

Multi-Cohort Exploration of Repetitive Element Transcription and DNA Methylation in Human Steatotic Liver Disease

 , , , , , and
, , , , , and 
Abstract
1. Introduction
2. Results
2.1. Examination of Transposable Element Transcripts in RNA-Seq from Pre-Cirrhotic Bariatric Surgery Patients (Cohort 1)
2.2. Examination of LINE and SINE Transcripts and DNA Methylation in FFPE Samples from Chronic Liver Disease and HCC Patients (Cohort 2)
2.3. Examination of LINE and SINE Transcripts in RNA Extracted from Fresh Liver Biopsies from ARLD Patients (Cohort 3)
3. Discussion, Limitations and Conclusions
3.1. Discussion
3.2. Limitations
3.3. Conclusions
4. Materials and Methods
4.1. RNA-Seq Analyses of Bariatric Surgery Cohort 1
4.2. Examination of Ex-Diagnostic Biopsy and Liver Resection FFPE Samples in MASLD/HCC Cohort 2
4.3. RT-qPCR on RNA from Fresh Liver Biopsy in ARLD Cohort 3
4.3.1. Fibroscan® Measurements
4.3.2. Transjugular Liver Biopsy and Tissue Sampling
4.3.3. RT-qPCR Methods
4.4. Statistical Analyses
4.4.1. RNA-Seq Analyses of Bariatric Surgery Cohort 1
4.4.2. Examination of Ex-Diagnostic Biopsy and Liver Resection FFPE Samples in MASLD/HCC Cohort 2
4.4.3. RT-qPCR on RNA from Fresh Liver Biopsy in ARLD Cohort 3
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kazazian, H.H., Jr.; Moran, J.V. Mobile DNA in Health and Disease. N. Engl. J. Med. 2017, 377, 361–370. [Google Scholar] [CrossRef]
- de Koning, A.P.; Gu, W.; Castoe, T.A.; Batzer, M.A.; Pollock, D.D. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011, 7, e1002384. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, E.; Beletskiy, A.; Kolosov, P. The Role of Transposable Elements of the Human Genome in Neuronal Function and Pathology. Int. J. Mol. Sci. 2022, 23, 5847. [Google Scholar] [CrossRef] [PubMed]
- Larouche, J.D.; Trofimov, A.; Hesnard, L.; Ehx, G.; Zhao, Q.; Vincent, K.; Durette, C.; Gendron, P.; Laverdure, J.P.; Bonneil, E.; et al. Widespread and tissue-specific expression of endogenous retroelements in human somatic tissues. Genome Med. 2020, 12, 40. [Google Scholar] [CrossRef] [PubMed]
- Assante, G.; Chandrasekaran, S.; Ng, S.; Tourna, A.; Chung, C.H.; Isse, K.A.; Banks, J.L.; Soffientini, U.; Filippi, C.; Dhawan, A.; et al. Acetyl-CoA metabolism drives epigenome change and contributes to carcinogenesis risk in fatty liver disease. Genome Med. 2022, 14, 67. [Google Scholar] [CrossRef]
- Kim, M.; Delgado, E.; Ko, S. DNA methylation in cell plasticity and malignant transformation in liver diseases. Pharmacol. Ther. 2023, 241, 108334. [Google Scholar] [CrossRef]
- Rodriguez-Sanabria, J.S.; Escutia-Gutierrez, R.; Rosas-Campos, R.; Armendariz-Borunda, J.S.; Sandoval-Rodriguez, A. An Update in Epigenetics in Metabolic-Associated Fatty Liver Disease. Front. Med. 2021, 8, 770504. [Google Scholar] [CrossRef]
- Habash, N.W.; Sehrawat, T.S.; Shah, V.H.; Cao, S. Epigenetics of alcohol-related liver diseases. JHEP Rep. 2022, 4, 100466. [Google Scholar] [CrossRef]
- Li, X.; Egervari, G.; Wang, Y.; Berger, S.L.; Lu, Z. Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol. 2018, 19, 563–578. [Google Scholar] [CrossRef]
- Ferrari, A.; Longo, R.; Silva, R.; Mitro, N.; Caruso, D.; De Fabiani, E.; Crestani, M. Epigenome modifiers and metabolic rewiring: New frontiers in therapeutics. Pharmacol. Ther. 2019, 193, 178–193. [Google Scholar] [CrossRef]
- Moghe, A.; Joshi-Barve, S.; Ghare, S.; Gobejishvili, L.; Kirpich, I.; McClain, C.J.; Barve, S. Histone modifications and alcohol-induced liver disease: Are altered nutrients the missing link? World, J. Gastroenterol. 2011, 17, 2465–2472. [Google Scholar] [CrossRef] [PubMed]
- Hylemon, P.B.; Takabe, K.; Dozmorov, M.; Nagahashi, M.; Zhou, H. Bile acids as global regulators of hepatic nutrient metabolism. Liver Res. 2017, 1, 10–16. [Google Scholar] [CrossRef]
- Chen, G.; Broseus, J.; Hergalant, S.; Donnart, A.; Chevalier, C.; Bolanos-Jimenez, F.; Gueant, J.L.; Houlgatte, R. Identification of master genes involved in liver key functions through transcriptomics and epigenomics of methyl donor deficiency in rat: Relevance to nonalcoholic liver disease. Mol. Nutr. Food Res. 2015, 59, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.J.; Peng, L.; Cline, G.W.; Petersen, K.F.; Shulman, G.I. A Non-invasive Method to Assess Hepatic Acetyl-CoA In Vivo. Cell Metab. 2017, 25, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Tryndyak, V.P.; Bagnyukova, T.V.; Melnyk, S.; Montgomery, B.; Ross, S.A.; Latendresse, J.R.; Rusyn, I.; Beland, F.A. Hepatic epigenetic phenotype predetermines individual susceptibility to hepatic steatosis in mice fed a lipogenic methyl-deficient diet. J. Hepatol. 2009, 51, 176–186. [Google Scholar] [CrossRef]
- Wahl, D.; Cavalier, A.N.; Smith, M.; Seals, D.R.; LaRocca, T.J. Healthy Aging Interventions Reduce Repetitive Element Transcripts. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 805–810. [Google Scholar] [CrossRef]
- Szabo, L.; Molnar, R.; Tomesz, A.; Deutsch, A.; Darago, R.; Varjas, T.; Ritter, Z.; Szentpeteri, J.L.; Andreidesz, K.; Mathe, D.; et al. Olive Oil Improves While Trans Fatty Acids Further Aggravate the Hypomethylation of LINE-1 Retrotransposon DNA in an Environmental Carcinogen Model. Nutrients 2022, 14, 908. [Google Scholar] [CrossRef]
- Chew, N.W.S.; Ng, C.H.; Tan, D.J.H.; Kong, G.; Lin, C.; Chin, Y.H.; Lim, W.H.; Huang, D.Q.; Quek, J.; Fu, C.E.; et al. The global burden of metabolic disease: Data from 2000 to 2019. Cell Metab. 2023, 35, 414–428.e413. [Google Scholar] [CrossRef]
- Ikejima, K.; Kon, K.; Yamashina, S. Nonalcoholic fatty liver disease and alcohol-related liver disease: From clinical aspects to pathophysiological insights. Clin. Mol. Hepatol. 2020, 26, 728–735. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- de Mello, V.D.; Matte, A.; Perfilyev, A.; Mannisto, V.; Ronn, T.; Nilsson, E.; Kakela, P.; Ling, C.; Pihlajamaki, J. Human liver epigenetic alterations in non-alcoholic steatohepatitis are related to insulin action. Epigenetics 2017, 12, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.K.; Yang, H.; Moylan, C.A.; Pang, H.; Dellinger, A.; Abdelmalek, M.F.; Garrett, M.E.; Ashley-Koch, A.; Suzuki, A.; Tillmann, H.L.; et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 1076–1087. [Google Scholar] [CrossRef]
- Mwinyi, J.; Bostrom, A.E.; Pisanu, C.; Murphy, S.K.; Erhart, W.; Schafmayer, C.; Hampe, J.; Moylan, C.; Schioth, H.B. NAFLD is associated with methylation shifts with relevance for the expression of genes involved in lipoprotein particle composition. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 314–323. [Google Scholar] [CrossRef] [PubMed]
- Schioth, H.B.; Bostrom, A.; Murphy, S.K.; Erhart, W.; Hampe, J.; Moylan, C.; Mwinyi, J. A targeted analysis reveals relevant shifts in the methylation and transcription of genes responsible for bile acid homeostasis and drug metabolism in non-alcoholic fatty liver disease. BMC Genomics 2016, 17, 462. [Google Scholar] [CrossRef] [PubMed]
- Schauer, S.N.; Carreira, P.E.; Shukla, R.; Gerhardt, D.J.; Gerdes, P.; Sanchez-Luque, F.J.; Nicoli, P.; Kindlova, M.; Ghisletti, S.; Santos, A.D.; et al. L1 retrotransposition is a common feature of mammalian hepatocarcinogenesis. Genome Res. 2018, 28, 639–653. [Google Scholar] [CrossRef]
- Lai, Z.; Chen, J.; Ding, C.; Wong, K.; Chen, X.; Pu, L.; Huang, Q.; Chen, X.; Cheng, Z.; Liu, Y.; et al. Association of Hepatic Global DNA Methylation and Serum One-Carbon Metabolites with Histological Severity in Patients with NAFLD. Obesity 2020, 28, 197–205. [Google Scholar] [CrossRef]
- Pantano, L.; Agyapong, G.; Shen, Y.; Zhuo, Z.; Fernandez-Albert, F.; Rust, W.; Knebel, D.; Hill, J.; Boustany-Kari, C.M.; Doerner, J.F.; et al. Molecular characterization and cell type composition deconvolution of fibrosis in NAFLD. Sci. Rep. 2021, 11, 18045. [Google Scholar] [CrossRef]
- Jin, Y.; Tam, O.H.; Paniagua, E.; Hammell, M. TEtranscripts: A package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics 2015, 31, 3593–3599. [Google Scholar] [CrossRef]
- Salavrakos, M.; Piessevaux, H.; Komuta, M.; Lanthier, N.; Starkel, P. Fibroscan Reliably Rules Out Advanced Liver Fibrosis and Significant Portal Hypertension in Alcoholic Patients. J. Clin. Gastroenterol. 2019, 53, 772–778. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Wong, G.; Anstee, Q.M.; Henry, L. The Global Burden of Liver Disease. Clin. Gastroenterol. Hepatol. 2023, 21, 1978–1991. [Google Scholar] [CrossRef]
- Cespiati, A.; Youngson, N.A.; Tourna, A.; Valenti, L. Genetics and Epigenetics in the Clinic: Precision Medicine in the Management of Fatty Liver Disease. Curr. Pharm. Des. 2020, 26, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martin, B.; Alvarez, E.G.; Baez-Ortega, A.; Zamora, J.; Supek, F.; Demeulemeester, J.; Santamarina, M.; Ju, Y.S.; Temes, J.; Garcia-Souto, D.; et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat. Genet. 2020, 52, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Bulut-Karslioglu, A.; De La Rosa-Velazquez, I.A.; Ramirez, F.; Barenboim, M.; Onishi-Seebacher, M.; Arand, J.; Galan, C.; Winter, G.E.; Engist, B.; Gerle, B.; et al. Suv39h-dependent H3K9me3 marks intact retrotransposons and silences LINE elements in mouse embryonic stem cells. Mol. Cell 2014, 55, 277–290. [Google Scholar] [CrossRef]
- Ishak, C.A.; Marshall, A.E.; Passos, D.T.; White, C.R.; Kim, S.J.; Cecchini, M.J.; Ferwati, S.; MacDonald, W.A.; Howlett, C.J.; Welch, I.D.; et al. An RB-EZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol. Cell 2016, 64, 1074–1087. [Google Scholar] [CrossRef]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, W.; Mao, M.; Gao, R.; Shi, W.; Li, D.; Calderone, R.; Sui, B.; Tian, X.; Meng, X. Similarities and Differences: A Comparative Review of the Molecular Mechanisms and Effectors of NAFLD and AFLD. Front. Physiol. 2021, 12, 710285. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Arteel, G.E. Effect of ethanol on lipid metabolism. J. Hepatol. 2019, 70, 237–248. [Google Scholar] [CrossRef]
- Miousse, I.R.; Koturbash, I. The Fine LINE: Methylation Drawing the Cancer Landscape. Biomed. Res. Int. 2015, 2015, 131547. [Google Scholar] [CrossRef]
- Kelly, M.; Lihua, S.; Zhe, Z.; Li, S.; Yoselin, P.; Michelle, P.; Sullivan Kathleen, E. Transposable element dysregulation in systemic lupus erythematosus and regulation by histone conformation and Hsp90. Clin. Immunol. 2018, 197, 6–18. [Google Scholar] [CrossRef]
- Li, T.H.; Schmid, C.W. Differential stress induction of individual Alu loci: Implications for transcription and retrotransposition. Gene 2001, 276, 135–141. [Google Scholar] [CrossRef]
- Takai, D.; Yagi, Y.; Habib, N.; Sugimura, T.; Ushijima, T. Hypomethylation of LINE1 retrotransposon in human hepatocellular carcinomas, but not in surrounding liver cirrhosis. Jpn. J. Clin. Oncol. 2000, 30, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.D.W.; George, J.; Qiao, L. From MAFLD to hepatocellular carcinoma and everything in between. Chin. Med. J. 2022, 135, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Elzehery, R.; Effat, N.; El Farahaty, R.; Elsayed Farag, R.; Abo-Hashem, E.M.; Elhelaly, R. Circulating Cell-Free DNA and DNA Integrity as Molecular Diagnostic Tools in Hepatocellular Carcinoma. Am. J. Clin. Pathol. 2022, 158, 254–262. [Google Scholar] [CrossRef]
- Reggiardo, R.E.; Maroli, S.V.; Peddu, V.; Davidson, A.E.; Hill, A.; LaMontagne, E.; Aaraj, Y.A.; Jain, M.; Chan, S.Y.; Kim, D.H. Profiling of repetitive RNA sequences in the blood plasma of patients with cancer. Nat. Biomed. Eng. 2023, 7, 1627–1635. [Google Scholar] [CrossRef]
- Pal, D.; Patel, M.; Boulet, F.; Sundarraj, J.; Grant, O.A.; Branco, M.R.; Basu, S.; Santos, S.D.M.; Zabet, N.R.; Scaffidi, P.; et al. H4K16ac activates the transcription of transposable elements and contributes to their cis-regulatory function. Nat. Struct. Mol. Biol. 2023, 30, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Galaxy, C. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef]
- Jones, R.B.; Song, H.; Xu, Y.; Garrison, K.E.; Buzdin, A.A.; Anwar, N.; Hunter, D.V.; Mujib, S.; Mihajlovic, V.; Martin, E.; et al. LINE-1 retrotransposable element DNA accumulates in HIV-1-infected cells. J. Virol. 2013, 87, 13307–13320. [Google Scholar] [CrossRef]
- Papasotiriou, I.; Pantopikou, K.; Apostolou, P. L1 retrotransposon expression in circulating tumor cells. PLoS ONE 2017, 12, e0171466. [Google Scholar] [CrossRef]
- Nguyen-Khac, E.; Thiele, M.; Voican, C.; Nahon, P.; Moreno, C.; Boursier, J.; Mueller, S.; de Ledinghen, V.; Starkel, P.; Gyune Kim, S.; et al. Non-invasive diagnosis of liver fibrosis in patients with alcohol-related liver disease by transient elastography: An individual patient data meta-analysis. Lancet Gastroenterol. Hepatol. 2018, 3, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
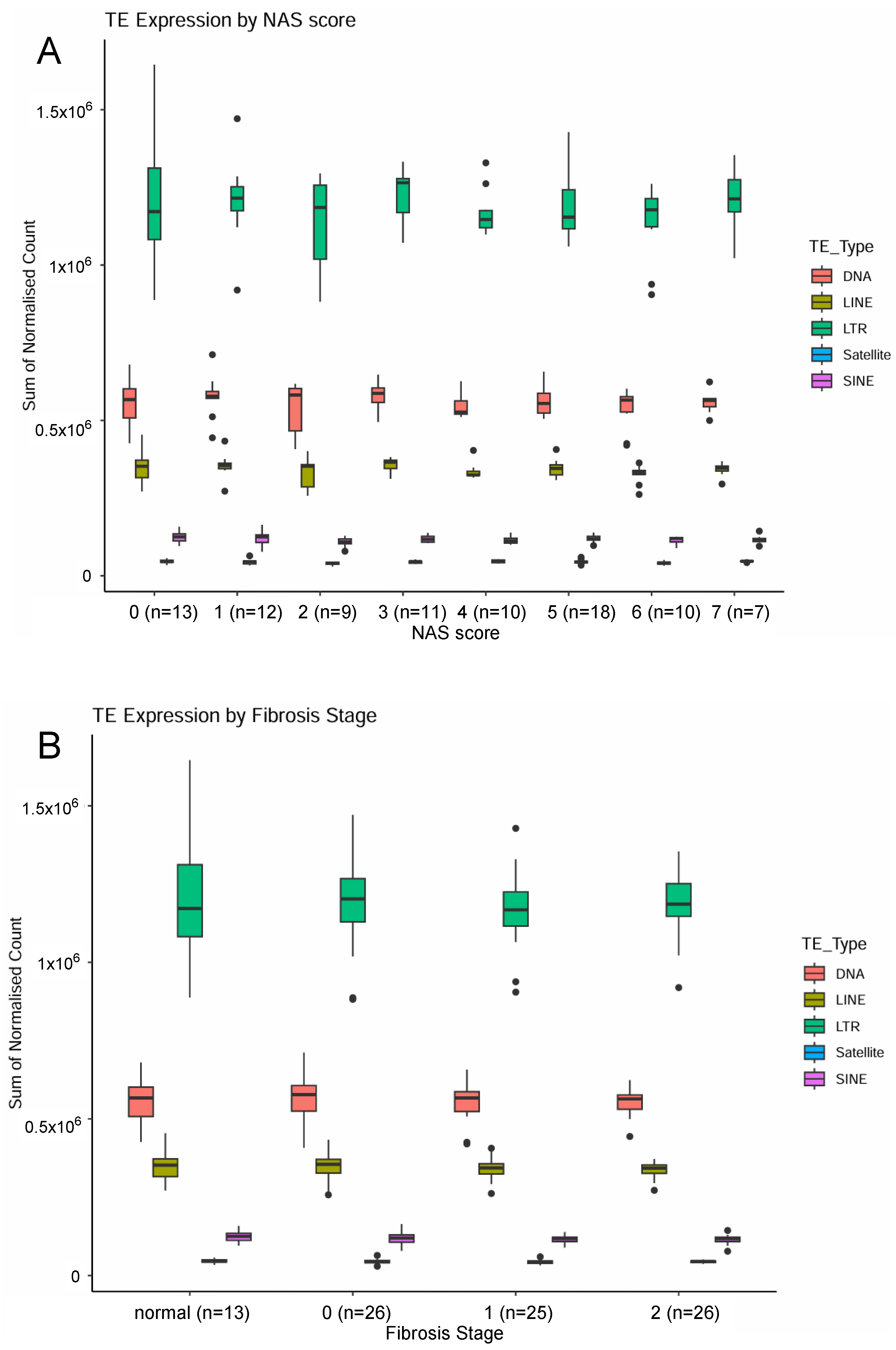
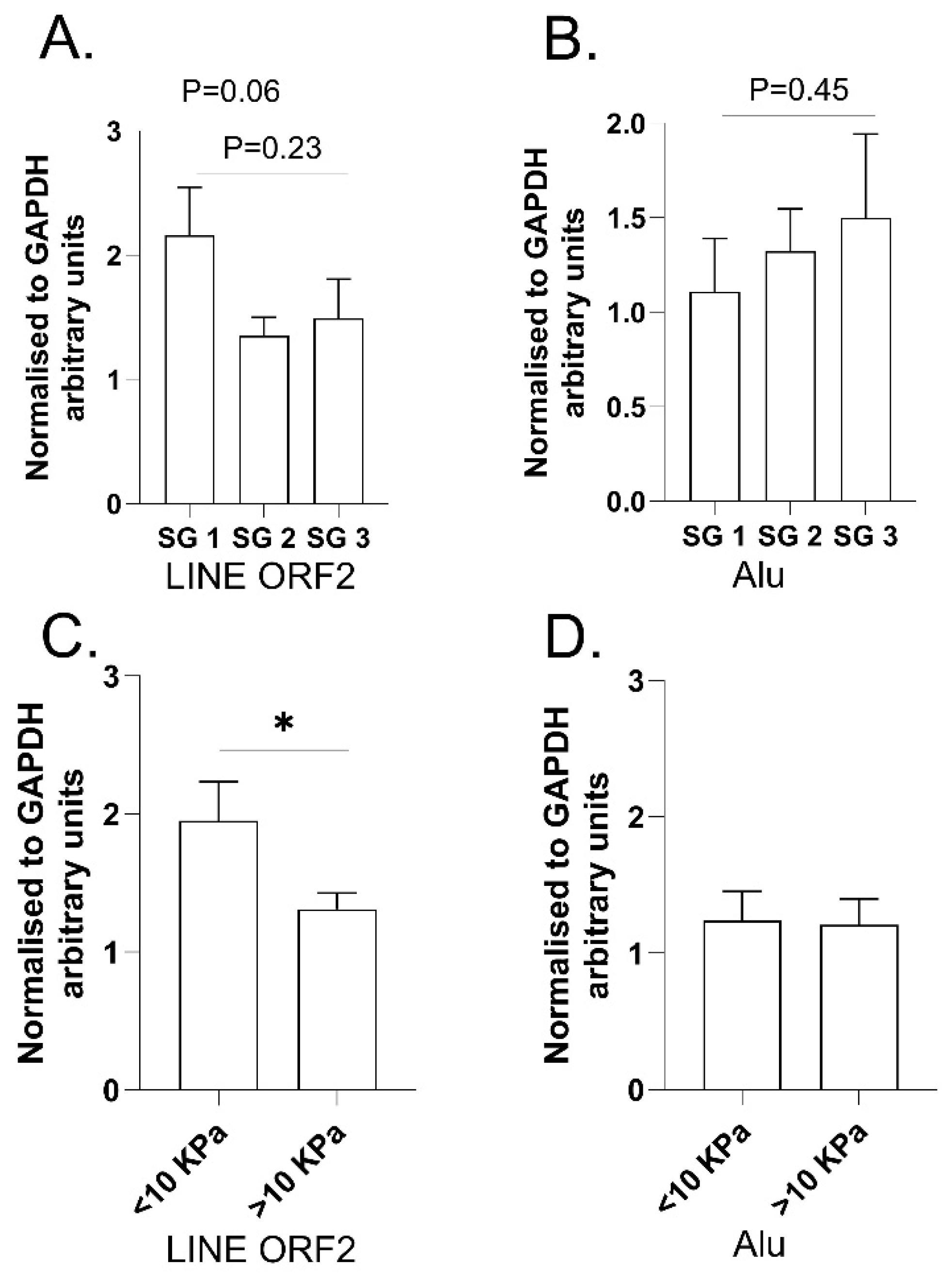

{kind=link}
{kind=link}
| log2 Fold Change | lfcSE | padj | |
|---|---|---|---|
| Fibrosis normal vs. 0 | |||
| MIRc:MIR:SINE | −0.58 | 0.18 | 0.03 |
| Charlie22a:hAT-Charlie:DNA | −0.54 | 0.17 | 0.03 |
| MER96:hAT-Tip100:DNA | −0.66 | 0.21 | 0.03 |
| Fibrosis normal vs. 1 | |||
| MER50C:ERV1:LTR | −1.06 | 0.31 | 0.01 |
| HERV35I-int:ERV1:LTR | −0.63 | 0.19 | 0.02 |
| Charlie4:hAT-Charlie:DNA | −0.59 | 0.19 | 0.03 |
| MIRc:MIR:SINE | −0.58 | 0.18 | 0.03 |
| NAS low (0–3) vs. NAS high (4–6) | |||
| CR1-13_AMi:CR1:LINE | 1.39 | 0.39 | 0.01 |
| MER50B:ERV1:LTR | 0.79 | 0.26 | 0.03 |
| MLT1C-int:ERVL-MaLR:LTR | 0.85 | 0.28 | 0.03 |
| Non-Alcoholic Fatty Liver Activity Score (NAS) in MASLD Patients | |||
|---|---|---|---|
| Normal Liver | 1–3 | ≥5 | |
| Alu | (n = 12) 0.006490 (0–0.023670) | (n = 25) 0.007753 (0–0.154509) | (n = 12) 0.006865 (0–0.154500) |
| L1 | (n = 12) 0.000269 (0–0.003355) | (n = 25) 0.000163 (0–0.003606) | (n = 12) 0.000298 (0.000014–0.004378) |
| L1 Methylation | (n = 21) 50.6 ± 4.9 | (n = 6) 58.8 ± 10.9 | (n = 10) 34.6 ± 7.0 1,2 |
| Steatosis Grade in MASLD Patients | ||||
|---|---|---|---|---|
| Normal Liver | 1 | 2 | 3 | |
| Alu | n = 12 0.006490 (0–0.023670) | (n = 8) 0.006752 (0–0.107100) | (n = 22) 0.006865 (0–0.154500) | (n = 7) 0.000702 1,2,3 (0–0.004928) |
| L1 | (n = 12) 0.000269 (0–0.003355) | (n = 8) 0.000069 (0–0.003606) | (n = 22) 0.000195 (0–0.00438) | (n = 7) 0.000456 (0–0.001186) |
| L1 Methylation | (n = 21) 50.6 ± 4.9 | # | (n = 6) 60.1 ± 10.8 | (n = 8) 30.2 ± 7.9 4,5 |
| Fibrosis Stage in MASLD Patients | |||
|---|---|---|---|
| Normal Liver | 0–2 | 3–4 | |
| Alu | (n = 12) 0.006490 (0–0.023670) | (n = 24) 0.006064 (0–0.069150) | (n = 13) 0.004928 (0–0.107100) |
| L1 | (n = 12) 0.000269 (0–0.003355) | (n = 24) 0.0002072 (0–0.004378) | (n = 13) 0.000203 (0.000014–0.003606) |
| L1 Methylation | (n = 21) 43.7 (13.0–96.1) | (n = 6) 54.4 (7.6–81.3) | (n = 10) 46.3 (8.9–79.5) |
| HCC Presence Reduces Alu Transcript Levels in Background Liver Tissue in MASLD Patients (HCC Tissue from Same Patients in Last Column for Comparison) | |||
|---|---|---|---|
| Liver Tissue in MASLD Patients Without HCC | Liver Tissue in MASLD Patients with HCC | MASLD-Associated HCC Tissue | |
| Alu | (n = 30) 0.007846 (0.000115–0.154500) | (n = 7) 0.0001524 (0–0.040070) 1 | (n = 11) 0.003860 (0–0.015690) 4 |
| Alu (excluding Steatosis Grade 3) | (n = 25) 0.007753 (0.000115–0.154500) | (n = 5) 0.0001524 (0–0.040070) 2 | N/A |
| L1 | (n = 30) 0.000273 (0–0.004378) | (n = 7) 0.000168 (0–0.002724) | (n = 12) 0.000282 (0–0.006193) |
| L1 Methylation | (n = 4) 12.4 (8.9–48.2) # | (n = 12) 54.2 (7.6–81.3) 3 | (n = 13) 35.6 (6.2–80.6) |
| r | p | |
|---|---|---|
| Overall (n = 28) | −0.62 | 0.0004 |
| Normal (n = 7) | −0.56 | 0.21 |
| MASLD (n = 10) | −0.78 | 0.01 |
| HCC (n = 11) | −0.52 | 0.11 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Youngson, N.A.; Tourna, A.; Chalmers, T.; Prates, K.V.; Argemi, J.; Bataller, R.; Haghighi, K.S.; Wu, L.E.; Chokshi, S.; Starkel, P.; et al. Multi-Cohort Exploration of Repetitive Element Transcription and DNA Methylation in Human Steatotic Liver Disease. Int. J. Mol. Sci. 2025, 26, 5494. https://doi.org/10.3390/ijms26125494
Youngson NA, Tourna A, Chalmers T, Prates KV, Argemi J, Bataller R, Haghighi KS, Wu LE, Chokshi S, Starkel P, et al. Multi-Cohort Exploration of Repetitive Element Transcription and DNA Methylation in Human Steatotic Liver Disease. International Journal of Molecular Sciences. 2025; 26(12):5494. https://doi.org/10.3390/ijms26125494
Chicago/Turabian StyleYoungson, Neil A., Aikaterini Tourna, Timothy Chalmers, Kelly V. Prates, Josepmaria Argemi, Ramon Bataller, Koroush S. Haghighi, Lindsay E. Wu, Shilpa Chokshi, Peter Starkel, and et al. 2025. "Multi-Cohort Exploration of Repetitive Element Transcription and DNA Methylation in Human Steatotic Liver Disease" International Journal of Molecular Sciences 26, no. 12: 5494. https://doi.org/10.3390/ijms26125494
APA StyleYoungson, N. A., Tourna, A., Chalmers, T., Prates, K. V., Argemi, J., Bataller, R., Haghighi, K. S., Wu, L. E., Chokshi, S., Starkel, P., Western, P. S., Morris, M. J., & Riordan, S. M. (2025). Multi-Cohort Exploration of Repetitive Element Transcription and DNA Methylation in Human Steatotic Liver Disease. International Journal of Molecular Sciences, 26(12), 5494. https://doi.org/10.3390/ijms26125494