
Characterisation of the ABO Blood Group Phenotypes Using Third-Generation Sequencing

 , , and
, , and
Abstract
1. Introduction
2. Results and Discussion
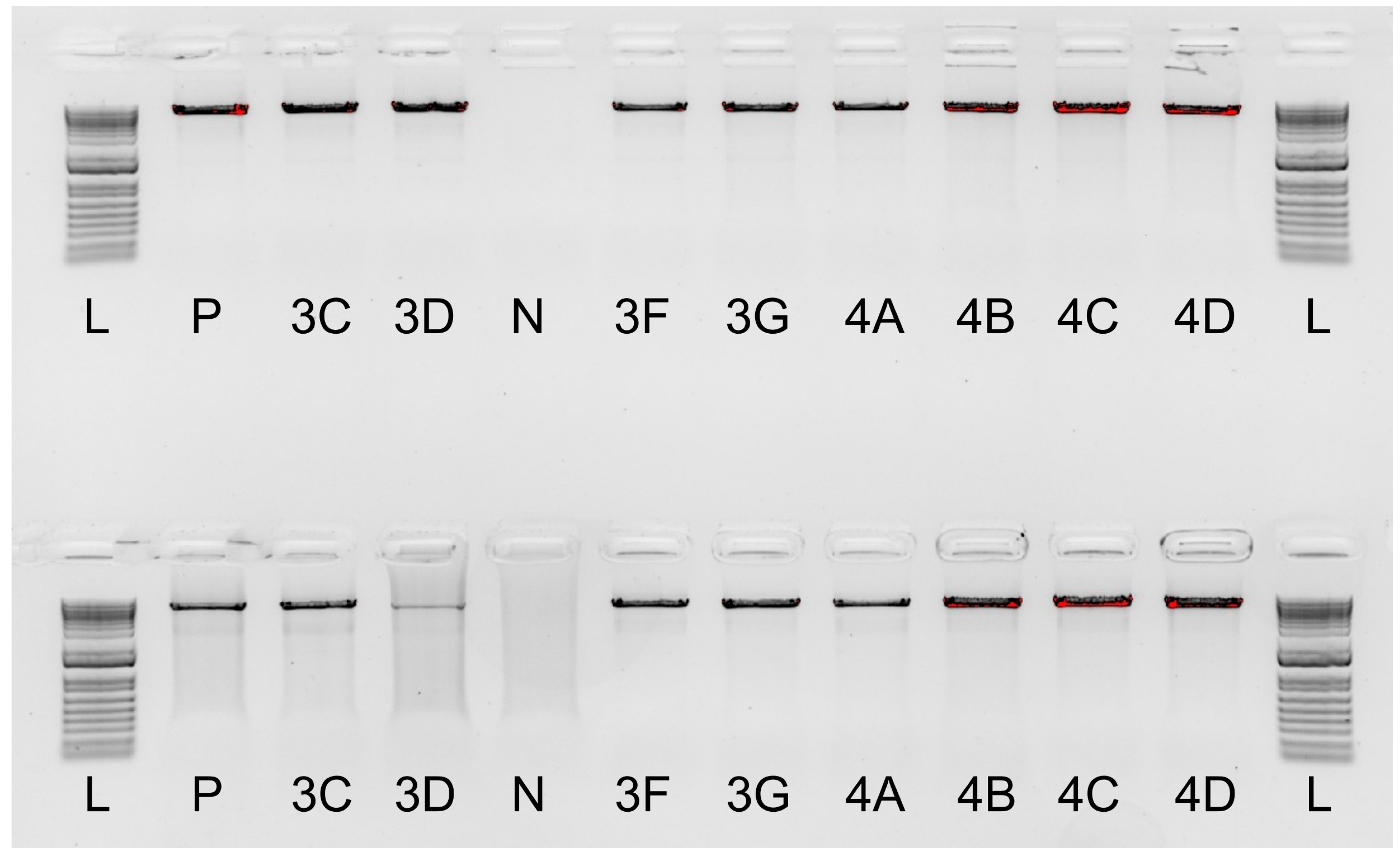
2.1. Optimising Laboratory Assays
2.2. DNA Sequencing and ABO Phenotyping
2.3. Orchestration and Benchmarking Against the Validation Criteria
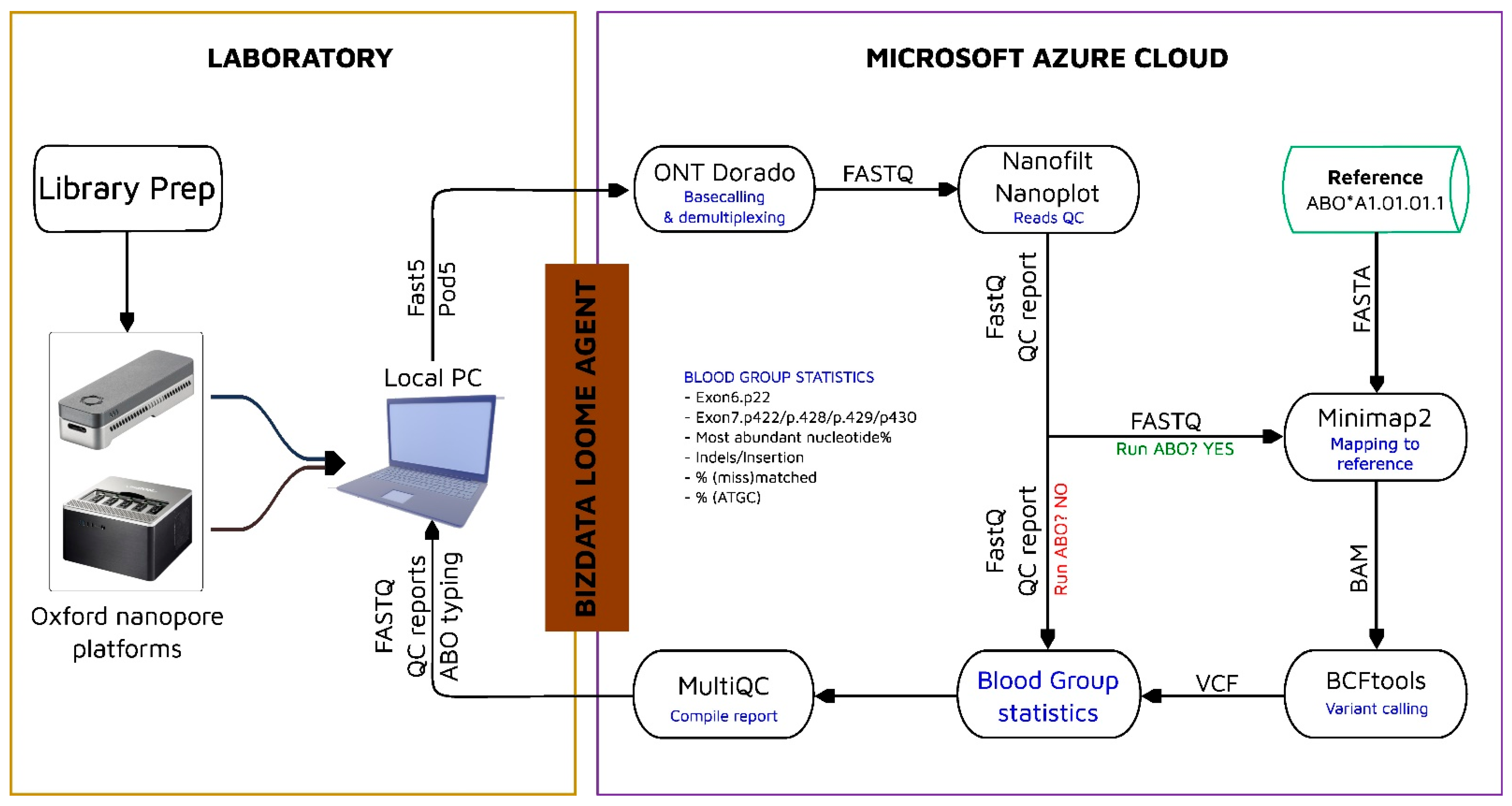
3. Materials and Methods
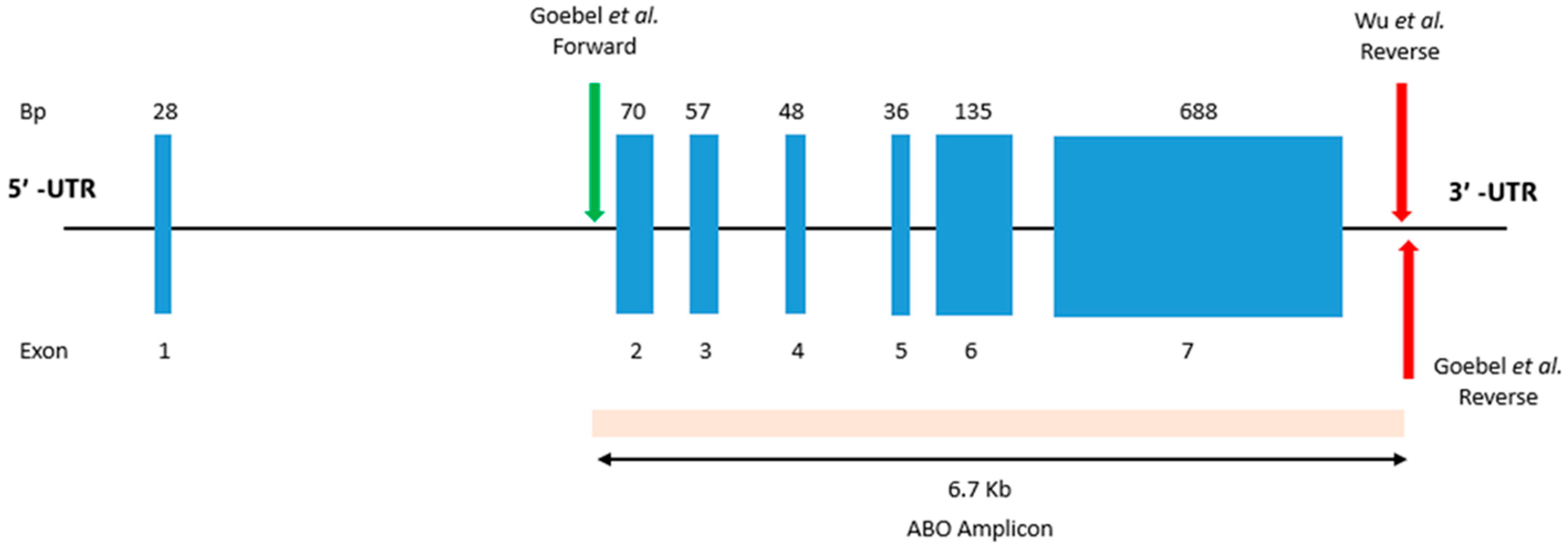
3.1. Primer Validation and Library Preparation
3.2. Sequencing and Base Calling
3.3. Reads Mapping and Variants Detection
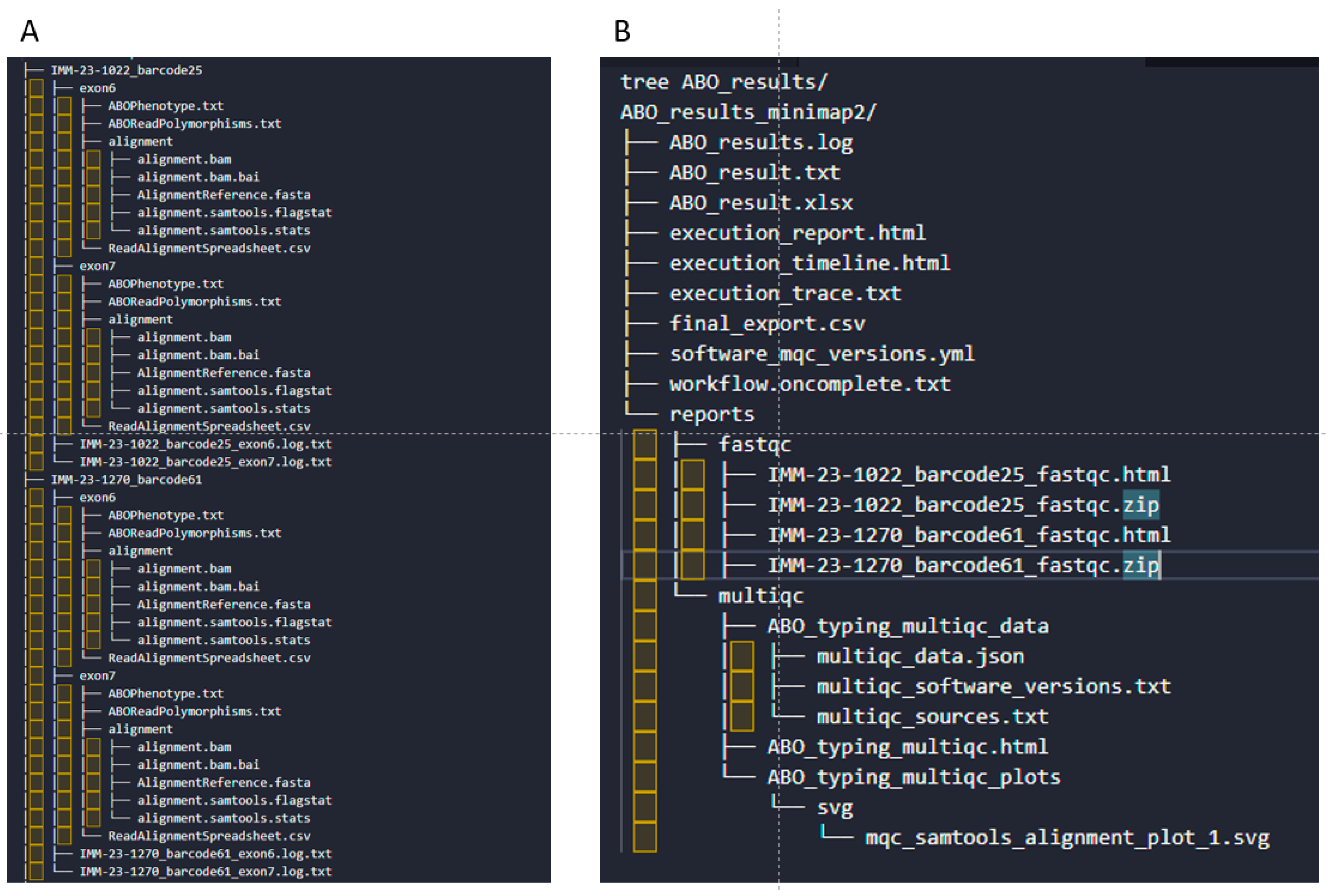
3.4. ABO Variant Quantification, ABO Phenotyping, and Reporting
3.5. The Core Code, Pipelining, and Deployment
4. Conclusions
Limitations and Future Directions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Storry, J.R.; Olsson, M.L. The ABO blood group system revisited: A review and update. Immunohematology 2009, 25, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Pushpa, B.; Shanmugha Priya, R.A.; Kumar, U.S.; Saminathan, J. Understanding the Immunological Significance of Blood Groups in Organ Transplantation. In Innate Immunity—New Perspectives and Therapeutic Opportunities; Lasfar, A., Ed.; IntechOpen: Rijeka, Croatia, 2024. [Google Scholar] [CrossRef]
- Miserre, L.; Wienzek-Lischka, S.; Mann, A.; Cooper, N.; Santoso, S.; Ehrhardt, H.; Sachs, U.J.; Bein, G. ABO Incompatibility between the Mother and Fetus Does Not Protect against Anti-Human Platelet Antigen-1a Immunization by Pregnancy. J. Clin. Med. 2022, 11, 6811. [Google Scholar] [CrossRef] [PubMed]
- Kollman, C.; Spellman, S.R.; Zhang, M.J.; Hassebroek, A.; Anasetti, C.; Antin, J.H.; Champlin, R.E.; Confer, D.L.; DiPersio, J.F.; Fernandez-Vina, M.; et al. The effect of donor characteristics on survival after unrelated donor transplantation for hematologic malignancy. Blood 2016, 127, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Goebel, M.; Halm-Heinrich, I.; Parkner, A.; Rink, G.; Heim, M.U.; Bugert, P. A Novel ABO Gene Variant Leads to Discrepant Results in Forward/Reverse and Molecular Blood Grouping. Transfus. Med. Hemother. 2013, 40, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, F.; Clausen, H.; White, T.; Marken, J.; Hakomori, S. Molecular genetic basis of the histo-blood group ABO system. Nature 1990, 345, 229–233. [Google Scholar] [CrossRef]
- Yamamoto, F.; McNeill, P.D.; Hakomori, S. Genomic organization of human histo-blood group ABO genes. Glycobiology 1995, 5, 51–58. [Google Scholar] [CrossRef]
- Chester, M.A.; Olsson, M.L. The ABO blood group gene: A locus of considerable genetic diversity. Transfus. Med. Rev. 2001, 15, 177–200. [Google Scholar] [CrossRef]
- Matern, B.; Groeneweg, M.; Slangen, T.; Tilanus, M.G.; Voorter, C. P095 ABO blood group typing with the Oxford nanopore minion. Hum. Immunol. 2017, 78, 122. [Google Scholar] [CrossRef]
- Thun, G.A.; Gueuning, M.; Mattle-Greminger, M. Long-Read Sequencing in Blood Group Genetics. Transfus. Med. Hemother. 2023, 50, 184–197. [Google Scholar] [CrossRef]
- Wu, P.C.; Lin, Y.H.; Tsai, L.F.; Chen, M.H.; Chen, P.L.; Pai, S.C. ABO genotyping with next-generation sequencing to resolve heterogeneity in donors with serology discrepancies. Transfusion 2018, 58, 2232–2242. [Google Scholar] [CrossRef]
- Gueuning, M.; Thun, G.A.; Wittig, M.; Galati, A.L.; Meyer, S.; Trost, N.; Gourri, E.; Fuss, J.; Sigurdardottir, S.; Merki, Y.; et al. Haplotype sequence collection of ABO blood group alleles by long-read sequencing reveals putative A1-diagnostic variants. Blood Adv. 2023, 7, 878–892. [Google Scholar] [CrossRef]
- Lang, K.; Wagner, I.; Schone, B.; Schofl, G.; Birkner, K.; Hofmann, J.A.; Sauter, J.; Pingel, J.; Bohme, I.; Schmidt, A.H.; et al. ABO allele-level frequency estimation based on population-scale genotyping by next generation sequencing. BMC Genom. 2016, 17, 374. [Google Scholar] [CrossRef]
- Martinez, K.L.; Klein, A.; Martin, J.R.; Sampson, C.U.; Giles, J.B.; Beck, M.L.; Bhakta, K.; Quatraro, G.; Farol, J.; Karnes, J.H. Disparities in ABO blood type determination across diverse ancestries: A systematic review and validation in the All of Us Research Program. J. Am. Med. Inform. Assoc. 2024, 31, 3022–3031. [Google Scholar] [CrossRef]
- Rieneck, K.; Egeberg Hother, C.; Clausen, F.B.; Jakobsen, M.A.; Bergholt, T.; Hellmuth, E.; Gronbeck, L.; Dziegiel, M.H. Next Generation Sequencing-Based Fetal ABO Blood Group Prediction by Analysis of Cell-Free DNA from Maternal Plasma. Transfus. Med. Hemother. 2020, 47, 45–53. [Google Scholar] [CrossRef]
- Yip, S.P. Sequence variation at the human ABO locus. Ann. Hum. Genet. 2002, 66, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Storry, J.R. Nomenclature for red blood cell blood group alleles. Transfusion 2013, 53, 2844–2846. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef]
- Di Tommaso, P.; Chatzou, M.; Floden, E.W.; Barja, P.P.; Palumbo, E.; Notredame, C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef]
- Langer, B.E.; Amaral, A.; Baudement, M.-O.; Bonath, F.; Charles, M.; Chitneedi, P.K.; Clark, E.L.; Di Tommaso, P.; Djebali, S.; Ewels, P.A.; et al. Empowering bioinformatics communities with Nextflow and nf-core. bioRxiv 2024. [Google Scholar] [CrossRef]
- Ewels, P.A.; Peltzer, A.; Fillinger, S.; Patel, H.; Alneberg, J.; Wilm, A.; Garcia, M.U.; Di Tommaso, P.; Nahnsen, S. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Andrews, S.; Krueger, F.; Segonds-Pichon, A.; Biggins, L.; Krueger, C.; Wingett, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; Babraham Institute: Babraham, UK, 2010. [Google Scholar]
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Truong, L.; Ayora, F.; D’Orsogna, L.; Martinez, P.; De Santis, D. Nanopore sequencing data analysis using Microsoft Azure cloud computing service. PLoS ONE 2022, 17, e0278609. [Google Scholar] [CrossRef]
- Montgomery, M.C.; Petraroia, R.; Weimer, E.T. Buccal swab genomic DNA fragmentation predicts likelihood of successful HLA genotyping by next-generation sequencing. Hum. Immunol. 2017, 78, 634–641. [Google Scholar] [CrossRef]
- El-Lagta, N.; Truong, L.; Ayora, F.; Mobegi, F.; Bruce, S.; Martinez, P.; D’Orsogna, L.; De Santis, D. Revolutionising High Resolution HLA Genotyping for Transplant Assessment: Validation, Implementation and Challenges of Oxford Nanopore Technologies’ Q20(+) Sequencing. HLA 2024, 104, e15725. [Google Scholar] [CrossRef]
- Samarakoon, H.; Ferguson, J.M.; Jenner, S.P.; Amos, T.G.; Parameswaran, S.; Gamaarachchi, H.; Deveson, I.W. Flexible and efficient handling of nanopore sequencing signal data with slow5tools. Genome Biol. 2023, 24, 69. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- De Coster, W.; Rademakers, R. NanoPack2: Population-scale evaluation of long-read sequencing data. Bioinformatics 2023, 39, btad311. [Google Scholar] [CrossRef] [PubMed]
- The International Society of Blood Transfusion. Names for ABO (ISBT 001) Blood Group Alleles; The International Society of Blood Transfusion: Amsterdam, The Netherlands, 2017; Volume v1.2, pp. 1–17. [Google Scholar]
- Bonfield, J.K.; Marshall, J.; Danecek, P.; Li, H.; Ohan, V.; Whitwham, A.; Keane, T.; Davies, R.M. HTSlib: C library for reading/writing high-throughput sequencing data. Gigascience 2021, 10, giab007. [Google Scholar] [CrossRef] [PubMed]
- Merkel, D. Docker: Lightweight linux containers for consistent development and deployment. Linux J. 2014, 239, 2. [Google Scholar]
- Kadri, S.; Sboner, A.; Sigaras, A.; Roy, S. Containers in Bioinformatics: Applications, Practical Considerations, and Best Practices in Molecular Pathology. J. Mol. Diagn. 2022, 24, 442–454. [Google Scholar] [CrossRef]
- Bisen, S.S.; Getsin, S.N.; Chiang, P.Y.; Herrick-Reynolds, K.; Zeiser, L.B.; Yu, S.; Desai, N.M.; Al Ammary, F.; Jackson, K.R.; Segev, D.L.; et al. Patient and Graft Survival After A1/A2-incompatible Living Donor Kidney Transplantation. Transplant. Direct 2022, 8, e1388. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PrimerID a | Gene Location | Chromosome Location | Sequence (5′-3′) |
|---|---|---|---|
| ABO IN1 Forward Geobel | ABO Intron 1 | 9q34.2 | GACCATCTTGGCAGATGAAGG |
| ABO 3UTR Reverse Geobel | ABO 3′ UTR | 9q34.2 | GCCTAGGCTTCAGTTACTCAC |
| ABO 3UTR Reverse Wu | ABO 3′ UTR | 9q34.2 | GGGCCTAGGCTTCAGTTACTC |
| Purpose | Tool | Version |
|---|---|---|
| Reads QC | FastQC | 0.12.1 |
| Local alignment of FASTQ to reference | BWA | 0.7.18 |
| Minimap2 | 2.28 | |
| Processing BAM files | BCFtools | 1.21 |
| SAMtools | 1.21 | |
| htslib | 1.21 | |
| Quantifying variants | Python + pip packages | 3.10 |
| Bio | 1.7.1 | |
| BioPython | 1.83 | |
| openpyxl | 3.1.5 | |
| pandas | 2.2.3 | |
| pysam | 0.22.1 | |
| xlsxwriter | 3.2.2 | |
| Reporting | MultiQC | 1.25.1 |
| Reference SNP ID | rs8176719 | rs8176746 | rs41302905 | rs8176747 | rs782782485 | |
|---|---|---|---|---|---|---|
| Exon | 6 | 7 | 7 | 7 | 7 | |
| Position exon (gene) | 22(261) | 422(796) | 428(802) | 429(803) | 431(805) | |
| Position GRCh38.p14 | chr9:133257521-133257522 | chr9:133255935 | chr9:133255929 | chr9:133255928 | chr9:133255898 | |
| Amino acid change | p.Thr88Profs | p.Leu266Met | p.Gly268Arg | p.Gly268Ala/p.Gly268Val | p.Phe269Valfs*124 | |
| Nucleotide change | c.261delG/c.260_262insG | c.796C>A | c.802G>A/c.802G>C | c.803G>C/c.803G>T | c.804dupG | |
| Consequence | frameshift | missense | missense | missense | frameshift | |
| Phenotype | O1 | Deletion | C | G | G | T |
| O2 | G | C | A | G | T | |
| O3 | G | C | G | G | G | |
| A | G | C | G | G | T | |
| AB | G | C/A | G | G/C | T | |
| B | G | A | G | C | T | |
| Genotype | O1A | Del 50%/G 50% | C 100% | G 100% | G 100% | T 100% |
| O2A | G 100% | C 100% | A 50%/G 50% | G 100% | T 100% | |
| O3A | G 100% | C 100% | G 100% | G 100% | G 50%/T 50% | |
| O1B | Del 50%/G 50% | C 50%/A 50% | G 100% | G 50%/C 50% | T 100% | |
| O2B | G 100% | C 50%/A 50% | A 50%/G 50% | G 50%/C 50% | T 100% | |
| O3B | G 100% | C 50%/A 50% | G 100% | G 50%/C 50% | G 50%/T 50% | |
| O1O2 | Del 50%/G 50% | C 100% | A 50%/G 50% | G 100% | T 100% | |
| O1O3 | Del 50%/G 50% | C 100% | G 100% | G 100% | G 50%/T 50% | |
| O2O3 | G 100% | C 100% | A 50%/G 50% | G 100% | G 50%/T 50% | |
| O1O1 | Del 100% | C 100% | G 100% | G 100% | T 100% | |
| O2O2 | G 100% | C 100% | A 100% | G 100% | T 100% | |
| O3O3 | G 100% | C 100% | G 100% | G 100% | G 100% | |
| AA | G 100% | C 100% | G 100% | G 100% | T 100% | |
| BB | G 100% | A 100% | G 100% | C 100% | T 100% | |
| AB | G 100% | C 50%/A 50% | G 100% | G 50%/C 50% | T 100% | |
| Sample | Azure Production Pipeline | Azure Test Pipeline | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Reads | Phenotype | Genotype | Extended Genotype | Reads | Phenotype | Genotype | Extended Genotype | Reliability | |
| Sample0040 | 8 | O | OO | O1O1 | 8 | O | OO | O1O1 | Very Low (≤20 reads) |
| Sample0044 | 72 | O | OO | O1O1 | 72 | O | OO | O1O1 | |
| Sample0048 | 16 | O | OO | O1O1 | 6 | O | OO | O1O1 | Very Low (≤20 reads) |
| Sample0049 | 732 | O | OO | O1O1 | 727 | O | OO | O1O1 | Robust (≥500 reads) |
| Sample0085 | 2678 | O | OO | O1O1 | 2677 | O | OO | O1O1 | Robust (≥500 reads) |
| Sample0087 | 947 | A | AO | AO1 | 943 | A | AO | AO1 | Robust (≥500 reads) |
| Sample0088 | 2547 | B | BO | BO1 | 2539 | B | BO | BO1 | Robust (≥500 reads) |
| Sample0089 | 1298 | O | OO | O1O1 | 1298 | O | OO | O1O1 | Robust (≥500 reads) |
| Sample0091 | 848 | O | OO | O1O1 | 851 | O | OO | O1O1 | Robust (≥500 reads) |
| Sample0092 | 1448 | O | OO | O1O1 | 1447 | O | OO | O1O1 | Robust (≥500 reads) |
| Sample0093 | 406 | A | AO | AO1 | 405 | A | AO | AO1 | |
| Sample0097 | 3512 | O | OO | O1O1 | 3521 | O | OO | O1O1 | Robust (≥500 reads) |
| Sample0098 | 1707 | O | OO | O1O1 | 1712 | O | OO | O1O1 | Robust (≥500 reads) |
| Sample0099 | 239 | O | OO | O1O1 | 240 | O | OO | O1O1 | |
| Sample0100 | 1852 | A | AO | AO1 | 1854 | A | AO | AO1 | Robust (≥500 reads) |
| Sample0101 | 1203 | O | OO | O1O2 | 1204 | O | OO | O1O2 | Robust (≥500 reads) |
| Sample0102 | 194 | A | AO | AO1 | 193 | A | AO | AO1 | |
| Sample0103 | 651 | O | OO | O1O1 | 651 | O | OO | O1O1 | Robust (≥500 reads) |
| Sample0105 | 1173 | O | OO | O1O2 | 1172 | O | OO | O1O2 | Robust (≥500 reads) |
| Sample0106 | 35 | B | BO | BO1 | 35 | B | BO | BO1 | Low (≤50 reads) |
| Sample0107 | 18 | O | OO | O1O1 | 18 | O | OO | O1O1 | Very Low (≤20 reads) |
| Sample0110 | 1191 | B | BO | BO1 | 1191 | B | BO | BO1 | Robust (≥500 reads) |
| Sample0111 | 26 | O | OO | O1O1 | 26 | O | OO | O1O1 | Very Low (≤20 reads) |
| Sample0112 | 357 | A | AO | AO1 | 358 | A | AO | AO1 | |
| Sample0113 | 943 | A | AA | AA | 945 | A | AA | AA | Robust (≥500 reads) |
| Sample0114 | 314 | O | OO | O1O1 | 313 | O | OO | O1O1 | |
| Sample0115 | 1286 | O | OO | O1O1 | 1286 | O | OO | O1O1 | Robust (≥500 reads) |
| Sample0116 | 32 | A | AO | AO1 | 32 | A | AO | AO1 | Low (≤50 reads) |
| Sample0117 | 13 | O | OO | O1O1 | 13 | O | OO | O1O1 | Very Low (≤20 reads) |
| Sample0119 | 115 | O | OO | O1O1 | 113 | O | OO | O1O1 | |
| Sample0120 | 1037 | O | OO | O1O1 | 1039 | O | OO | O1O1 | Robust (≥500 reads) |
| Sample | Reads | Exon 6 (pos 22) | Exon 7 (pos 422) | Exon 7 (pos 428) | Exon 7 (pos 429) | Exon 7 (pos 431) | 3SNV Phenotype | 3SNV Genotype | 5SNV Phenotype | 5SNV Genotype | Expected | Concordance |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample0054 | 824 | 50%G/50%Del | 100%C | 50%G/50%A | 100%G | 100%T | A | AO | O | OO | O | Yes |
| Sample0061 | 1684 | 50%G/50%Del | 100%C | 50%G/50%A | 100%G | 100%T | A | AO | O | OO | O | Yes |
| Sample0071 | 432 | 100%G | 50%C/50%A | 50%G/50%A | 50%G/50%C | 100%T | AB | AB | B | OB | B | Yes |
| Sample0081 | 117 | 50%G/50%Del | 100%C | 50%G/50%A | 100%G | 100%T | A | AO | O | OO | O | Yes |
| Sample0101 | 575 | 50%G/50%Del | 100%C | 50%G/50%A | 100%G | 100%T | A | AO | O | OO | O | Yes |
| Sample0105 | 529 | 50%G/50%Del | 100%C | 50%G/50%A | 100%G | 100%T | A | AO | O | OO | O | Yes |
| Sample | Histogenetics (Molecular) | In-House (Molecular) | Serology | Concordance a | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sample0124 | OO | O | - | O- | No result | No result | No result | No result | No result | Very Low (≤20 reads) |
| Sample0121 | AO | A | + | A+ | No result | No result | No result | No result | No result | Very Low (≤20 reads) |
| Sample0118 | AO | A | + | A+ | No result | No result | No result | No result | No result | Very Low (≤20 reads) |
| Sample0117 | OO | O | - | O- | No result | No result | O | No result | No result | Very Low (≤20 reads) |
| Sample0107 | OO | O | + | O+ | No result | No result | O | No result | No result | Very Low (≤20 reads) |
| Sample0096 | OO | O | + | O+ | No result | No result | O | No result | No result | Very Low (≤20 reads) |
| Sample0090 | AO | A | + | A+ | No result | No result | A | No result | No result | Very Low (≤20 reads) |
| Sample0082 | No result | No result | No result | No result | AA | A | A | No result | No result | Robust (≥500 reads) |
| Sample0080 | No result | No result | No result | No result | OO | O | No result | No result | No result | |
| Sample1030 | AA | A | + | A+ | No result | No result | A | No result | No result | |
| Sample0113 | AO | A | + | A+ | AA | A | A | FALSE | TRUE | Robust (≥500 reads) |
| Sample0116 | AO | A | + | A+ | AO | A | No result | TRUE | TRUE | Low (≤50 reads) |
| Sample0115 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | Robust (≥500 reads) |
| Sample0114 | OO | O | - | O- | OO | O | O | TRUE | TRUE | |
| Sample0112 | AO | A | + | A+ | AO | A | A | TRUE | TRUE | |
| Sample0111 | OO | O | - | O- | OO | O | O | TRUE | TRUE | |
| Sample0110 | BO | B | + | B+ | BO | B | A | TRUE | TRUE | Robust (≥500 reads) |
| Sample0109 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | Low (≤50 reads) |
| Sample0108 | AO | A | + | A+ | AO | A | A | TRUE | TRUE | |
| Sample0104 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | |
| Sample0103 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | Robust (≥500 reads) |
| Sample0102 | AO | A | + | A+ | AO | A | No result | TRUE | TRUE | |
| Sample0101 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | Robust (≥500 reads) |
| Sample0100 | AO | A | + | A+ | AO | A | A | TRUE | TRUE | Robust (≥500 reads) |
| Sample0099 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | |
| Sample0098 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | Robust (≥500 reads) |
| Sample0097 | OO | O | - | O- | OO | O | O | TRUE | TRUE | Robust (≥500 reads) |
| Sample0095 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | |
| Sample0094 | OO | O | + | O+ | OO | O | No result | TRUE | TRUE | |
| Sample0093 | AO | A | + | A+ | AO | A | No result | TRUE | TRUE | |
| Sample0092 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | Robust (≥500 reads) |
| Sample0091 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | Robust (≥500 reads) |
| Sample0089 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | Robust (≥500 reads) |
| Sample0088 | BO | B | + | B+ | BO | B | B | TRUE | TRUE | Robust (≥500 reads) |
| Sample0087 | AO | A | + | A+ | AO | A | No result | TRUE | TRUE | Robust (≥500 reads) |
| Sample0086 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | |
| Sample0085 | OO | O | + | O+ | OO | O | No result | TRUE | TRUE | Robust (≥500 reads) |
| Sample0084 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | |
| Sample0083 | AA | A | - | A- | AA | A | A | TRUE | TRUE | Robust (≥500 reads) |
| Sample0081 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | |
| Sample0079 | AO | A | + | A+ | AO | A | A | TRUE | TRUE | |
| Sample0078 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | |
| Sample0077 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | |
| Sample0076 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | Robust (≥500 reads) |
| Sample0075 | OO | O | - | O- | OO | O | O | TRUE | TRUE | Low (≤50 reads) |
| Sample0074 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | |
| Sample0061 | OO | O | + | O+ | OO | O | O | TRUE | TRUE | Robust (≥500 reads) |
| Sample0054 | OO | O | - | O- | OO | O | O | TRUE | TRUE | Robust (≥500 reads) |
| Sample0020 | OO | O | - | O- | OO | O | O | TRUE | TRUE | |
| Sample0018 | BO | B | + | B+ | BO | B | B | TRUE | TRUE | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mobegi, F.M.; Bruce, S.; El-Lagta, N.; Ayora, F.; Matern, B.M.; Groeneweg, M.; D’Orsogna, L.J.; De Santis, D. Characterisation of the ABO Blood Group Phenotypes Using Third-Generation Sequencing. Int. J. Mol. Sci. 2025, 26, 5443. https://doi.org/10.3390/ijms26125443
Mobegi FM, Bruce S, El-Lagta N, Ayora F, Matern BM, Groeneweg M, D’Orsogna LJ, De Santis D. Characterisation of the ABO Blood Group Phenotypes Using Third-Generation Sequencing. International Journal of Molecular Sciences. 2025; 26(12):5443. https://doi.org/10.3390/ijms26125443
Chicago/Turabian StyleMobegi, Fredrick M., Samuel Bruce, Naser El-Lagta, Felipe Ayora, Benedict M. Matern, Mathijs Groeneweg, Lloyd J. D’Orsogna, and Dianne De Santis. 2025. "Characterisation of the ABO Blood Group Phenotypes Using Third-Generation Sequencing" International Journal of Molecular Sciences 26, no. 12: 5443. https://doi.org/10.3390/ijms26125443
APA StyleMobegi, F. M., Bruce, S., El-Lagta, N., Ayora, F., Matern, B. M., Groeneweg, M., D’Orsogna, L. J., & De Santis, D. (2025). Characterisation of the ABO Blood Group Phenotypes Using Third-Generation Sequencing. International Journal of Molecular Sciences, 26(12), 5443. https://doi.org/10.3390/ijms26125443