare, H.; Raben, N.; Ballabio, A.; et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 2016, 12, 1240–1258. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Felszeghy, S.; Viiri, J.; Paterno, J.J.; Hyttinen, J.M.; Koskela, A.; Chen, M.; Leinonen, H.; Tanila, H.; Kivinen, N.; Koistinen, A.; et al. Loss of NRF-2 and PGC-1α genes leads to retinal pigment epithelium damage resembling dry age-related macular degeneration. Redox Biol. 2019, 20, 1–12. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ishikawa, K.; Kannan, R.; Hinton, D.R. Molecular mechanisms of subretinal fibrosis in age-related macular degeneration. Exp. Eye Res. 2016, 142, 19–25. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hirasawa, M.; Noda, K.; Noda, S.; Suzuki, M.; Ozawa, Y.; Shinoda, K.; Inoue, M.; Ogawa, Y.; Tsubota, K.; Ishida, S. Transcriptional factors associated with epithelial-mesenchymal transition in choroidal neovascularization. Mol. Vis. 2011, 17, 1222–1230. [Google Scholar] [PubMed] [PubMed Central]
- Hansman, D.S.; Ma, Y.; Thomas, D.; Smith, J.R.; Casson, R.J.; Peet, D.J. Metabolic reprogramming of the retinal pigment epithelium by cytokines associated with age-related macular degeneration. Biosci. Rep. 2024, 44, BSR20231904. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Thiery, J.P. Epithelial-mesenchymal transitions: Insights from development. Development 2012, 139, 3471–3486. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Geathers, J.S.; Grillo, S.L.; Weber, S.R.; Wang, W.; Zhao, Y.; Sundstrom, J.M. Role of Epithelial-Mesenchymal Transition in Retinal Pigment Epithelium Dysfunction. Front. Cell Dev. Biol. 2020, 8, 501. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Santibañez, J.F. JNK mediates TGF-beta1-induced epithelial mesenchymal transdifferentiation of mouse transformed keratinocytes. FEBS Lett. 2006, 580, 5385–5391. [Google Scholar] [CrossRef] [PubMed]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Radeke, M.J.; Radeke, C.M.; Shih, Y.-H.; Hu, J.; Bok, D.; Johnson, L.V.; Coffey, P.J. Restoration of mesenchymal retinal pigmented epithelial cells by TGFβ pathway inhibitors: Implications for age-related macular degeneration. Genome Med. 2015, 7, 58. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Boles, N.C.; Fernandes, M.; Swigut, T.; Srinivasan, R.; Schiff, L.; Rada-Iglesias, A.; Wang, Q.; Saini, J.S.; Kiehl, T.; Stern, J.H.; et al. Epigenomic and transcriptomic changes during human RPE EMT in a stem cell model of epiretinal membrane pathogenesis and prevention by nicotinamide. Stem Cell Rep. 2020, 14, 631–647. [Google Scholar] [CrossRef]
- Kuroda, T.; Ando, S.; Takeno, Y.; Kishino, A.; Kimura, T. Robust induction of retinal pigment epithelium cells from human induced pluripotent stem cells by inhibiting FGF/MAPK signaling. Stem Cell Res. 2019, 39, 101514. [Google Scholar] [CrossRef] [PubMed]
- Sripathi, S.R.; Hu, M.-W.; Liu, M.M.; Wan, J.; Cheng, J.; Duan, Y.; Mertz, J.L.; Wahlin, K.J.; Maruotti, J.; Berlinicke, C.A.; et al. Transcriptome Landscape of Epithelial to Mesenchymal Transition of Human Stem Cell-Derived RPE. Investig. Ophthalmol. Vis. Sci. 2021, 62, 1. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Strebinger, D.; Deluz, C.; Friman, E.T.; Govindan, S.; Alber, A.B.; Suter, D.M. Endogenous fluctuations of OCT4 and SOX2 bias pluripotent cell fate decisions. Mol. Syst. Biol. 2019, 15, e9002. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lukauskas, S.; Tvardovskiy, A.; Nguyen, N.V.; Stadler, M.; Faull, P.; Ravnsborg, T.; Aygenli, B.Ö.; Dornauer, S.; Flynn, H.; Lindeboom, R.G.H.; et al. Decoding chromatin states by proteomic profiling of nucleosome readers. Nature 2024, 627, 671–679, Erratum in Nature 2024, 628, E6. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abyadeh, M.; Gupta, V.; Paulo, J.A.; Sheriff, S.; Shadfar, S.; Fitzhenry, M.; Amirkhani, A.; Gupta, V.; Salekdeh, G.H.; A Haynes, P.; et al. Apolipoprotein ε in Brain and Retinal Neurodegenerative Diseases. Aging Dis. 2023, 14, 1311–1330. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, X.; Zhu, Z.; Huang, Y.; Shang, X.; O’Brien, T.J.; Kwan, P.; Ha, J.; Wang, W.; Liu, S.; Zhang, X.; et al. Shared genetic aetiology of Alzheimer’s disease and age-related macular degeneration by APOC1 and APOE genes. BMJ Neurol. Open 2024, 6, e000570. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hyttinen, J.M.T.; Kannan, R.; Felszeghy, S.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. The Regulation of NFE2L2 (NRF2) Signalling and Epithelial-to-Mesenchymal Transition in Age-Related Macular Degeneration Pathology. Int. J. Mol. Sci. 2019, 20, 5800. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chhunchha, B.; Kubo, E.; Singh, D.P. Switching of Redox Signaling by Prdx6 Expression Decides Cellular Fate by Hormetic Phenomena Involving Nrf2 and Reactive Oxygen Species. Cells 2022, 11, 1266. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, X.; Tzekov, R.; Su, M.; Zhu, Y.; Han, A.; Li, W. Hydrogen peroxide-induced oxidative damage and protective role of peroxiredoxin 6 protein via EGFR/ERK signaling pathway in RPE cells. Front. Aging Neurosci. 2023, 15, 1169211. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mack, A.A.; Kroboth, S.; Rajesh, D.; Wang, W.B. Generation of induced pluripotent stem cells from CD34+ cells across blood drawn from multiple donors with non-integrating episomal vectors. PLoS ONE 2011, 6, e27956. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic andadult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.; et al. Amore efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Gulbranson, D.R.; Hou, Z.; Bolin, J.M.; Ruotti, V.; Probasco, M.D.; Smuga-Otto, K.; Howden, S.E.; Diol, N.R.; E Propson, N.; et al. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods 2011, 8, 424–429. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Okita, K.; Yamakawa, T.; Matsumura, Y.; Sato, Y.; Amano, N.; Watanabe, A.; Goshima, N.; Yamanaka, S. An efficient nonviral method to generate integration-free human-induced pluripotent stem cellsfrom cord blood and peripheral blood cells. Stem Cells 2013, 31, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Osakada, F.; Jin, Z.B.; Hirami, Y.; Ikeda, H.; Danjyo, T.; Watanabe, K.; Sasai, Y.; Takahashi, M. In vitro differentiation of retinal cells from human pluripotent stem cells by small-molecule induction. J. Cell Sci. 2009, 122, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Schaub, N.J.; Hotaling, N.A.; Manescu, P.; Padi, S.; Wan, Q.; Sharma, R.; George, A.; Chalfoun, J.; Simon, M.; Ouladi, M.; et al. Deep learning predicts function of live retinalpigment epithelium from quantitative microscopy. J. Clin. Investig. 2020, 130, 1010–1023. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Samavarchi-Tehrani, P.; Golipour, A.; David, L.; Sung, H.K.; Beyer, T.A.; Datti, A.; Woltjen, K.; Nagy, A.; Wrana, J.L. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell Stem Cell 2010, 7, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Guo, X.; Hong, W.; Liu, Q.; Wei, T.; Lu, C.; Gao, L.; Ye, D.; Zhou, Y.; Chen, J.; et al. Critical regulation of miR-200/ZEB2 pathway in Oct4/Sox2-induced mesenchymal-to-epithelial transition and induced pluripotent stem cell generation. Proc. Natl. Acad. Sci. USA 2013, 110, 2858–2863. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maruotti, J.; Wahlin, K.; Gorrell, D.; Bhutto, I.; Lutty, G.; Zack, D.J. A simple and scalable process for the differentiation of retinal pigment epithelium from human pluripotent stem cells. Stem Cells Transl. Med. 2013, 2, 341–354. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maruotti, J.; Sripathi, S.R.; Bharti, K.; Fuller, J.; Wahlin, K.J.; Ranganathan, V.; Sluch, V.M.; Berlinicke, C.A.; Davis, J.; Kim, C.; et al. Small-molecule-directed, efficient generation of retinal pigment epithelium from human pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2015, 112, 10950–10955. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Singh, R.; Phillips, M.J.; Kuai, D.; Meyer, J.; Martin, J.M.; Smith, M.A.; Perez, E.T.; Shen, W.; Wallace, K.A.; Capowski, E.E.; et al. Functional analysis of serially expanded human iPS cell-derived RPE cultures. Investig. Opthalmol. Vis. Sci. 2013, 54, 6767–6778. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kamao, H.; Mandai, M.; Okamoto, S.; Sakai, N.; Suga, A.; Sugita, S.; Kiryu, J.; Takahashi, M. Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Rep. 2014, 2, 205–218. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- D’antonio-Chronowska, A.; D’antonio, M.; Frazer, K.A. In vitro Differentiation of Human iPSC-derived Retinal Pigment Epithelium Cells (iPSC-RPE). Bio-protocol 2019, 9, e3469. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Salas, A.; Duarri, A.; Fontrodona, L.; Ramírez, D.M.; Badia, A.; Isla-Magrané, H.; Ferreira-de-Souza, B.; Zapata, M.Á.; Raya, Á.; Veiga, A.; et al. Cell therapy with hiPSC-derived RPE cells and RPCs prevents visual function loss in a rat model of retinal degeneration. Mol. Ther. Methods Clin. Dev. 2021, 20, 688–702. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nishida, M.; Tanaka, Y.; Tanaka, Y.; Amaya, S.; Tanaka, N.; Uyama, H.; Masuda, T.; Onishi, A.; Sho, J.; Yokota, S.; et al. Human iPS cell derived RPE strips for secure delivery of graft cells at a target place with minimal surgical invasion. Sci. Rep. 2021, 11, 21421. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blenkinsop, T.A.; Saini, J.S.; Maminishkis, A.; Bharti, K.; Wan, Q.; Banzon, T.; Lotfi, M.; Davis, J.; Singh, D.; Rizzolo, L.J.; et al. Human Adult Retinal Pigment Epithelial Stem Cell-Derived RPE Monolayers Exhibit Key Physiological Characteristics of Native Tissue. Investig. Opthalmol. Vis. Sci. 2015, 56, 7085–7099. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Brandl, C. Generation of Functional Retinal Pigment Epithelium from Human Induced Pluripotent Stem Cells. Methods Mol. Biol. 2019, 1834, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Picard Toolkit. Broad Institute, GitHub Repository. 2018. Available online: http://broadinstitute.github.io/picard/ (accessed on 19 March 2019).
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578, Erratum in Nat. Protoc. 2014, 9, 2513. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Van der Auwera, G.A.; O’Connor, B.D. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra, 1st ed.; O’Reilly Media: Sebastopol, CA, USA, 2000. [Google Scholar]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. bioRxiv 2018, 201178. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Feng, J.; Liu, T.; Qin, B.; Zhang, Y.; Liu, X.S. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012, 7, 1728–1740. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef]
- Castro-Mondragon, J.A.; Riudavets-Puig, R.; Rauluseviciute, I.; Lemma, R.B.; Turchi, L.; Blanc-Mathieu, R.; Lucas, J.; Boddie, P.; Khan, A.; Manosalva Pérez, N.; et al. JASPAR 2022: The 9th release of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2022, 50, D165–D173. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tanigawa, Y.; Dyer, E.S.; Bejerano, G. WhichTF is functionally important in your open chromatin data? PLoS Comput. Biol. 2022, 18, e1010378. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lalanne, J.B.; Regalado, S.G.; Domcke, S.; Calderon, D.; Martin, B.K.; Li, X.; Li, T.; Suiter, C.C.; Lee, C.; Trapnell, C.; et al. Multiplex profiling of developmental cis-regulatory elements with quantitative single-cell expression reporters. Nat. Methods 2024, 21, 983–993. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Grünewald, J.; Zhou, R.; Lareau, C.A.; Garcia, S.P.; Iyer, S.; Miller, B.R.; Langner, L.M.; Hsu, J.Y.; Aryee, M.J.; Joung, J.K. A dual-deaminase CRISPR base editor enables concurrent adenine and cytosine editing. Nat. Biotechnol. 2020, 38, 861–864. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hsiung, C.C.; Wilson, C.M.; Sambold, N.A.; Dai, R.; Chen, Q.; Teyssier, N.; Misiukiewicz, S.; Arab, A.; O’loughlin, T.; Cofsky, J.C.; et al. Engineered CRISPR-Cas12a for higher-order combinatorial chromatin perturbations. Nat. Biotechnol. 2024, 43, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Sherwood, R.I.; Hashimoto, T.; O’Donnell, C.W.; Lewis, S.; Barkal, A.A.; van Hoff, J.P.; Karun, V.; Jaakkola, T.; Gifford, D.K. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat. Biotechnol. 2014, 32, 171–178. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Baek, S.; Goldstein, I.; Hager, G.L. Bivariate Genomic Footprinting Detects Changes in Transcription Factor Activity. Cell Rep. 2017, 19, 1710–1722. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kulakovskiy, I.V.; Medvedeva, Y.A.; Schaefer, U.; Kasianov, A.S.; Vorontsov, I.E.; Bajic, V.B.; Makeev, V.J. HOCOMOCO: A comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 2013, 41, D195–D202. [Google Scholar] [CrossRef]
- Bartke, T.; Vermeulen, M.; Xhemalce, B.; Robson, S.C.; Mann, M.; Kouzarides, T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 2010, 143, 470–484. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Makowski, M.M.; Gräwe, C.; Foster, B.M.; Nguyen, N.V.; Bartke, T.; Vermeulen, M. Global profiling of protein-DNA and protein-nucleosome binding affinities using quantitative mass spectrometry. Nat. Commun. 2018, 9, 1653. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, W.H.; Zheng, J.; Feldman, J.L.; Klein, M.A.; Kuznetsov, V.I.; Peterson, C.L.; Griffin, P.R.; Denu, J.M. Multivalent interactions drive nucleosome binding and efficient chromatin deacetylation by SIRT6. Nat. Commun. 2020, 11, 5244. [Google Scholar] [CrossRef]
- Stützer, A.; Welp, L.M.; Raabe, M.; Sachsenberg, T.; Kappert, C.; Wulf, A.; Lau, A.M.; David, S.-S.; Chernev, A.; Kramer, K.; et al. Analysis of protein-DNA interactions in chromatin by UV induced cross-linking and mass spectrometry. Nat. Commun. 2020, 11, 5250. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zibetti, C. Molecular Determinants of the Human Retinal Pigment Epithelium Cell Fate and Potential Pharmacogenomic Targets for Precision Medicine. Int. J. Mol. Sci. 2025, 26, 5817. https://doi.org/10.3390/ijms26125817
Zibetti C. Molecular Determinants of the Human Retinal Pigment Epithelium Cell Fate and Potential Pharmacogenomic Targets for Precision Medicine. International Journal of Molecular Sciences. 2025; 26(12):5817. https://doi.org/10.3390/ijms26125817
Chicago/Turabian StyleZibetti, Cristina. 2025. "Molecular Determinants of the Human Retinal Pigment Epithelium Cell Fate and Potential Pharmacogenomic Targets for Precision Medicine" International Journal of Molecular Sciences 26, no. 12: 5817. https://doi.org/10.3390/ijms26125817
APA StyleZibetti, C. (2025). Molecular Determinants of the Human Retinal Pigment Epithelium Cell Fate and Potential Pharmacogenomic Targets for Precision Medicine. International Journal of Molecular Sciences, 26(12), 5817. https://doi.org/10.3390/ijms26125817