
Reversing Epigenetic Dysregulation in Neurodegenerative Diseases: Mechanistic and Therapeutic Considerations
 , , and
, , and
Abstract
1. Introduction
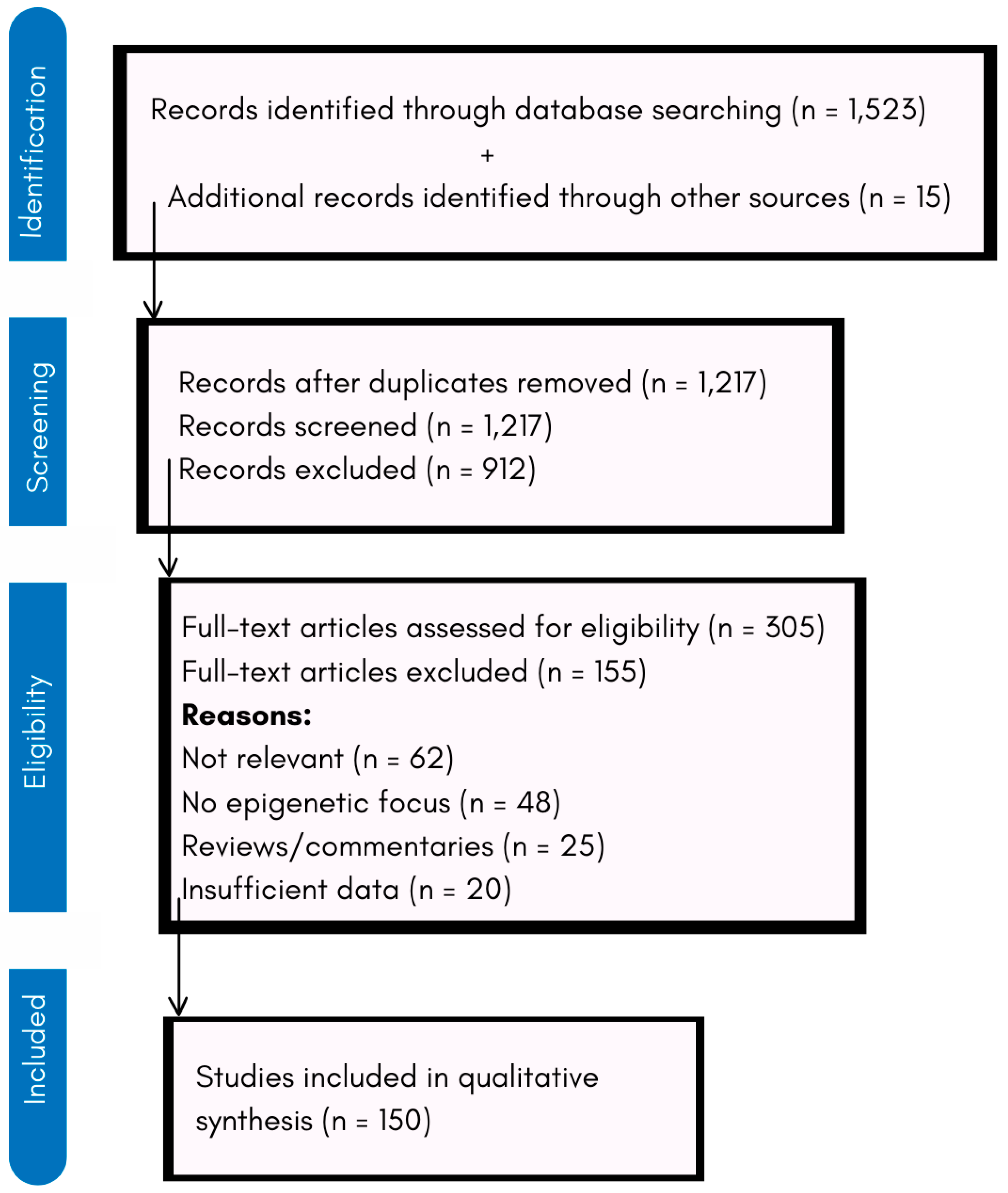
2. Methodology
2.1. Literature Search Strategy
2.2. Inclusion and Exclusion Criteria
- Inclusion Criteria
- ○
- Studies investigating the role of epigenetic mechanisms in NDDs.
- ○
- Preclinical (animal and cell culture) and clinical studies on HDAC and DNMT inhibitors.
- ○
- Research on environmental influences on epigenetic dysregulation in NDDs.
- ○
- Studies assessing biomarkers for epigenetic therapy efficacy.
- ○
- Articles published in English in peer-reviewed journals.
- Exclusion Criteria
- ○
- Studies with a primary focus on genetic mutations unrelated to epigenetics.
- ○
- Research on non-neurological diseases or non-mammalian models.
- ○
- Reviews, commentaries, or opinion articles without experimental data.
- ○
- Articles with limited methodological transparency or small sample sizes without statistical significance.
2.3. Data Extraction and Synthesis
3. Epigenetic Dysregulation in Neurodegeneration
3.1. Mechanisms of Epigenetic Dysregulation
3.2. Role of Environmental Toxins
4. Epigenetic Modulators as Therapeutic Agents
4.1. HDAC Inhibitors
4.2. DNMT Inhibitors
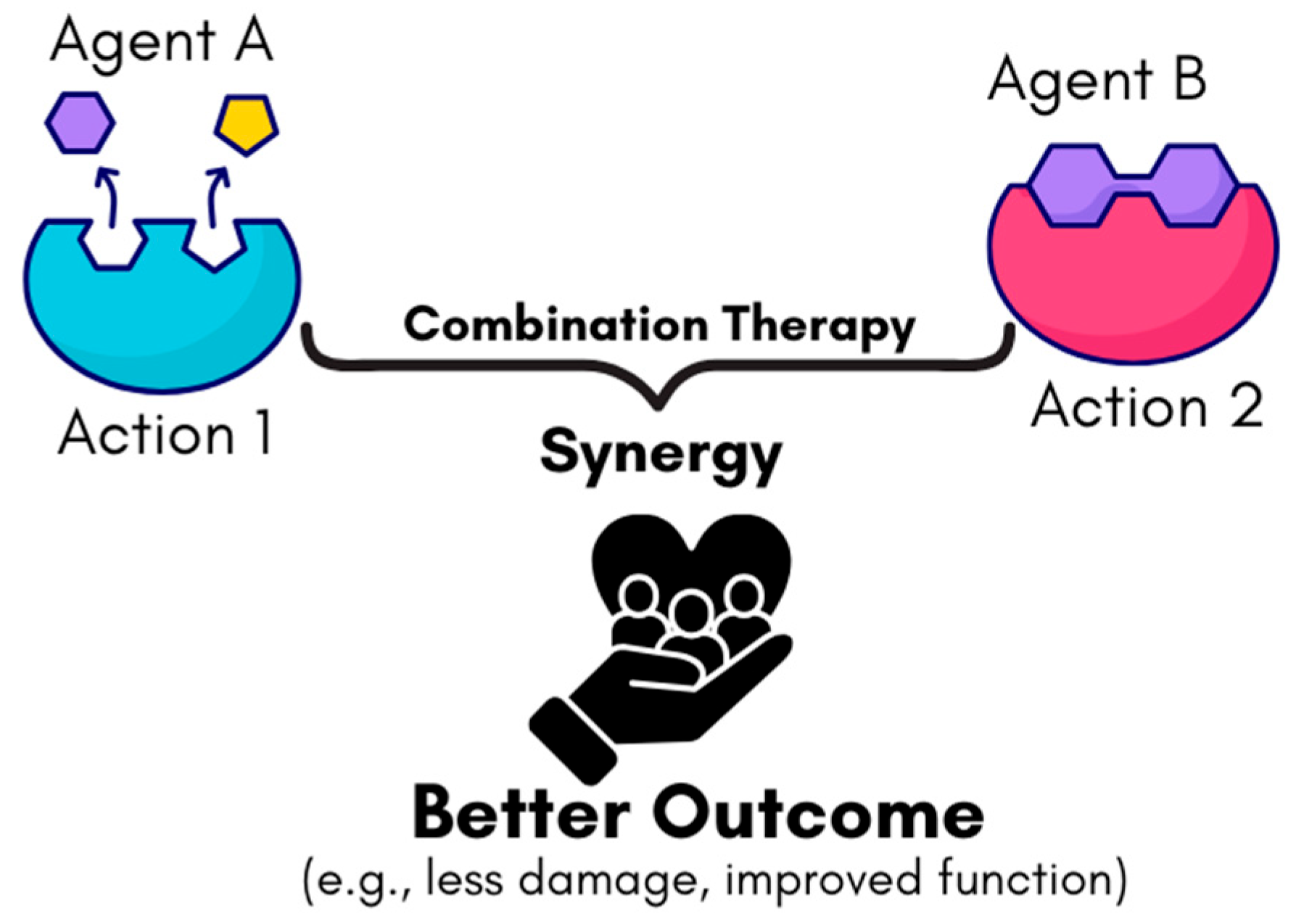
4.3. Combination Therapies
5. Preclinical and Clinical Trials
5.1. Preclinical Trials
5.2. Clinical Trials
6. One-Carbon Metabolism and Epigenetic Modulation in Neurodegeneration
7. Natural Products as Epigenetic Modulators in Neurodegeneration
7.1. Resveratrol
7.2. Curcumin
7.3. Epigallocatechin-3-Gallate (EGCG)
8. Challenges in Epigenetic Therapeutics
8.1. Tissue-Specific Targeting
8.2. Reversibility of Epigenetic Changes
8.3. Safety and Efficacy
9. Future Directions
9.1. Development of Brain-Specific Delivery Systems
9.2. Precision Medicine Approaches
9.3. Combination Therapies
9.4. Biomarker Development
10. Limitations of the Review
11. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alkahtani, S.; Al-Johani, N.S.; Alarifi, S. Mechanistic Insights, Treatment Paradigms, and Clinical Progress in Neurological Disorders: Current and Future Prospects. Int. J. Mol. Sci. 2023, 24, 1340. [Google Scholar] [CrossRef] [PubMed]
- Kakoti, B.B.; Bezbaruah, R.; Ahmed, N. Therapeutic drug repositioning with special emphasis on neurodegenerative diseases: Threats and issues. Front. Pharmacol. 2022, 13, 1007315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Y.; Wang, J.; Xia, Y.; Zhang, J.; Chen, L. Recent advances in Alzheimer’s disease: Mechanisms, clinical trials and new drug development strategies. Signal. Transduct. Target. Ther. 2024, 9, 211. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Xu, K.F.; Shu, F.X.; Zhang, F. Neurotropic virus infection and neurodegenerative diseases: Potential roles of autophagy pathway. CNS Neurosci. Ther. 2024, 30, e14548. [Google Scholar] [CrossRef]
- Migliore, L.; Coppedè, F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat. Res. 2009, 667, 82–97. [Google Scholar] [CrossRef]
- Boyce, W.T.; Kobor, M.S. Development and the epigenome: The ‘synapse’ of gene-environment interplay. Dev. Sci. 2015, 18, 1–23. [Google Scholar] [CrossRef]
- Gibney, E.R.; Nolan, C.M. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Bure, I.V.; Nemtsova, M.V.; Kuznetsova, E.B. Histone Modifications and Non-Coding RNAs: Mutual Epigenetic Regulation and Role in Pathogenesis. Int. J. Mol. Sci. 2022, 23, 5801. [Google Scholar] [CrossRef]
- Maze, I.; Noh, K.M.; Allis, C.D. Histone regulation in the CNS: Basic principles of epigenetic plasticity. Neuropsychopharmacology 2013, 38, 3–22. [Google Scholar] [CrossRef]
- Wendimu, M.Y.; Hooks, S.B. Microglia Phenotypes in Aging and Neurodegenerative Diseases. Cells 2022, 11, 2091. [Google Scholar] [CrossRef]
- De Plano, L.M.; Saitta, A.; Oddo, S.; Caccamo, A. Epigenetic Changes in Alzheimer’s Disease: DNA Methylation and Histone Modification. Cells 2024, 13, 719. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Dogaru, B.G.; Munteanu, C. The Role of Hydrogen Sulfide (H2S) in Epigenetic Regulation of Neurodegenerative Diseases: A Systematic Review. Int. J. Mol. Sci. 2023, 24, 12555. [Google Scholar] [CrossRef] [PubMed]
- Olufunmilayo, E.O.; Holsinger, R.M.D. Roles of Non-Coding RNA in Alzheimer’s Disease Pathophysiology. Int. J. Mol. Sci. 2023, 24, 12498. [Google Scholar] [CrossRef] [PubMed]
- Ravel-Godreuil, C.; Znaidi, R.; Bonnifet, T.; Joshi, R.L.; Fuchs, J. Transposable elements as new players in neurodegenerative diseases. FEBS Lett. 2021, 595, 2733–2755. [Google Scholar] [CrossRef]
- Angelopoulou, E.; Paudel, Y.N.; Papageorgiou, S.G.; Piperi, C. Environmental Impact on the Epigenetic Mechanisms Underlying Parkinson’s Disease Pathogenesis: A Narrative Review. Brain Sci. 2022, 12, 175. [Google Scholar] [CrossRef]
- Miller, C.W.T. Epigenetics and Neural Circuitry Landscape of Psychotherapeutic Interventions. Psychiatry J. 2017, 2017, 5491812. [Google Scholar] [CrossRef]
- Wang, K.; Liu, H.; Hu, Q.; Wang, L.; Liu, J.; Zheng, Z.; Zhang, W.; Ren, J.; Zhu, F.; Liu, G.H. Epigenetic regulation of aging: Implications for interventions of aging and diseases. Signal Transduct. Target. Ther. 2022, 7, 374. [Google Scholar] [CrossRef]
- Majchrzak-Celińska, A.; Warych, A.; Szoszkiewicz, M. Novel Approaches to Epigenetic Therapies: From Drug Combinations to Epigenetic Editing. Genes 2021, 12, 208. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Ramos-Campo, D.J.; Belinchón-deMiguel, P.; Martinez-Guardado, I.; Dalamitros, A.A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. [Google Scholar] [CrossRef]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef] [PubMed]
- Toader, C.; Dobrin, N.; Brehar, F.M.; Popa, C.; Covache-Busuioc, R.A.; Glavan, L.A.; Costin, H.P.; Bratu, B.G.; Corlatescu, A.D.; Popa, A.A.; et al. From Recognition to Remedy: The Significance of Biomarkers in Neurodegenerative Disease Pathology. Int. J. Mol. Sci. 2023, 24, 16119. [Google Scholar] [CrossRef]
- Shi, F.; He, Y.; Chen, Y.; Yin, X.; Sha, X.; Wang, Y. Comparative Analysis of Multiple Neurodegenerative Diseases Based on Advanced Epigenetic Aging Brain. Front. Genet. 2021, 12, 657636. [Google Scholar] [CrossRef]
- Wang, W.; Bu, B.; Xie, M.; Zhang, M.; Yu, Z.; Tao, D. Neural cell cycle dysregulation and central nervous system diseases. Prog. Neurobiol. 2009, 89, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Karpova, N.N.; Sales, A.J.; Joca, S.R. Epigenetic Basis of Neuronal and Synaptic Plasticity. Curr. Top. Med. Chem. 2017, 17, 771–793. [Google Scholar] [CrossRef]
- Singh, M.B.; Sartor, G.C. BET bromodomains as novel epigenetic targets for brain health and disease. Neuropharmacology 2020, 181, 108306. [Google Scholar] [CrossRef]
- Chen, Y.; Mateski, J.; Gerace, L.; Wheeler, J.; Burl, J.; Prakash, B.; Svedin, C.; Amrick, R.; Adams, B.D. Non-coding RNAs and neuroinflammation: Implications for neurological disorders. Exp. Biol. Med. 2024, 249, 10120. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Wang, B.; Wang, Z.; Zhang, X.; Wang, X. Epigenetic Regulation of NK Cell-Mediated Antitumor Immunity. Front. Immunol. 2021, 12, 672328. [Google Scholar] [CrossRef]
- Li, C.; Ren, J.; Zhang, M.; Wang, H.; Yi, F.; Wu, J.; Tang, Y. The heterogeneity of microglial activation and its epigenetic and non-coding RNA regulations in the immunopathogenesis of neurodegenerative diseases. Cell. Mol. Life Sci. 2022, 79, 511. [Google Scholar] [CrossRef]
- Rasmi, Y.; Shokati, A.; Hassan, A.; Aziz, S.G.; Bastani, S.; Jalali, L.; Moradi, F.; Alipour, S. The role of DNA methylation in progression of neurological disorders and neurodegenerative diseases as well as the prospect of using DNA methylation inhibitors as therapeutic agents for such disorders. IBRO Neurosci. Rep. 2022, 14, 28–37. [Google Scholar] [CrossRef]
- Mohd Murshid, N.; Aminullah Lubis, F.; Makpol, S. Epigenetic Changes and Its Intervention in Age-Related Neurodegenerative Diseases. Cell. Mol. Neurobiol. 2022, 42, 577–595. [Google Scholar] [CrossRef] [PubMed]
- Tabatabaeian, H.; Bai, Y.; Huang, R.; Chaurasia, A.; Darido, C. Navigating therapeutic strategies: HPV classification in head and neck cancer. Br. J. Cancer. 2024, 131, 220–230. [Google Scholar] [CrossRef]
- Mastroeni, D.; Grover, A.; Delvaux, E.; Whiteside, C.; Coleman, P.D.; Rogers, J. Epigenetic changes in Alzheimer’s disease: Decrements in DNA methylation and dynamin-like protein 1 isoforms. Neurobiol. Aging. 2010, 31, 2025–2037. [Google Scholar] [CrossRef]
- de Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef]
- Rossetto, D.; Avvakumov, N.; Côté, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell. Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef] [PubMed]
- Neal, M.; Richardson, J.R. Epigenetic regulation of astrocyte function in neuroinflammation and neurodegeneration. Biochim. Biophys. Acta. Mol. Basis. Dis. 2018, 1864, 432–443. [Google Scholar] [CrossRef]
- Valor, L.M. Transcription, epigenetics and ameliorative strategies in Huntington’s Disease: A genome-wide perspective. Mol. Neurobiol. 2015, 51, 406–423. [Google Scholar] [CrossRef] [PubMed]
- Gräff, J.; Rei, D.; Guan, J.S.; Wang, W.Y.; Seo, J.; Hennig, K.M.; Nieland, T.J.; Fass, D.M.; Kao, P.F.; Kahn, M.; et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 2012, 483, 222–226. [Google Scholar] [CrossRef]
- Klein, H.U.; McCabe, C.; Gjoneska, E.; Sullivan, S.E.; Kaskow, B.J.; Tang, A.; Smith, R.V.; Xu, J.; Pfenning, A.R.; Bernstein, B.E.; et al. Epigenome-wide study uncovers large-scale changes in histone acetylation in the Alzheimer’s disease entorhinal cortex. Nat. Neurosci. 2019, 22, 37–47. [Google Scholar] [CrossRef]
- Li, G.F.; Li, Z.B.; Zhuang, S.J.; Li, G.C. Inhibition of microRNA-34a protects against propofol anesthesia-induced neurotoxicity and cognitive dysfunction via the MAPK/ERK signaling pathway. Neurosci. Lett. 2018, 675, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Martinez, B.; Peplow, P.V. MicroRNAs in Parkinson’s disease and emerging therapeutic targets. Neural. Regen. Res. 2017, 12, 1945–1959. [Google Scholar] [CrossRef]
- Zovoilis, A.; Agbemenyah, H.Y.; Agis-Balboa, R.C.; Stilling, R.M.; Edbauer, D.; Rao, P.; Farinelli, L.; Delalle, I.; Schmitt, A.; Falkai, P.; et al. microRNA-34c is a novel target to treat dementias. EMBO J. 2011, 30, 4299–4308. [Google Scholar] [CrossRef]
- Maciotta, S.; Meregalli, M.; Torrente, Y. The involvement of microRNAs in neurodegenerative diseases. Front. Cell. Neurosci. 2013, 7, 265. [Google Scholar] [CrossRef]
- Johnson, R.; Zuccato, C.; Belyaev, N.D.; Guest, D.J.; Cattaneo, E.; Buckley, N.J. A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol. Dis. 2008, 29, 438–445. [Google Scholar] [CrossRef]
- Nabi, M.; Tabassum, N. Role of Environmental Toxicants on Neurodegenerative Disorders. Front. Toxicol. 2022, 4, 837579. [Google Scholar] [CrossRef] [PubMed]
- Köhler, N. PubMed Commons: Beschreibung und Analyse von PubMeds neuer Kommentarfunktion. Bibliometr.–Prax. Und Forschung 2014, 3. [Google Scholar] [CrossRef]
- Du, X.; Tian, M.; Wang, X.; Zhang, J.; Huang, Q.; Liu, L.; Shen, H. Cortex and hippocampus DNA epigenetic response to a long-term arsenic exposure via drinking water. Environ. Pollut. 2018, 234, 590–600. [Google Scholar] [CrossRef]
- Karri, V.; Ramos, D.; Martinez, J.B.; Odena, A.; Oliveira, E.; Coort, S.L.; Evelo, C.T.; Mariman, E.C.M.; Schuhmacher, M.; Kumar, V. Differential protein expression of hippocampal cells associated with heavy metals (Pb, As, and MeHg) neurotoxicity: Deepening into the molecular mechanism of neurodegenerative diseases. J. Proteom. 2018, 187, 106–125. [Google Scholar] [CrossRef]
- Khani, L.; Martin, L.; Pułaski, Ł. Cellular and physiological mechanisms of halogenated and organophosphorus flame retardant toxicity. Sci. Total. Environ. 2023, 897, 165272. [Google Scholar] [CrossRef]
- Wang, R.; Sun, H.; Wang, G.; Ren, H. Imbalance of Lysine Acetylation Contributes to the Pathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 7182. [Google Scholar] [CrossRef]
- Huang, M.; Bargues-Carot, A.; Riaz, Z.; Wickham, H.; Zenitsky, G.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Impact of Environmental Risk Factors on Mitochondrial Dysfunction, Neuroinflammation, Protein Misfolding, and Oxidative Stress in the Etiopathogenesis of Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 10808. [Google Scholar] [CrossRef]
- Adegbola, P.I.; Adetutu, A. Genetic and epigenetic modulations in toxicity: The two-sided roles of heavy metals and polycyclic aromatic hydrocarbons from the environment. Toxicol. Rep. 2024, 12, 502–519. [Google Scholar] [CrossRef]
- Tran, N.Q.V.; Miyake, K. Neurodevelopmental Disorders and Environmental Toxicants: Epigenetics as an Underlying Mechanism. Int. J. Genom. 2017, 2017, 7526592. [Google Scholar] [CrossRef]
- Qin, S.J.; Zeng, Q.G.; Zeng, H.X.; Li, S.P.; Andersson, J.; Zhao, B.; Oudin, A.; Kanninen, K.M.; Jalava, P.; Jin, N.X.; et al. Neurotoxicity of fine and ultrafine particulate matter: A comprehensive review using a toxicity pathway-oriented adverse outcome pathway framework. Sci. Total. Environ. 2024, 947, 174450. [Google Scholar] [CrossRef]
- Migliore, L.; Coppedè, F. Gene-environment interactions in Alzheimer disease: The emerging role of epigenetics. Nat. Rev. Neurol. 2022, 18, 643–660. [Google Scholar] [CrossRef]
- Löscher, W.; Brandt, C. Prevention or modification of epileptogenesis after brain insults: Experimental approaches and translational research. Pharmacol. Rev. 2010, 62, 668–700. [Google Scholar] [CrossRef]
- Babar, Q.; Saeed, A.; Tabish, T.A.; Pricl, S.; Townley, H.; Thorat, N. Novel epigenetic therapeutic strategies and targets in cancer. Biochim. Biophys. Acta. Mol. Basis. Dis. 2022, 1868, 166552. [Google Scholar] [CrossRef]
- Didonna, A.; Opal, P. The promise and perils of HDAC inhibitors in neurodegeneration. Ann. Clin. Transl. Neurol. 2015, 2, 79–101. [Google Scholar] [CrossRef]
- Ye, L.; Li, W.; Tang, X.; Xu, T.; Wang, G. Emerging Neuroprotective Strategies: Unraveling the Potential of HDAC Inhibitors in Traumatic Brain Injury Management. Curr. Neuropharmacol. 2024, 22, 2298–2313. [Google Scholar] [CrossRef]
- Upadhyay, R.K. Drug delivery systems, CNS protection, and the blood brain barrier. Biomed. Res. Int. 2014, 2014, 869269. [Google Scholar] [CrossRef]
- Satapathy, M.K.; Yen, T.L.; Jan, J.S.; Tang, R.D.; Wang, J.Y.; Taliyan, R.; Yang, C.H. Solid Lipid Nanoparticles (SLNs): An Advanced Drug Delivery System Targeting Brain through BBB. Pharmaceutics 2021, 13, 1183. [Google Scholar] [CrossRef]
- Al-Jipouri, A.; Almurisi, S.H.; Al-Japairai, K.; Bakar, L.M.; Doolaanea, A.A. Liposomes or Extracellular Vesicles: A Comprehensive Comparison of Both Lipid Bilayer Vesicles for Pulmonary Drug Delivery. Polymers 2023, 15, 318. [Google Scholar] [CrossRef]
- Choudhury, H.; Pandey, M.; Chin, P.X.; Phang, Y.L.; Cheah, J.Y.; Ooi, S.C.; Mak, K.K.; Pichika, M.R.; Kesharwani, P.; Hussain, Z.; et al. Transferrin receptors-targeting nanocarriers for efficient targeted delivery and transcytosis of drugs into the brain tumors: A review of recent advancements and emerging trends. Drug Deliv. Transl. Res. 2018, 8, 1545–1563. [Google Scholar] [CrossRef]
- Rittiner, J.; Cumaran, M.; Malhotra, S.; Kantor, B. Therapeutic modulation of gene expression in the disease state: Treatment strategies and approaches for the development of next-generation of the epigenetic drugs. Front. Bioeng. Biotechnol. 2022, 10, 1035543. [Google Scholar] [CrossRef]
- Askenase, P.W. Ancient Evolutionary Origin and Properties of Universally Produced Natural Exosomes Contribute to Their Therapeutic Superiority Compared to Artificial Nanoparticles. Int. J. Mol. Sci. 2021, 22, 1429. [Google Scholar] [CrossRef]
- Boussios, S.; Devo, P.; Goodall, I.C.A.; Sirlantzis, K.; Ghose, A.; Shinde, S.D.; Papadopoulos, V.; Sanchez, E.; Rassy, E.; Ovsepian, S.V. Exosomes in the Diagnosis and Treatment of Renal Cell Cancer. Int. J. Mol. Sci. 2023, 24, 14356. [Google Scholar] [CrossRef]
- Boussios, S.; Ovsepian, S.V. Exosomes in Renal Cell Cancer: Diagnostic and Therapeutic Nanovehicles. Technol. Cancer Res. Treat. 2024, 23, 15330338241275403. [Google Scholar] [CrossRef]
- Hamid, Y.; Rabbani, R.D.; Afsara, R.; Nowrin, S.; Ghose, A.; Papadopoulos, V.; Sirlantzis, K.; Ovsepian, S.V.; Boussios, S. Exosomal Liquid Biopsy in Prostate Cancer: A Systematic Review of Biomarkers for Diagnosis, Prognosis, and Treatment Response. Int. J. Mol. Sci. 2025, 26, 802. [Google Scholar] [CrossRef]
- Rogge, G.A.; Wood, M.A. The role of histone acetylation in cocaine-induced neural plasticity and behavior. Neuropsychopharmacology 2013, 38, 94–110. [Google Scholar] [CrossRef]
- Kumari, S.; Dhapola, R.; Sharma, P.; Nagar, P.; Medhi, B.; HariKrishnaReddy, D. The impact of cytokines in neuroinflammation-mediated stroke. Cytokine Growth Factor Rev. 2024, 78, 105–119. [Google Scholar] [CrossRef]
- Shen, S.; Kozikowski, A.P. A patent review of histone deacetylase 6 inhibitors in neurodegenerative diseases (2014-2019). Expert Opin. Ther. Pat. 2020, 30, 121–136. [Google Scholar] [CrossRef]
- Romoli, M.; Mazzocchetti, P.; D’Alonzo, R.; Siliquini, S.; Rinaldi, V.E.; Verrotti, A.; Calabresi, P.; Costa, C. Valproic Acid and Epilepsy: From Molecular Mechanisms to Clinical Evidences. Curr. Neuropharmacol. 2019, 17, 926–946. [Google Scholar] [CrossRef]
- Kumar, V.; Kundu, S.; Singh, A.; Singh, S. Understanding the Role of Histone Deacetylase and their Inhibitors in Neurodegenerative Disorders: Current Targets and Future Perspective. Curr. Neuropharmacol. 2022, 20, 158–178. [Google Scholar] [CrossRef]
- Saute, J.A.M.; Jardim, L.B. Planning Future Clinical Trials for Machado-Joseph Disease. Adv. Exp. Med. Biol. 2018, 1049, 321–348. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Kilgore, M.; Miller, C.A.; Fass, D.M.; Hennig, K.M.; Haggarty, S.J.; Sweatt, J.D.; Rumbaugh, G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 2010, 35, 870–880. [Google Scholar] [CrossRef]
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D.; et al. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Treatment of cardiovascular pathology with epigenetically active agents: Focus on natural and synthetic inhibitors of DNA methylation and histone deacetylation. Int. J. Cardiol. 2017, 227, 66–82. [Google Scholar] [CrossRef]
- Ciechomska, M.; Roszkowski, L.; Maslinski, W. DNA Methylation as a Future Therapeutic and Diagnostic Target in Rheumatoid Arthritis. Cells 2019, 8, 953. [Google Scholar] [CrossRef]
- Teijido, O.; Cacabelos, R. Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases. Int. J. Mol. Sci. 2018, 19, 3199. [Google Scholar] [CrossRef]
- Luo, M.; Lee, L.K.C.; Peng, B.; Choi, C.H.J.; Tong, W.Y.; Voelcker, N.H. Delivering the Promise of Gene Therapy with Nanomedicines in Treating Central Nervous System Diseases. Adv. Sci. 2022, 9, e2201740. [Google Scholar] [CrossRef]
- Kular, L.; Jagodic, M. ‘DNA Methylation in Multiple Sclerosis’. In The DNA, RNA, and Histone Methylomes; Springer: Berlin/Heidelberg, Germany, 2019; pp. 181–214. [Google Scholar] [CrossRef]
- Gladkova, M.G.; Leidmaa, E.; Anderzhanova, E.A. Epidrugs in the Therapy of Central Nervous System Disorders: A Way to Drive on? Cells 2023, 12, 1464. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, A.; Wang, Z.J.; Cao, Q.; Wang, W.; Lin, L.; Ma, K.; Zhang, F.; Wei, J.; Matas, E.; et al. Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer’s disease. Brain 2019, 142, 787–807. [Google Scholar] [CrossRef]
- Wang, Z.; Leng, Y.; Wang, J.; Liao, H.M.; Bergman, J.; Leeds, P.; Kozikowski, A.; Chuang, D.M. Tubastatin A, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: Potential roles of α-tubulin acetylation and FGF-21 up-regulation. Sci. Rep. 2016, 6, 19626. [Google Scholar] [CrossRef]
- Mielcarek, M.; Landles, C.; Weiss, A.; Bradaia, A.; Seredenina, T.; Inuabasi, L.; Osborne, F.G.; Wadel, K.; Touller, C.; Butler, R.; et al. HDAC4 reduction: A novel therapeutic strategy to target cytoplasmic huntingtin and ameliorate neurodegeneration. PLoS Biol. 2013, 11, e1001717. [Google Scholar] [CrossRef]
- Toma, L.; Sanda, G.M.; Niculescu, L.S.; Deleanu, M.; Sima, A.V.; Stancu, C.S. Phenolic Compounds Exerting Lipid-Regulatory, Anti-Inflammatory and Epigenetic Effects as Complementary Treatments in Cardiovascular Diseases. Biomolecules 2020, 10, 641. [Google Scholar] [CrossRef]
- D’Egidio, F.; Castelli, V.; Cimini, A.; d’Angelo, M. Cell Rearrangement and Oxidant/Antioxidant Imbalance in Huntington’s Disease. Antioxidants 2023, 12, 571. [Google Scholar] [CrossRef]
- Chen, Z.; Rasheed, M.; Deng, Y. The epigenetic mechanisms involved in mitochondrial dysfunction: Implication for Parkinson’s disease. Brain Pathol. 2022, 32, e13012. [Google Scholar] [CrossRef]
- Landgrave-Gómez, J.; Mercado-Gómez, O.; Guevara-Guzmán, R. Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 58. [Google Scholar] [CrossRef]
- Koval, E.D.; Shaner, C.; Zhang, P.; du Maine, X.; Fischer, K.; Tay, J.; Chau, B.N.; Wu, G.F.; Miller, T.M. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 2013, 22, 4127–4135. [Google Scholar] [CrossRef]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef]
- Konopka, W.; Kiryk, A.; Novak, M.; Herwerth, M.; Parkitna, J.R.; Wawrzyniak, M.; Kowarsch, A.; Michaluk, P.; Dzwonek, J.; Arnsperger, T.; et al. MicroRNA loss enhances learning and memory in mice. J. Neurosci. 2010, 30, 14835–14842. [Google Scholar] [CrossRef]
- Ghosh, A.; Himaja, A.; Biswas, S.; Kulkarni, O.; Ghosh, B. Advances in the Delivery and Development of Epigenetic Therapeutics for the Treatment of Cancer. Mol. Pharm. 2023, 20, 5981–6009. [Google Scholar] [CrossRef]
- Griñán-Ferré, C.; Bellver-Sanchis, A.; Guerrero, A.; Pallàs, M. Advancing personalized medicine in neurodegenerative diseases: The role of epigenetics and pharmacoepigenomics in pharmacotherapy. Pharmacol. Res. 2024, 205, 107247. [Google Scholar] [CrossRef]
- Wood, I.C. The Contribution and Therapeutic Potential of Epigenetic Modifications in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 649. [Google Scholar] [CrossRef]
- Coneys, R.; Wood, I.C. Alzheimer’s disease: The potential of epigenetic treatments and current clinical candidates. Neurodegener. Dis. Manag. 2020, 10, 543–558. [Google Scholar] [CrossRef]
- Poon, C.H.; Liu, Y.; Pak, S.; Zhao, R.C.; Aquili, L.; Tipoe, G.L.; Leung, G.K.; Chan, Y.S.; Yang, S.; Fung, M.L.; et al. Prelimbic Cortical Stimulation with L-methionine Enhances Cognition through Hippocampal DNA Methylation and Neuroplasticity Mechanisms. Aging Dis. 2023, 14, 112–135. [Google Scholar] [CrossRef]
- Harrison, I.F.; Dexter, D.T. Epigenetic targeting of histone deacetylase: Therapeutic potential in Parkinson’s disease? Pharmacol. Ther. 2013, 140, 34–52. [Google Scholar] [CrossRef]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; de Álava, E.; Hajji, N.; García-Domínguez, D.J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 2020, 11, 578011. [Google Scholar] [CrossRef] [PubMed]
- Banik, D.; Moufarrij, S.; Villagra, A. Immunoepigenetics Combination Therapies: An Overview of the Role of HDACs in Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 2241. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Orsi, S.A.; Zhang, M.; Grill, R.J.; Pati, S.; Zhao, J.; Moore, A.N. Valproate administered after traumatic brain injury provides neuroprotection and improves cognitive function in rats. PLoS ONE 2010, 5, e11383. [Google Scholar] [CrossRef] [PubMed]
- Geissler, F.; Nesic, K.; Kondrashova, O.; Dobrovic, A.; Swisher, E.M.; Scott, C.L.; Wakefield, J.M. The role of aberrant DNA methylation in cancer initiation and clinical impacts. Ther. Adv. Med. Oncol. 2024, 16, 17588359231220511. [Google Scholar] [CrossRef]
- Wani, A.L.; Shadab, G.H.A. Brain, behavior and the journey towards neuroepigenetic therapeutics. Epigenomics 2019, 11, 969–981. [Google Scholar] [CrossRef]
- Amin, S.; Bathe, O.F. Response biomarkers: Re-envisioning the approach to tailoring drug therapy for cancer. BMC Cancer. 2016, 16, 850. [Google Scholar] [CrossRef]
- Coyle, K.M.; Boudreau, J.E.; Marcato, P. Genetic Mutations and Epigenetic Modifications: Driving Cancer and Informing Precision Medicine. Biomed. Res. Int. 2017, 2017, 9620870. [Google Scholar] [CrossRef] [PubMed]
- Padmakumar, S.; D’Souza, A.; Parayath, N.N.; Bleier, B.S.; Amiji, M.M. Nucleic acid therapies for CNS diseases: Pathophysiology, targets, barriers, and delivery strategies. J. Control. Release 2022, 352, 121–145. [Google Scholar] [CrossRef]
- Clarke, R.; Smith, A.D.; Jobst, K.A.; Refsum, H.; Sutton, L.; Ueland, P.M. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch. Neurol. 1998, 55, 1449–1455. [Google Scholar] [CrossRef]
- Miller, J.W.; Green, R.; Mungas, D.M.; Reed, B.R.; Jagust, W.J. Homocysteine, vitamin B6 and vascular disease in the era of folic acid fortification. Nutr. Rev. 2002, 58, 1471–1475. [Google Scholar] [CrossRef]
- Smith, A.D.; Smith, S.M.; de Jager, C.A.; Whitbread, P.; Johnston, C.; Agacinski, G.; Oulhaj, A.; Bradley, K.M.; Jacoby, R.; Refsum, H. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: A randomized controlled trial. PLoS ONE 2010, 5, e12244. [Google Scholar] [CrossRef] [PubMed]
- Douaud, G.; Refsum, H.; de Jager, C.A.; Jacoby, R.; Nichols, T.E.; Smith, S.M.; Smith, A.D. Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc. Natl. Acad. Sci. USA 2013, 110, 9523–9528. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F.; Tannorella, P.; Pezzini, I.; Migheli, F.; Ricci, G.; Caldarazzo lenco, E.; Piaceri, I.; Polini, A.; Nacmias, B.; Monzani, F.; et al. Folate, homocysteine, vitamin B12, and polymorphisms of genes participating in one-carbon metabolism in late-onset Alzheimer’s disease patients and healthy controls. Antioxid. Redox. Signal. 2012, 17, 195–204. [Google Scholar] [CrossRef]
- Liu, H.; Tian, R.; Wang, H.; Feng, S.; Li, H.; Xiao, Y.; Liu, Y.; Wang, Y.; Sun, Z. Folic acid deficiency enhances abeta accumulation in APP/PS1 mice brain and decreases amyloid-associated miRNAs expression. J. Nutr. Biochem. 2016, 35, 72–78. [Google Scholar] [CrossRef]
- Lee, S.; Lemere, C.A.; Frost, J.L.; Shea, T.B. Dietary supplementation with S-adenosyl methionine delayed amyloid-β and tau pathology in 3xTg-AD mice. J. Alzheimers Dis. 2018, 64, 835–847. [Google Scholar] [CrossRef]
- Blount, B.C.; Mack, M.M.; Wehr, C.M.; MacGregor, J.T.; Hiatt, R.A.; Wang, G.; Wickramasinghe, S.N.; Everson, R.B.; Ames, B.N. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: Implications for cancer and neuronal damage. Proc. Natl. Acad. Sci. USA 1997, 94, 3290–3295. [Google Scholar] [CrossRef]
- Mattson, M.P.; Shea, T.B. Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci. 2003, 26, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.I.; Kumaravel, T.S.; Lohani, A.; Pedersen, W.A.; Cutler, R.G.; Kruman, Y.; Mattson, M.P. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J. Neurosci. 2002, 22, 1752–1762. [Google Scholar] [CrossRef]
- Jadavji, N.M.; Emmerson, J.T.; Willmore, W.G.; MacFarlane, A.J.; Smith, P. B-vitamin and choline supplementation increases neuroplasticity and recovery after stroke. Neurobiol. Dis. 2017, 103, 89–100. [Google Scholar] [CrossRef]
- Chu, M.; Teng, J.; Guo, L.; Wang, Y.; Zhang, L.; Gao, J.; Liu, L. Mild hyperhomocysteinemia induces blood–brain barrier dysfunction but not neuroinflammation in the cerebral cortex and hippocampus of wild-type mice. Can. J. Physiol. Pharmacol. 2021, 99, 847–856. [Google Scholar] [CrossRef]
- Fuso, A.; Nicolia, V.; Pasqualato, A.; Fiorenza, M.T.; Cavallaro, R.A.; Scarpa, S. Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol. Aging 2011, 32, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.C.; Cheung, M.W.; Fu, E.; Win, H.H.; Zaw, M.H.; Ng, A. Is high homocysteine level a risk factor for cognitive decline in elderly? A systematic review, meta-analysis, and meta-regression. Am. J. Geriatr. Psychiatry 2011, 19, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Pogribna, M.; Melnyk, S.; Pogribny, I.P.; Chango, A.; Yi, P.; James, S.J. Homocysteine metabolism in children with Down syndrome: In vitro modulation. Am. J. Hum. Genet. 2001, 69, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Chouliaras, L.; Rutten, B.P.; Kenis, G.; Peerbooms, O.; Visser, P.J.; Verhey, F.; van Os, J.; Steinbusch, H.W.; Lunnon, K.; Mill, J.; et al. Epigenetic regulation in the pathophysiology of Alzheimer’s disease. Prog. Neurobiol. 2010, 90, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Giliberto, L.; Zhao, H.; Chandakkar, P.; Wu, Q.; Simon, J.E.; Janle, E.M.; Lobo, J.; Ferruzzi, M.G.; Davies, P.; et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J. Biol. Chem. 2010, 285, 9100–9113. [Google Scholar] [CrossRef]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Schneider, L.S. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, X.P.; Yang, S.G.; Wang, Y.J.; Zhang, X.; Du, X.T.; Sun, X.X.; Zhao, M.; Huang, L.; Liu, R.T. Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation. Neurotoxicology 2009, 31, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.P.; Chu, T.; Yang, F.; Beech, W.; Frautschy, S.A.; Cole, G.M. The curry spice curcumin reduces oxidative damage and amyloid pathology in an Alzheimer transgenic mouse. J. Neurosci. 2001, 21, 8370–8377. [Google Scholar] [CrossRef]
- Yang, Y.; Liu, Y.; Zhang, A.L.; Tang, S.F.; Ming, Q.; Ao, C.Y.; Liu, Y.; Li, C.Z.; Yu, C.; Zhao, H.; et al. Curcumin protects against manganese-induced neurotoxicity in rat by regulating oxidative stress-related gene expression via H3K27 acetylation. Ecotoxicol. Environ. Saf. 2022, 236, 113469. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Chen, Z.; Fei, G.; Pan, S.; Bao, W.; Ren, S.; Guan, Y.; Zhong, C. Long-term cognitive improvement after benfotiamine administration in patients with Alzheimer’s disease. Neurosci. Bull. 2016, 32, 591–596. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, S.; Li, Q.; Lang, W.; Li, W.; Jiang, X.; Wan, Z.; Chen, J.; Wang, H. Epigallocatechin-3-gallate: A phytochemical as a promising drug candidate for the treatment of Parkinson’s disease. Front. Pharmacol. 2022, 13, 977521. [Google Scholar] [CrossRef]
- Liu, D.; Wang, Z.; Gao, Z.; Xie, K.; Zhang, Q.; Jiang, H.; Pang, Q. Effects of curcumin on learning and memory deficits, BDNF, and ERK protein expression in rats exposed to chronic unpredictable stress. Behav. Brain Res. 2014, 271, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.A.; Mandal, A.K.A.; Khan, Z.A. Potential neuroprotective properties of epigallocatechin-3-gallate (EGCG). Nutr. J. 2016, 15, 60. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, A.; Andleeb, A.; Waris, T.S.; Bazzar, M.; Moradi, A.R.; Awan, N.R.; Yar, M. Neurodegenerative diseases and effective drug delivery: A review of challenges and novel therapeutics. J. Control. Release 2021, 330, 1152–1167. [Google Scholar] [CrossRef] [PubMed]
- Ihezie, S.A.; Mathew, I.E.; McBride, D.W.; Dienel, A.; Blackburn, S.L.; Thankamani Pandit, P.K. Epigenetics in blood-brain barrier disruption. Fluids Barriers CNS 2021, 18, 17. [Google Scholar] [CrossRef]
- Gräff, J.; Kim, D.; Dobbin, M.M.; Tsai, L.H. Epigenetic regulation of gene expression in physiological and pathological brain processes. Physiol. Rev. 2011, 91, 603–649. [Google Scholar] [CrossRef]
- Edelmann, M.J.; Maegawa, G.H.B. CNS-Targeting Therapies for Lysosomal Storage Diseases: Current Advances and Challenges. Front. Mol. Biosci. 2020, 7, 559804. [Google Scholar] [CrossRef]
- Santaló, J.; Berdasco, M. Ethical implications of epigenetics in the era of personalized medicine. Clin. Epigenetics 2022, 14, 44. [Google Scholar] [CrossRef]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours-past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef]
- Popovic, R.; Licht, J.D. Emerging epigenetic targets and therapies in cancer medicine. Cancer Discov. 2012, 2, 405–413. [Google Scholar] [CrossRef]
- Riccio, A. Dynamic epigenetic regulation in neurons: Enzymes, stimuli and signaling pathways. Nat. Neurosci. 2010, 13, 1330–1337. [Google Scholar] [CrossRef] [PubMed]
- Linares, C.A.; Varghese, A.; Ghose, A.; Shinde, S.D.; Adeleke, S.; Sanchez, E.; Sheriff, M.; Chargari, C.; Rassy, E.; Boussios, S. Hallmarks of the Tumour Microenvironment of Gliomas and Its Interaction with Emerging Immunotherapy Modalities. Int. J. Mol. Sci. 2023, 24, 13215. [Google Scholar] [CrossRef]
- Sulewska, A.; Pilz, L.; Manegold, C.; Ramlau, R.; Charkiewicz, R.; Niklinski, J. A Systematic Review of Progress toward Unlocking the Power of Epigenetics in NSCLC: Latest Updates and Perspectives. Cells 2023, 12, 905. [Google Scholar] [CrossRef] [PubMed]
- Coppede, F. Targeting the epigenome to treat neurodegenerative diseases or delay their onset: A perspective. Neural Regen. Res. 2022, 17, 1745–1747. [Google Scholar] [CrossRef]
- Kharod, S.C.; Kang, S.K.; Kadam, S.D. Off-Label Use of Bumetanide for Brain Disorders: An Overview. Front. Neurosci. 2019, 13, 310. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef] [PubMed]
- Boussios, S.; Ozturk, M.A.; Moschetta, M.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Katsanos, K.H.; Christodoulou, D.K.; Pavlidis, N. The Developing Story of Predictive Biomarkers in Colorectal Cancer. J. Pers. Med. 2019, 9, 12. [Google Scholar] [CrossRef]
- Shaman, J.A. The Future of Pharmacogenomics: Integrating Epigenetics, Nutrigenomics, and Beyond. J. Pers. Med. 2024, 14, 1121. [Google Scholar] [CrossRef]
- Olszewska, D.A.; Lang, A.E. The definition of precision medicine in neurodegenerative disorders and the one disease-many diseases tension. Handb. Clin. Neurol. 2023, 192, 3–20. [Google Scholar] [CrossRef]
- Marullo, C.; Di Minin, A.; De Marco, C.; Piccaluga, A. Is open innovation always the best for SMEs? An exploratory analysis at the project level. Creat. Innov. Manag. 2020, 29, 209–223. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Y.; Zhang, H. Dietary polyphenols for tumor therapy: Bioactivities, nano-therapeutic systems and delivery strategies. Food Funct. 2025, 16, 853–866. [Google Scholar] [CrossRef]
- Georgieva, J.V.; Hoekstra, D.; Zuhorn, I.S. Smuggling Drugs into the Brain: An Overview of Ligands Targeting Transcytosis for Drug Delivery across the Blood-Brain Barrier. Pharmaceutics 2014, 6, 557–583. [Google Scholar] [CrossRef]
- Ghose, A.; Lapitan, P.; Apte, V.; Ghosh, A.; Kandala, A.; Basu, S.; Parkes, J.; Shinde, S.D.; Boussios, S.; Sharma, A.; et al. Antibody Drug Conjugates in Urological Cancers: A Review of the Current Landscape. Curr. Oncol. Rep. 2024, 26, 633–646. [Google Scholar] [CrossRef]
- Singh, K.; Sethi, P.; Datta, S.; Chaudhary, J.S.; Kumar, S.; Jain, D.; Gupta, J.K.; Kumar, S.; Guru, A.; Panda, S.P. Advances in gene therapy approaches targeting neuro-inflammation in neurodegenerative diseases. Ageing Res. Rev. 2024, 98, 102321. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.C.; Wang, Z. Precision Medicine: Disease Subtyping and Tailored Treatment. Cancers 2023, 15, 3837. [Google Scholar] [CrossRef] [PubMed]
- Ovsepian, S.V.; Waxman, S.G. Gene therapy for chronic pain: Emerging opportunities in target-rich peripheral nociceptors. Nat. Rev. Neurosci. 2023, 24, 252–265. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, V.B.; Ovsepian, S.V.; Bodeker, M.; Dolly, J.O. Improved lentiviral transduction of ALS motoneurons in vivo via dual targeting. Mol. Pharm. 2013, 10, 4195–4206. [Google Scholar] [CrossRef]
- Ovsepian, S.V.; O’Leary, V.B.; Ntziachristos, V.; Dolly, J.O. Circumventing Brain Barriers: Nanovehicles for Retroaxonal Therapeutic Delivery. Trends Mol. Med. 2016, 22, 983–993. [Google Scholar] [CrossRef]
- Hogg, S.J.; Beavis, P.A.; Dawson, M.A.; Johnstone, R.W. Targeting the epigenetic regulation of antitumour immunity. Nat. Rev. Drug Discov. 2020, 19, 776–800. [Google Scholar] [CrossRef]
- Shinde, S.; Bigogno, C.M.; Simmons, A.; Kathuria, N.; Ghose, A.; Apte, V.; Lapitan, P.; Makker, S.; Caglayan, A.; Boussios, S. Precision oncology through next generation sequencing in hepatocellular carcinoma. Heliyon 2025, 11, e42054. [Google Scholar] [CrossRef]
- Ganatra, H.; Tan, J.K.; Simmons, A.; Bigogno, C.M.; Khurana, V.; Ghose, A.; Ghosh, A.; Mahajan, I.; Boussios, S.; Maniam, A.; et al. Applying whole-genome and whole-exome sequencing in breast cancer: A review of the landscape. Breast Cancer 2024, 31, 999–1009. [Google Scholar] [CrossRef]
- Hamamoto, R.; Komatsu, M.; Takasawa, K.; Asada, K.; Kaneko, S. Epigenetics Analysis and Integrated Analysis of Multiomics Data, Including Epigenetic Data, Using Artificial Intelligence in the Era of Precision Medicine. Biomolecules 2019, 10, 62. [Google Scholar] [CrossRef] [PubMed]
- David-Olawade, C.A.; Olawade, D.B.; Vanderbloemen, L.; Rotifa, O.B.; Fidelis, S.C.; Egbon, E.; Akpan, A.O.; Adeleke, S.; Ghose, A.; Boussios, S. AI-Driven Advances in Low-Dose Imaging and Enhancement—A Review. Diagnostics 2025, 15, 689. [Google Scholar] [CrossRef] [PubMed]
- Tapper, W.; Carneiro, G.; Mikropoulos, C.; Thomas, S.A.; Evans, P.M.; Boussios, S. The Application of Radiomics and AI to Molecular Imaging for Prostate Cancer. J. Pers. Med. 2024, 14, 287. [Google Scholar] [CrossRef] [PubMed]
- Olawade, D.B.; Weerasinghe, K.; Mathugamage, M.D.D.E.; Odetayo, A.; Aderinto, N.; Teke, J.; Boussios, S. Enhancing Ophthalmic Diagnosis and Treatment with Artificial Intelligence. Medicina 2025, 61, 433. [Google Scholar] [CrossRef]
- Olawade, D.B.; Clement David-Olawade, A.; Adereni, T.; Egbon, E.; Teke, J.; Boussios, S. Integrating AI into Cancer Immunotherapy—A Narrative Review of Current Applications and Future Directions. Diseases 2025, 13, 24. [Google Scholar] [CrossRef]
- Olawade, D.B.; Teke, J.; Adeleye, K.K.; Egbon, E.; Weerasinghe, K.; Ovsepian, S.V.; Boussios, S. AI-Guided Cancer Therapy for Patients with Coexisting Migraines. Cancers 2024, 16, 3690. [Google Scholar] [CrossRef]
- Carrera, I.; Martínez, O.; Cacabelos, R. Neuroprotection with Natural Antioxidants and Nutraceuticals in the Context of Brain Cell Degeneration: The Epigenetic Connection. Curr. Top. Med. Chem. 2019, 19, 2999–3011. [Google Scholar] [CrossRef]
- Sahafnejad, Z.; Ramazi, S.; Allahverdi, A. An Update of Epigenetic Drugs for the Treatment of Cancers and Brain Diseases: A Comprehensive Review. Genes 2023, 14, 873. [Google Scholar] [CrossRef]
- Chiu, F.Y.; Yen, Y. Imaging biomarkers for clinical applications in neuro-oncology: Current status and future perspectives. Biomark. Res. 2023, 11, 35. [Google Scholar] [CrossRef]
- Kocurova, G.; Ricny, J.; Ovsepian, S.V. Autoantibodies targeting neuronal proteins as biomarkers for neurodegenerative diseases. Theranostics 2022, 12, 3045–3056. [Google Scholar] [CrossRef] [PubMed]
- Pokorná, M.; Černá, M.; Boussios, S.; Ovsepian, S.V.; O’Leary, V.B. lncRNA Biomarkers of Glioblastoma Multiforme. Biomedicines 2024, 12, 932. [Google Scholar] [CrossRef]
- Kalsariya, R.A.; Kavila, D.; Shorter, S.; Negi, D.; Goodall, I.C.A.; Boussios, S.; Ovsepian, S.V. Molecular biomarkers of glial activation and injury in epilepsy. Drug Discov. Today 2025, 30, 104289. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.; McCann, L.; Makker, S.; Mukherjee, U.; Gullapalli, S.V.N.; Erekkath, J.; Shih, S.; Mahajan, I.; Sanchez, E.; Uccello, M.; et al. Diagnostic biomarkers in ovarian cancer: Advances beyond CA125 and HE4. Ther. Adv. Med. Oncol. 2024, 16, 17588359241233225. [Google Scholar] [CrossRef]
- Parent, P.; Marcq, G.; Adeleke, S.; Turpin, A.; Boussios, S.; Rassy, E.; Penel, N. Predictive biomarkers for immune checkpoint inhibitor response in urothelial cancer. Ther. Adv. Med. Oncol. 2023, 15, 17588359231192402. [Google Scholar] [CrossRef]
- Parikh, D.; Shah, M. A comprehensive study on epigenetic signatures to monitor disease progression and the response to therapy in breast cancer. Biomed. Anal. 2024, 1, 205–217. [Google Scholar] [CrossRef]
- Zakari, S.; Niels, N.K.; Olagunju, G.V.; Nnaji, P.C.; Ogunniyi, O.; Tebamifor, M.; Israel, E.N.; Atawodi, S.E.; Ogunlana, O.O. Emerging biomarkers for non-invasive diagnosis and treatment of cancer: A systematic review. Front. Oncol. 2024, 14, 1405267. [Google Scholar] [CrossRef]
- Hernaiz, A.; Toivonen, J.M.; Bolea, R.; Martín-Burriel, I. Epigenetic Changes in Prion and Prion-like Neurodegenerative Diseases: Recent Advances, Potential as Biomarkers, and Future Perspectives. Int. J. Mol. Sci. 2022, 23, 12609. [Google Scholar] [CrossRef]
- Ilieva, M.S. Non-Coding RNAs in Neurological and Neuropsychiatric Disorders: Unraveling the Hidden Players in Disease Pathogenesis. Cells 2024, 13, 1063. [Google Scholar] [CrossRef]
- Zimmer-Bensch, G.; Zempel, H. DNA Methylation in Genetic and Sporadic Forms of Neurodegeneration: Lessons from Alzheimer’s, Related Tauopathies and Genetic Tauopathies. Cells 2021, 10, 3064. [Google Scholar] [CrossRef]
- Sharma, V.; Nikolajeff, F.; Kumar, S. Employing nanoparticle tracking analysis of salivary neuronal exosomes for early detection of neurodegenerative diseases. Transl. Neurodegener. 2023, 12, 7. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Class/Reference | Target | Mechanism of Action | Examples | Application in NDDs |
|---|---|---|---|---|
| HDAC Inhibitors [25] | Histone deacetylases | Restore histone acetylation; reactivating gene transcription | VPA; Vorinostat | AD, HD |
| DNMT Inhibitors [25] | DNA methyltransferases | Reverse hypermethylation; restoring gene expression | Azacitidine; Decitabine | PD, HD |
| BET Inhibitors [26] | Bromodomain proteins | Disrupt the binding of proteins to acetylated histones | JQ1; OTX015 | ALS, AD |
| miRNA Modulators [27] | Non-coding RNAs | Alter microRNA expression to regulate target mRNA levels | Anti-miR-34a | PD, AD |
| Histone Methylation Modulators [28] | Histone methyltransferases/demethylases | Regulate chromatin states through methylation balance | EZH2 inhibitors | MS, HD |
| Delivery System/Reference | Mechanism | Advantages | Challenges | Examples |
|---|---|---|---|---|
| Nanoparticles [62] | Encapsulation of drugs | Enhanced BBB penetration; targeted delivery | Limited scalability; potential toxicity | Lipid-based nanoparticles |
| Liposomes [63] | Lipid bilayer carriers | Biocompatibility; controlled release | Stability issues; production cost | Doxil and epigenetic drug prototypes |
| Conjugated Peptides [64] | BBB receptor-mediated transport | High specificity; reduced off-target effects | Limited targeting peptides available | Transferrin-conjugated molecules |
| Viral Vectors [65] | Gene therapy-based delivery | Long-term expression; CNS specificity | Immune responses; insertional mutagenesis | AAV vectors for HDAC inhibitors |
| Exosome-Based Delivery [66,67,68,69] | Natural vesicle carriers | Biocompatible; minimal immune response | Difficult production; variability | Exosome-encapsulated small RNAs |
| Biomarker/Reference | Type | Disease Association | Diagnostic/Prognostic Use | Current Research Status |
|---|---|---|---|---|
| Global DNA Methylation [179] | Epigenetic modification | AD, PD | Monitor therapeutic efficacy | Preclinical validation |
| Histone Acetylation [38] | Epigenetic modification | HD | Assess treatment response | Limited clinical application |
| microRNA-34a [180] | Non-coding RNA | AD, PD | Diagnostic and therapeutic target | Ongoing clinical trials |
| BDNF Promoter Methylation [181] | Gene-specific DNA methylation | AD | Prognostic indicator | Experimental stage |
| Circulating Exosomal RNA [182] | RNA encapsulated in exosomes | Various NDDs | Non-invasive monitoring of CNS changes | Emerging research |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olawade, D.B.; Rashad, I.; Egbon, E.; Teke, J.; Ovsepian, S.V.; Boussios, S. Reversing Epigenetic Dysregulation in Neurodegenerative Diseases: Mechanistic and Therapeutic Considerations. Int. J. Mol. Sci. 2025, 26, 4929. https://doi.org/10.3390/ijms26104929
Olawade DB, Rashad I, Egbon E, Teke J, Ovsepian SV, Boussios S. Reversing Epigenetic Dysregulation in Neurodegenerative Diseases: Mechanistic and Therapeutic Considerations. International Journal of Molecular Sciences. 2025; 26(10):4929. https://doi.org/10.3390/ijms26104929
Chicago/Turabian StyleOlawade, David B., Intishar Rashad, Eghosasere Egbon, Jennifer Teke, Saak Victor Ovsepian, and Stergios Boussios. 2025. "Reversing Epigenetic Dysregulation in Neurodegenerative Diseases: Mechanistic and Therapeutic Considerations" International Journal of Molecular Sciences 26, no. 10: 4929. https://doi.org/10.3390/ijms26104929
APA StyleOlawade, D. B., Rashad, I., Egbon, E., Teke, J., Ovsepian, S. V., & Boussios, S. (2025). Reversing Epigenetic Dysregulation in Neurodegenerative Diseases: Mechanistic and Therapeutic Considerations. International Journal of Molecular Sciences, 26(10), 4929. https://doi.org/10.3390/ijms26104929