
Patterns of Recombination in Coronaviruses


Abstract
1. Introduction
2. Results
2.1. Recombination Inferences
2.2. Constrained vs. Unconstrained Same-Species Phylogenetic Trees
2.3. Constrained vs. Unconstrained Different-Species Phylogenetic Trees
2.4. Non-Synonymous Divergence (Ka)
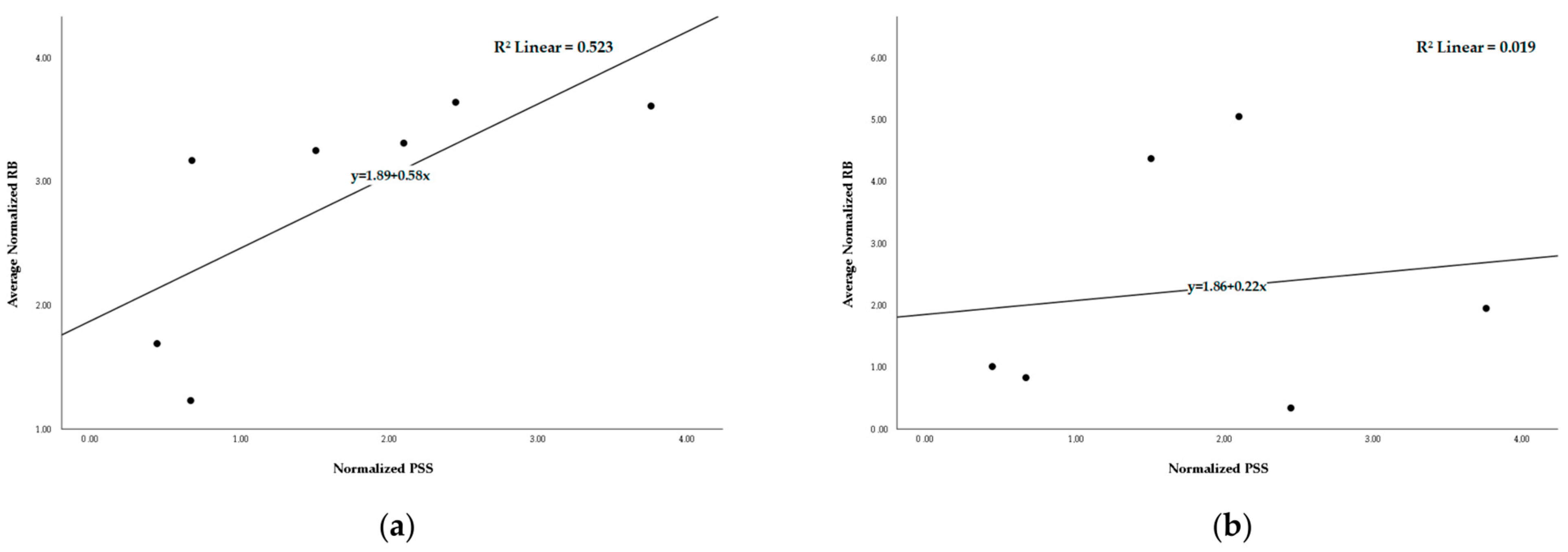
2.5. Recombination and Selection
3. Discussion
4. Materials and Methods
4.1. Sequence Retrieval
4.2. Recombination Inferences
4.3. Phylogenetic Analysis
4.4. Non-Synonymous Rate (Ka) of Evolution
4.5. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- LaTourrette, K.; Garcia-Ruiz, H. Determinants of Virus Variation, Evolution, and Host Adaptation. Pathogens 2022, 11, 1039. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; de Groot, R.J.; Haagmans, B.; Lau, S.K.P.; Neuman, B.W.; Perlman, S.; Sola, I.; van der Hoek, L.; Wong, A.C.P.; Yeh, S.-H. ICTV Virus Taxonomy Profile: Coronaviridae 2023. J. Gen. Virol. 2023, 104, 001843. [Google Scholar] [CrossRef]
- Zhou, Z.; Qiu, Y.; Ge, X. The taxonomy, host range and pathogenicity of coronaviruses and other viruses in the Nidovirales order. Anim. Dis. 2021, 1, 5. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Yip, C.C.; Huang, Y.; Tsoi, H.W.; Chan, K.H.; Yuen, K.Y. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J. Virol. 2006, 80, 7136–7145. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Xiao, Y.; Zhang, J.; Wang, Y.; Chen, L.; Paranhos-Baccalà, G.; Ren, L.; Wang, J. Genotype shift in human coronavirus OC43 and emergence of a novel genotype by natural recombination. J. Infect. 2015, 70, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Pyrc, K.; Dijkman, R.; Deng, L.; Jebbink, M.F.; Ross, H.A.; Berkhout, B.; van der Hoek, L. Mosaic Structure of Human Coronavirus NL63, One Thousand Years of Evolution. J. Mol. Biol. 2006, 364, 964–973. [Google Scholar] [CrossRef]
- Sabir, J.S.; Lam, T.T.; Ahmed, M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Jackson, B.; Boni, M.F.; Bull, M.J.; Colleran, A.; Colquhoun, R.M.; Darby, A.C.; Haldenby, S.; Hill, V.; Lucaci, A.; McCrone, J.T.; et al. Generation and transmission of interlineage recombinants in the SARS-CoV-2 pandemic. Cell 2021, 184, 5179–5188.e5178. [Google Scholar] [CrossRef]
- Posthuma, C.C.; Nedialkova, D.D.; Zevenhoven-Dobbe, J.C.; Blokhuis, J.H.; Gorbalenya, A.E.; Snijder, E.J. Site-directed mutagenesis of the Nidovirus replicative endoribonuclease NendoU exerts pleiotropic effects on the arterivirus life cycle. J. Virol. 2006, 80, 1653–1661. [Google Scholar] [CrossRef]
- Posthuma, C.C.; Pedersen, K.W.; Lu, Z.; Joosten, R.G.; Roos, N.; Zevenhoven-Dobbe, J.C.; Snijder, E.J. Formation of the arterivirus replication/transcription complex: A key role for nonstructural protein 3 in the remodeling of intracellular membranes. J. Virol. 2008, 82, 4480–4491. [Google Scholar] [CrossRef]
- Wells, H.L.; Bonavita, C.M.; Navarrete-Macias, I.; Vilchez, B.; Rasmussen, A.L.; Anthony, S.J. The coronavirus recombination pathway. Cell Host Microbe 2023, 31, 874–889. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Jackwood, M.W.; Hall, D.; Handel, A. Molecular evolution and emergence of avian gammacoronaviruses. Infect. Genet. Evol. 2012, 12, 1305–1311. [Google Scholar] [CrossRef]
- Lu, G.; Wang, Q.; Gao, G.F. Bat-to-human: Spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015, 23, 468–478. [Google Scholar] [CrossRef]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Sidarovich, A.; Arora, P.; Krüger, N.; Nehlmeier, I.; Kempf, A.; Graichen, L.; Winkler Martin, S.; Niemeyer, D.; Goffinet, C.; et al. Evidence for an ACE2-Independent Entry Pathway That Can Protect from Neutralization by an Antibody Used for COVID-19 Therapy. mBio 2022, 13, e0036422. [Google Scholar] [CrossRef] [PubMed]
- Baggen, J.; Jacquemyn, M.; Persoons, L.; Vanstreels, E.; Pye, V.E.; Wrobel, A.G.; Calvaresi, V.; Martin, S.R.; Roustan, C.; Cronin, N.B.; et al. TMEM106B is a receptor mediating ACE2-independent SARS-CoV-2 cell entry. Cell 2023, 186, 3427–3442.e3422. [Google Scholar] [CrossRef]
- Nikolaidis, M.; Markoulatos, P.; Van de Peer, Y.; Oliver, S.G.; Amoutzias, G.D. The Neighborhood of the Spike Gene Is a Hotspot for Modular Intertypic Homologous and Nonhomologous Recombination in Coronavirus Genomes. Mol. Biol. Evol. 2022, 39, msab292. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Huang, Y.; Tsoi, H.W.; Chan, K.H.; Yuen, K.Y. Phylogenetic and recombination analysis of coronavirus HKU1, a novel coronavirus from patients with pneumonia. Arch. Virol. 2005, 150, 2299–2311. [Google Scholar] [CrossRef]
- Patiño-Galindo, J.; García-Sastre, A.; Kuhn, J.H.; Rabadan, R.; Palacios, G. Recombination across distant coronavirid species and genera is a rare event with distinct genomic features. J. Virol. 2024, 98, e0110024. [Google Scholar] [CrossRef] [PubMed]
- Thor, S.W.; Hilt, D.A.; Kissinger, J.C.; Paterson, A.H.; Jackwood, M.W. Recombination in avian gamma-coronavirus infectious bronchitis virus. Viruses 2011, 3, 1777–1799. [Google Scholar] [CrossRef] [PubMed]
- Cooper, P.D.; Steiner-Pryor, A.; Scotti, P.D.; Delong, D. On the Nature of Poliovirus Genetic Recombinants. J. Gen. Virol. 1974, 23, 41–49. [Google Scholar] [CrossRef]
- Nagy, P.D.; Simon, A.E. New insights into the mechanisms of RNA recombination. Virology 1997, 235, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef]
- de Klerk, A.; Swanepoel, P.; Lourens, R.; Zondo, M.; Abodunran, I.; Lytras, S.; MacLean, O.A.; Robertson, D.; Kosakovsky Pond, S.L.; Zehr, J.D.; et al. Conserved recombination patterns across coronavirus subgenera. Virus Evol. 2022, 8, veac054. [Google Scholar] [CrossRef]
- Gallagher, T.M.; Buchmeier, M.J.; Perlman, S. Cell receptor-independent infection by a neurotropic murine coronavirus. Virology 1992, 191, 517–522. [Google Scholar] [CrossRef]
- McRoy, W.C.; Baric, R.S. Amino acid substitutions in the S2 subunit of mouse hepatitis virus variant V51 encode determinants of host range expansion. J. Virol. 2008, 82, 1414–1424. [Google Scholar] [CrossRef]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef]
- Tao, Y.; Shi, M.; Chommanard, C.; Queen, K.; Zhang, J.; Markotter, W.; Kuzmin, I.V.; Holmes, E.C.; Tong, S. Surveillance of Bat Coronaviruses in Kenya Identifies Relatives of Human Coronaviruses NL63 and 229E and Their Recombination History. J. Virol. 2017, 91, e01953-16. [Google Scholar] [CrossRef]
- Lau, S.K.; Lee, P.; Tsang, A.K.; Yip, C.C.; Tse, H.; Lee, R.A.; So, L.Y.; Lau, Y.L.; Chan, K.H.; Woo, P.C.; et al. Molecular epidemiology of human coronavirus OC43 reveals evolution of different genotypes over time and recent emergence of a novel genotype due to natural recombination. J. Virol. 2011, 85, 11325–11337. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, S.A.; Brown, J.; Pedersen, B.S.; Quinlan, A.R.; Elde, N.C. Extensive Recombination-driven Coronavirus Diversification Expands the Pool of Potential Pandemic Pathogens. Genome Biol. Evol. 2022, 14, evac161. [Google Scholar] [CrossRef]
- Forni, D.; Cagliani, R.; Sironi, M. Recombination and Positive Selection Differentially Shaped the Diversity of Betacoronavirus Subgenera. Viruses 2020, 12, 1313. [Google Scholar] [CrossRef]
- Vakulenko, Y.; Deviatkin, A.; Drexler, J.F.; Lukashev, A. Modular Evolution of Coronavirus Genomes. Viruses 2021, 13, 1270. [Google Scholar] [CrossRef]
- Ferreira, P.; Soares, R.; López-Fernández, H.; Vazquez, N.; Reboiro-Jato, M.; Vieira, C.P.; Vieira, J. Multiple Lines of Evidence Support 199 SARS-CoV-2 Positively Selected Amino Acid Sites. Int. J. Mol. Sci. 2024, 25, 2428. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, W.; Hall, A.B.; Jiang, X. Characterizing Transcriptional Regulatory Sequences in Coronaviruses and Their Role in Recombination. Mol. Biol. Evol. 2021, 38, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Esquivel Gomez, L.R.; Weber, A.; Kocher, A.; Kühnert, D. Recombination-aware phylogenetic analysis sheds light on the evolutionary origin of SARS-CoV-2. Sci. Rep. 2024, 14, 541. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. The effect of recombination on the accuracy of phylogeny estimation. J. Mol. Evol. 2002, 54, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Schierup, M.H.; Hein, J. Consequences of recombination on traditional phylogenetic analysis. Genetics 2000, 156, 879–891. [Google Scholar] [CrossRef]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved Algorithmic Complexity for the 3SEQ Recombination Detection Algorithm. Mol. Biol. Evol. 2018, 35, 247–251. [Google Scholar] [CrossRef]
- Brierley, I.; Boursnell, M.E.; Binns, M.M.; Bilimoria, B.; Blok, V.C.; Brown, T.D.; Inglis, S.C. An efficient ribosomal frame-shifting signal in the polymerase-encoding region of the coronavirus IBV. EMBO J. 1987, 6, 3779–3785. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Leeson, A.; Ballantine, M.; Andonov, A.; Baker, L.; Dobie, F.; Li, Y.; Bastien, N.; Feldmann, H.; Strocher, U.; et al. Characterization of protein-protein interactions between the nucleocapsid protein and membrane protein of the SARS coronavirus. Virus Res. 2004, 105, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Yan, C.; Qin, F.; Zheng, L.; Zhang, X.E. The intraviral protein-protein interaction of SARS-CoV-2 reveals the key role of N protein in virus-like particle assembly. Int. J. Biol. Sci. 2021, 17, 3889–3897. [Google Scholar] [CrossRef]
- de Haan, C.A.; Rottier, P.J. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar]
- Wang, Y.; Zhang, X. The nucleocapsid protein of coronavirus mouse hepatitis virus interacts with the cellular heterogeneous nuclear ribonucleoprotein A1 in vitro and in vivo. Virology 1999, 265, 96–109. [Google Scholar] [CrossRef]
- Kopecky-Bromberg, S.A.; Martínez-Sobrido, L.; Frieman, M.; Baric, R.A.; Palese, P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007, 81, 548–557. [Google Scholar] [CrossRef]
- Sui, L.; Zhao, Y.; Wang, W.; Wu, P.; Wang, Z.; Yu, Y.; Hou, Z.; Tan, G.; Liu, Q. SARS-CoV-2 Membrane Protein Inhibits Type I Interferon Production Through Ubiquitin-Mediated Degradation of TBK1. Front. Immunol. 2021, 12, 662989. [Google Scholar] [CrossRef]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Boson, B.; Legros, V.; Zhou, B.; Siret, E.; Mathieu, C.; Cosset, F.-L.; Lavillette, D.; Denolly, S. The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J. Biol. Chem. 2021, 296, 100111. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef]
- López-Fernández, H.; Ferreira, P.; Reboiro-Jato, M.; Vieira, C.P.; Vieira, J. The pegi3s Bioinformatics Docker Images Project. In Practical Applications of Computational Biology & Bioinformatics, Proceedings of the 15th International Conference (PACBB 2021), Salamanca, Spain, 6–8 October 2021; Springer International Publishing: Cham, Switzerland, 2022; pp. 31–40. [Google Scholar]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Fernandez, H.; Duque, P.; Vazquez, N.; Fdez-Riverola, F.; Reboiro-Jato, M.; Vieira, C.P.; Vieira, J. SEDA: A Desktop Tool Suite for FASTA Files Processing. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 1850–1860. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Mai, U.; Sayyari, E.; Mirarab, S. Minimum variance rooting of phylogenetic trees and implications for species tree reconstruction. PLoS ONE 2017, 12, e0182238. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, J.; Zhao, X.Q.; Wang, J.; Wong, G.K.; Yu, J. KaKs_Calculator: Calculating Ka and Ks through model selection and model averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef]

{kind=link}
| Method | Gene | Total RB | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ORF1a | ORF1b | S | E | M | N | Accessory | |||
| 3SEQ | RB | 174 | 271 | 220 | 7 | 22 | 34 | 158 | 886 |
| %RB | 19.64 | 30.59 | 24.83 | 0.79 | 2.48 | 3.84 | 17.83 | 0.82 | |
| %RB/%Alignment size | 0.47 | 0.92 | 1.97 | 0.20 | 1.17 | 0.67 | 1.79 | - | |
| Geneconv | RB | 379 | 314 | 129 | 8 | 41 | 50 | 237 | 1158 |
| %RB | 32.73 | 27.12 | 11.14 | 0.69 | 3.54 | 4.32 | 20.47 | 1.08 | |
| %RB/%Alignment size | 0.58 | 0.82 | 1.07 | 1.34 | 1.36 | 1.67 | 2.25 | - | |
| RDP | RB | 40118 | 24086 | 11094 | 488 | 1788 | 9148 | 18140 | 104862 |
| %RB | 38.26 | 22.97 | 10.58 | 0.47 | 1.71 | 8.72 | 17.30 | 97.46 | |
| %RB/%Alignment size | 0.60 | 0.52 | 0.90 | 1.21 | 0.86 | 2.38 | 4.08 | - | |
| Bootscan | RB | 116 | 105 | 123 | 7 | 12 | 14 | 313 | 690 |
| %RB | 16.81 | 15.22 | 17.83 | 1.01 | 1.74 | 2.03 | 45.53 | 0.64 | |
| %RB/%Alignment size | 0.25 | 0.45 | 0.96 | 1.40 | 0.45 | 0.51 | 5.40 | - | |
| Total per gene | RB | 40787 | 24776 | 11566 | 510 | 1863 | 9246 | 18848 | 107596 |
| %RB | 37.91 | 23.03 | 10.75 | 0.47 | 1.73 | 8.59 | 17.52 | - | |
| %RB/%Alignment size | 0.47 | 0.68 | 1.23 | 1.04 | 0.96 | 1.31 | 3.38 | - | |
| Genomic Region | Intratypic | Intertypic | |||
|---|---|---|---|---|---|
| Observed/Expected | Average | Observed/Expected | Average | ||
| ORF1a | ORF1a | 0.83 | 1.23 | 2.04 | 0.59 |
| ORF1b | 2.02 | 1.35 | |||
| S | 1.25 | 0.59 | |||
| E | 0.64 | 0.00 | |||
| M | 0.97 | 0.00 | |||
| N | 2.03 | 0.00 | |||
| Accessory | 0.83 | 0.12 | |||
| ORF1b | ORF1b | 1.01 | 1.69 | 13.19 | 2.25 |
| S | 2.49 | 0.22 | |||
| E | 1.60 | 0.00 | |||
| M | 1.18 | 0.00 | |||
| N | 1.22 | 0.00 | |||
| Accessory | 2.28 | 1.00 | |||
| S | S | 5.05 | 3.31 | 8.07 | 1.70 |
| E | 2.37 | 0.00 | |||
| M | 4.72 | 0.00 | |||
| N | 3.39 | 0.54 | |||
| Accessory | 3.94 | 2.50 | |||
| E | E | NA | 3.17 | 16.73 | 2.43 |
| M | 7.91 | 1.95 | |||
| N | 6.18 | 0.00 | |||
| Accessory | 3.50 | 0.73 | |||
| M | M | 0.34 | 3.64 | 2.38 | 2.04 |
| N | 6.51 | 9.75 | |||
| Accessory | 3.86 | 0.17 | |||
| N | N | 1.95 | 3.61 | 3.80 | 2.03 |
| Accessory | 3.98 | 0.15 | |||
| Accessory | Accessory | 4.37 | 3.25 | 0.17 | 0.69 |
| Gene | Statistically Significant Trees | Statistically Insignificant Trees | # of Converged Trees | Total |
| ORF1a | 20 (95.24%) | 1 (4.76%) | 19 (85.71%) | 21 |
| ORF1b | 18 (85.71%) | 3 (14.29%) | 21 (100%) | |
| E | 13 (61.90%) | 8 (38,10%) | 21 (100%) | |
| M | 19 (90.48%) | 2 (9.52%) | 20 (95.24%) | |
| N | 17 (80.95%) | 4 (19.05%) | 21 (100%) | |
| Whole Genome | 21 (100%) | 0 (0%) | 20 (95.24%) | |
| Total | 108 (85.71%) | 18 (14.29%) | 122 (96.83%) | 126 |
| Dataset | Genus | Number of Sequences |
|---|---|---|
| hCoV-229E | Alphacoronavirus | 152 |
| HKU2 | Alphacoronavirus | 10 |
| HKU10 | Alphacoronavirus | 25 |
| hCoV-NL63 | Alphacoronavirus | 151 |
| PEDV | Alphacoronavirus | 851 |
| TGEV | Alphacoronavirus | 62 |
| GCCDC1 | Betacoronavirus | 9 |
| hCoV-HKU1 | Betacoronavirus | 63 |
| HKU4 | Betacoronavirus | 10 |
| HKU9 | Betacoronavirus | 10 |
| MERS-CoV (animal hosts) | Betacoronavirus | 334 |
| MERS-CoV (human host) | Betacoronavirus | 286 |
| MERS-CoV-related | Betacoronavirus | 631 |
| Murine-CoV | Betacoronavirus | 38 |
| hCoV-OC43 | Betacoronavirus | 342 |
| PHEV | Betacoronavirus | 18 |
| SARS-CoV | Betacoronavirus | 13 |
| SARS-CoV-2 | Betacoronavirus | 2794 |
| SARS-CoV-related | Betacoronavirus | 276 |
| HKU15 | Deltacoronavirus | 200 |
| IBV | Gammacoronavirus | 667 |
| 37 species with fewer than eight sequences | - | 92 |
| Total | - | 7034 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soares, R.; Vieira, C.P.; Vieira, J. Patterns of Recombination in Coronaviruses. Int. J. Mol. Sci. 2025, 26, 5595. https://doi.org/10.3390/ijms26125595
Soares R, Vieira CP, Vieira J. Patterns of Recombination in Coronaviruses. International Journal of Molecular Sciences. 2025; 26(12):5595. https://doi.org/10.3390/ijms26125595
Chicago/Turabian StyleSoares, Ricardo, Cristina P. Vieira, and Jorge Vieira. 2025. "Patterns of Recombination in Coronaviruses" International Journal of Molecular Sciences 26, no. 12: 5595. https://doi.org/10.3390/ijms26125595
APA StyleSoares, R., Vieira, C. P., & Vieira, J. (2025). Patterns of Recombination in Coronaviruses. International Journal of Molecular Sciences, 26(12), 5595. https://doi.org/10.3390/ijms26125595