
Machine Learning on Toxicogenomic Data Reveals a Strong Association Between the Induction of Drug-Metabolizing Enzymes and Centrilobular Hepatocyte Hypertrophy in Rats


Abstract
1. Introduction
2. Results
2.1. Dataset
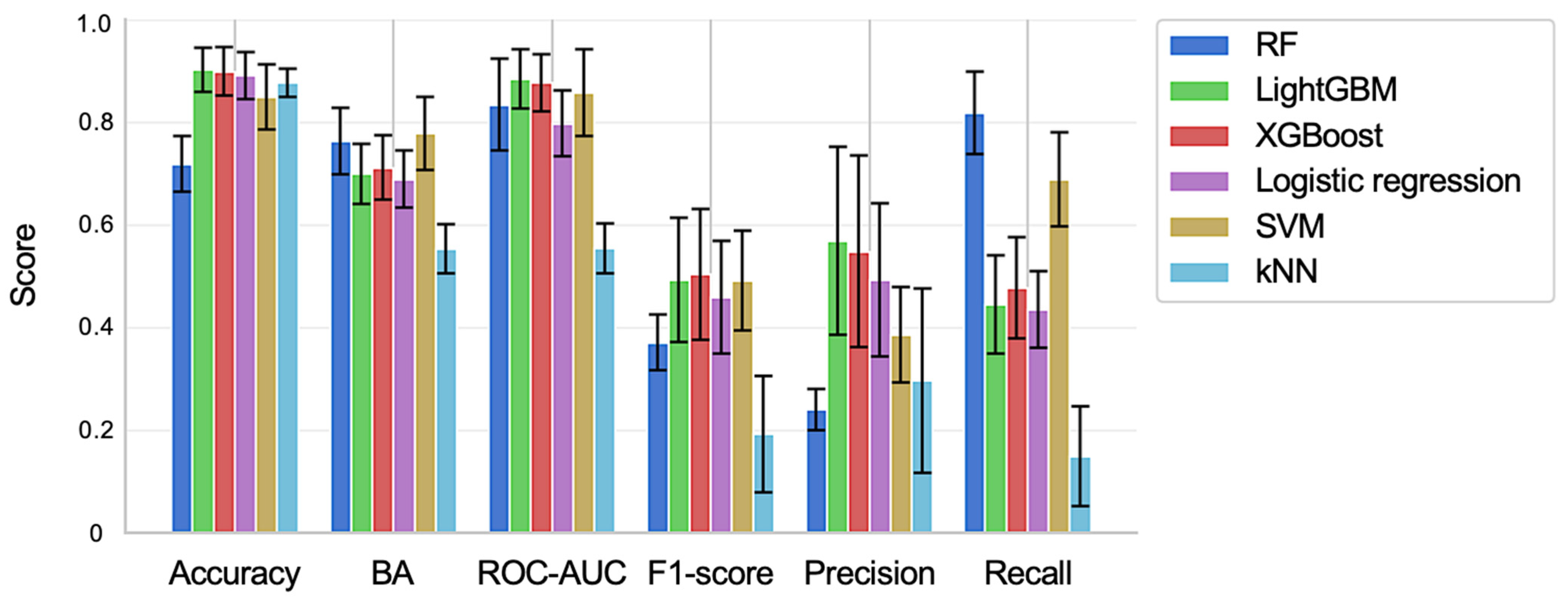
2.2. Training and Validation of Prediction Models for Centrilobular Hepatocyte Hypertrophy
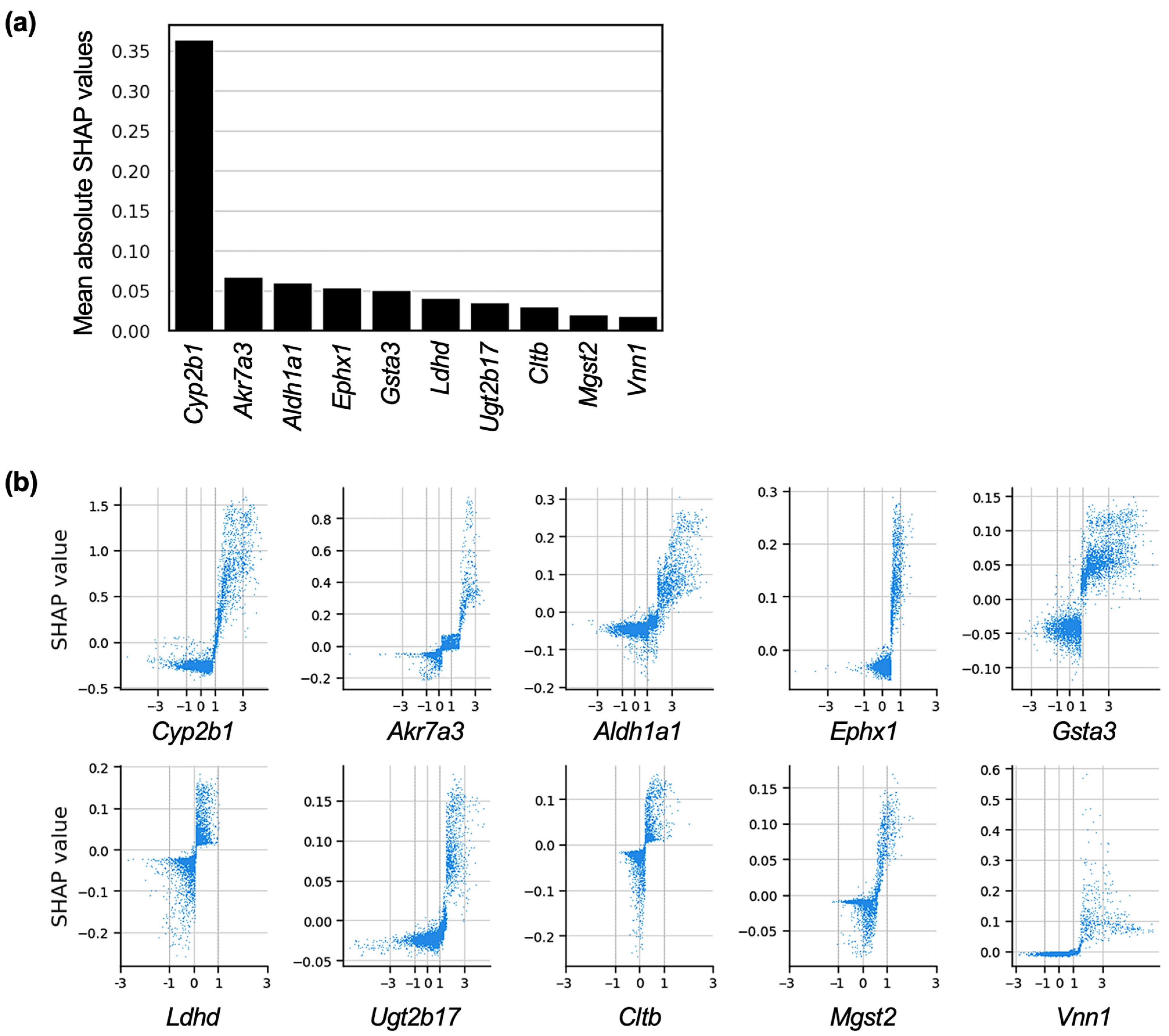
2.3. Genes Contributing to the Prediction of Centrilobular Hepatocyte Hypertrophy
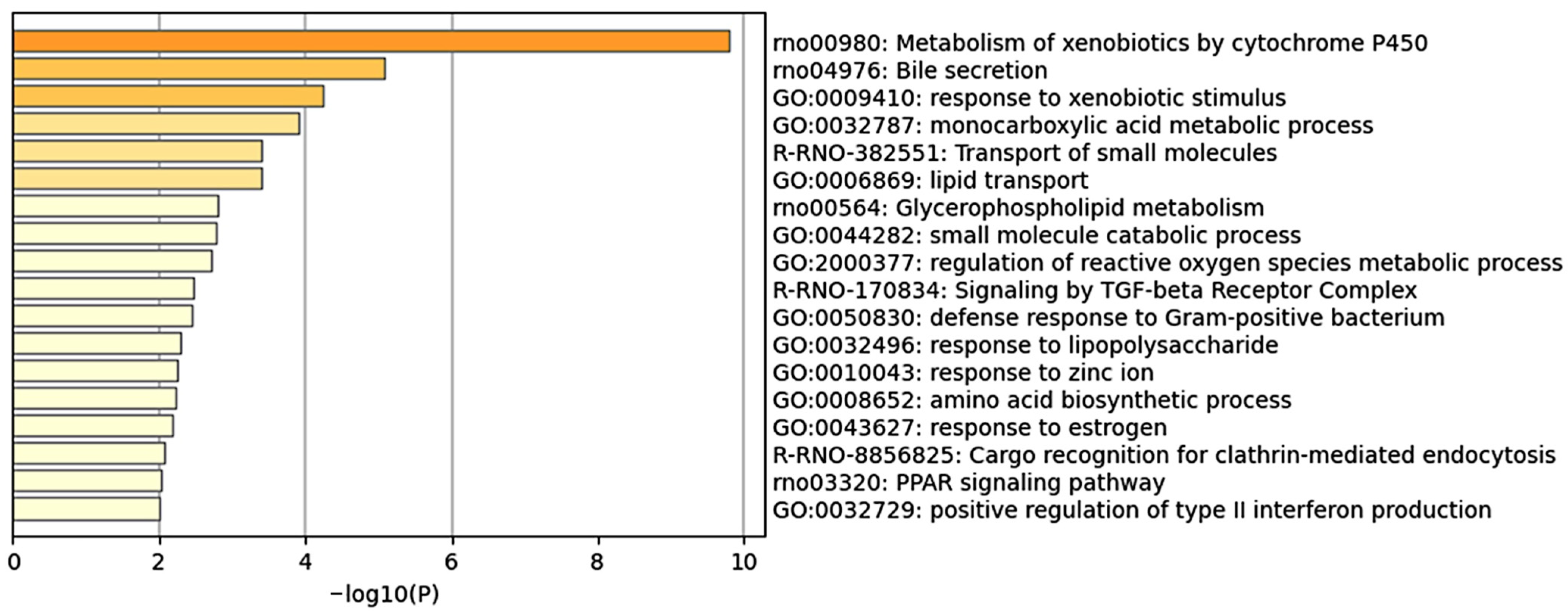
2.4. Enrichment Analysis Using the Top 100 Genes That Contributed to the Prediction
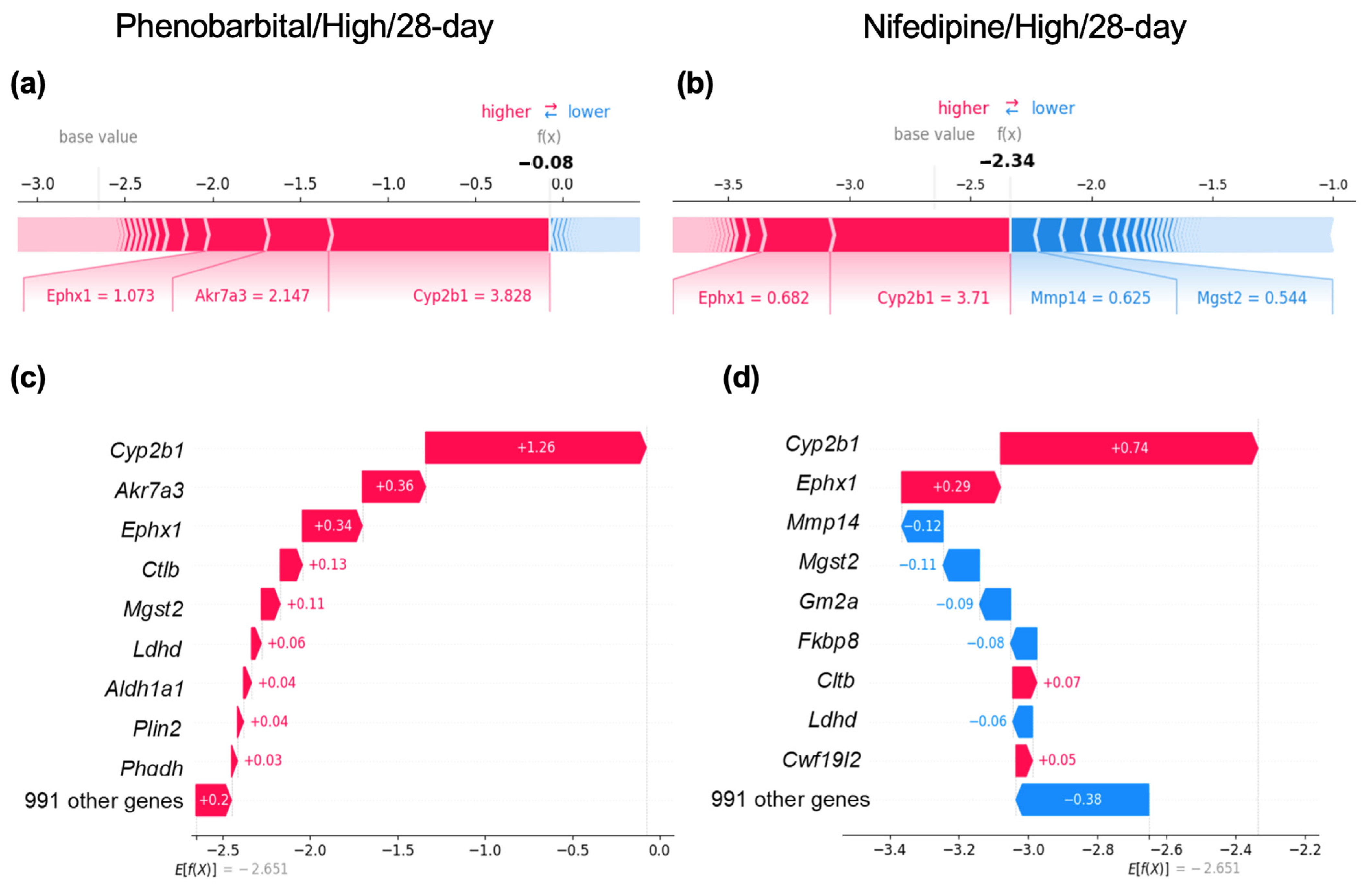
2.5. Case Studies of Samples Showing CYP2B1 Induction with or Without Centrilobular Hepatocyte Hypertrophy
3. Discussion
4. Materials and Methods
4.1. Dataset
4.2. Development of Prediction Models for Centrilobular Hepatocyte Hypertrophy
4.3. Evaluation of Predictive Models
4.4. Calculation of SHAP Values
4.5. Enrichment Analysis
4.6. Computational Environment and Program Codes
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Takeshita, J.; Goto, Y.; Yamamoto, S.; Sasaki, T.; Yoshinari, K. Comprehensive analysis of the toxicity-related findings from repeated-dose subacute toxicity studies of industrial chemicals in male rats. Crit. Rev. Toxicol. 2024, 54, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Umemura, T.; Kojima, H.; Inoue, K.; Takahashi, M.; Uramaru, N.; Kitamura, S.; Abe, K.; Tohkin, M.; Ozawa, S.; et al. Basic principles of interpretation of hepatocellular hypertrophy in risk assessment in Japan. Shokuhin Eiseigaku Zasshi 2015, 56, 42–48. [Google Scholar] [CrossRef]
- Thoolen, B.; Maronpot, R.R.; Harada, T.; Nyska, A.; Rousseaux, C.; Nolte, T.; Malarkey, D.E.; Kaufmann, W.; Kuttler, K.; Deschl, U.; et al. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol. Pathol. 2010, 38 (Suppl. S7), 5S–81S. [Google Scholar] [CrossRef]
- Clayton, N.P.; Yoshizawa, K.; Kissling, G.E.; Burka, L.T.; Chan, P.C.; Nyska, A. Immunohistochemical analysis of expressions of hepatic cytochrome P450 in F344 rats following oral treatment with kava extract. Exp. Toxicol. Pathol. 2007, 58, 223–236. [Google Scholar] [CrossRef]
- Hall, A.P.; Elcombe, C.R.; Foster, J.R.; Harada, T.; Kaufmann, W.; Knippel, A.; Kuttler, K.; Malarkey, D.E.; Maronpot, R.R.; Nishikawa, A.; et al. Liver hypertrophy: A review of adaptive (adverse and non-adverse) changes--conclusions from the 3rd International ESTP Expert Workshop. Toxicol. Pathol. 2012, 40, 971–994. [Google Scholar] [CrossRef] [PubMed]
- Maronpot, R.R.; Yoshizawa, K.; Nyska, A.; Harada, T.; Flake, G.; Mueller, G.; Singh, B.; Ward, J.M. Hepatic enzyme induction: Histopathology. Toxicol. Pathol. 2010, 38, 776–795. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Pearse, G.; Haseman, J.K.; Maronpot, R.R. Prediction of rodent carcinogenesis: An evaluation of prechronic liver lesions as forecasters of liver tumors in NTP carcinogenicity studies. Toxicol. Pathol. 2004, 32, 393–401. [Google Scholar] [CrossRef]
- Carmichael, N.G.; Enzmann, H.; Pate, I.; Waechter, F. The significance of mouse liver tumor formation for carcinogenic risk assessment: Results and conclusions from a survey of ten years of testing by the agrochemical industry. Environ. Health Perspect. 1997, 105, 1196–1203. [Google Scholar] [CrossRef]
- Liu, S.; Kawamoto, T.; Morita, O.; Yoshinari, K.; Honda, H. Discriminating between adaptive and carcinogenic liver hypertrophy in rat studies using logistic ridge regression analysis of toxicogenomic data: The mode of action and predictive models. Toxicol. Appl. Pharmacol. 2017, 318, 79–87. [Google Scholar] [CrossRef]
- Mutoh, S.; Sobhany, M.; Moore, R.; Perera, L.; Pedersen, L.; Sueyoshi, T.; Negishi, M. Phenobarbital indirectly activates the constitutive active androstane receptor (CAR) by inhibition of epidermal growth factor receptor signaling. Sci. Signal 2013, 6, ra31. [Google Scholar] [CrossRef]
- Shizu, R.; Makida, N.; Sobe, K.; Ishimura, M.; Takeshita, A.; Hosaka, T.; Kanno, Y.; Sasaki, T.; Yoshinari, K. Interaction with YAP underlies the species differences between humans and rodents in CAR-dependent hepatocyte proliferation. Toxicol. Sci. 2024, 198, 101–112. [Google Scholar] [CrossRef]
- Yoshinari, K. Role of nuclear receptors PXR and CAR in xenobiotic-induced hepatocyte proliferation and chemical carcinogenesis. Biol. Pharm. Bull. 2019, 42, 1243–1252. [Google Scholar] [CrossRef]
- Shizu, R.; Yoshinari, K. Nuclear receptor CAR-mediated liver cancer and its species differences. Expert Opin. Drug Metab. Toxicol. 2020, 16, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, K.; Shizu, R. Distinct Roles of the Sister Nuclear Receptors PXR and CAR in Liver Cancer Development. Drug Metab. Dispos. 2022, 50, 1019–1026. [Google Scholar] [CrossRef]
- Peters, J.M.; Cattley, R.C.; Gonzalez, F.J. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis 1997, 18, 2029–2033. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, Y.; Ebato, K.; Kato, H.; Arakawa, S.; Shimizu, S.; Miyajima, A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr. Biol. 2012, 22, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Ruz-Maldonado, I.; Gonzalez, J.T.; Zhang, H.; Sun, J.; Bort, A.; Kabir, I.; Kibbey, R.G.; Suarez, Y.; Greif, D.M.; Fernandez-Hernando, C. Heterogeneity of hepatocyte dynamics restores liver architecture after chemical, physical or viral damage. Nat. Commun. 2024, 15, 1247. [Google Scholar] [CrossRef]
- Ronis, M.J.; Lumpkin, C.K.; Thomas, P.E.; Ingelman-Sundberg, M.; Badger, T.M. The microsomal monooxygenase system of regenerating liver. An examination of the role of estradiol in the demasculinization of drug metabolism produced by 2/3 partial hepatectomy. Biochem. Pharmacol. 1992, 43, 567–573. [Google Scholar] [CrossRef]
- Ishizuka, M.; Yoshino, S.; Yamamoto, Y.; Yamamoto, H.; Imaoka, S.; Funae, Y.; Masuda, M.; Iwata, H.; Kazusaka, A.; Fujita, S. Isozyme selective alterations of the expression of cytochrome P450 during regeneration of male rat liver following partial hepatectomy. Xenobiotica 1997, 27, 923–931. [Google Scholar] [CrossRef]
- Igarashi, Y.; Nakatsu, N.; Yamashita, T.; Ono, A.; Ohno, Y.; Urushidani, T.; Yamada, H. Open TG-GATEs: A large-scale toxicogenomics database. Nucleic Acids Res. 2015, 43, D921–D927. [Google Scholar] [CrossRef]
- Nyström-Persson, J.; Igarashi, Y.; Ito, M.; Morita, M.; Nakatsu, N.; Yamada, H.; Mizuguchi, K. Toxygates: Interactive toxicity analysis on a hybrid microarray and linked data platform. Bioinformatics 2013, 29, 3080–3086. [Google Scholar] [CrossRef]
- Nagata, K.; Washio, T.; Kawahara, Y.; Unami, A. Toxicity prediction from toxicogenomic data based on class association rule mining. Toxicol. Rep. 2014, 1, 1133–1142. [Google Scholar] [CrossRef]
- Ke, G.; Meng, Q.; Finley, T.; Wang, T.; Chen, W.; Ma, W.; Ye, Q.; Liu, T.-Y. LightGBM: A Highly Efficient Gradient Boosting Decision Tree. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017. [Google Scholar]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In The 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; ACM: San Francisco, CA, USA, 2016; Volume 11, pp. 785–794. [Google Scholar]
- Lundberg, S.M.; Lee, S.-I. A unified approach to interpreting model predictions. In Proceedings of the 31st Conference on Neural Information Processing Systems (NIPS 2017), Long Beach, CA, USA, 4–9 December 2017; ACM: Long Beach, CA, USA, 2017; pp. 4768–4777. [Google Scholar]
- Mastrolorito, F.; Togo, M.V.; Gambacorta, N.; Trisciuzzi, D.; Giannuzzi, V.; Bonifazi, F.; Liantonio, A.; Imbrici, P.; De Luca, A.; Altomare, C.D.; et al. TISBE: A Public Web Platform for the Consensus-Based Explainable Prediction of Developmental Toxicity. Chem. Res. Toxicol. 2024, 37, 323–339. [Google Scholar] [CrossRef]
- Pramudito, M.A.; Fuadah, Y.N.; Qauli, A.I.; Marcellinus, A.; Lim, K.M. Explainable artificial intelligence (XAI) to find optimal in-silico biomarkers for cardiac drug toxicity evaluation. Sci. Rep. 2024, 14, 24045. [Google Scholar] [CrossRef] [PubMed]
- Timsit, Y.E.; Negishi, M. CAR and PXR: The xenobiotic-sensing receptors. Steroids 2007, 72, 231–246. [Google Scholar] [CrossRef]
- Kodama, S.; Negishi, M. Phenobarbital confers its diverse effects by activating the orphan nuclear receptor car. Drug Metab. Rev. 2006, 38, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Moore, R.; Goldsworthy, T.L.; Negishi, M.; Maronpot, R.R. The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer Res. 2004, 64, 7197–7200. [Google Scholar] [CrossRef]
- Kowalik, M.A.; Saliba, C.; Pibiri, M.; Perra, A.; Ledda-Columbano, G.M.; Sarotto, I.; Ghiso, E.; Giordano, S.; Columbano, A. Yes-associated protein regulation of adaptive liver enlargement and hepatocellular carcinoma development in mice. Hepatology 2011, 53, 2086–2096. [Google Scholar] [CrossRef] [PubMed]
- Hakkola, J.; Bernasconi, C.; Coecke, S.; Richert, L.; Andersson, T.B.; Pelkonen, O. Cytochrome P450 Induction and Xeno-Sensing Receptors Pregnane X Receptor, Constitutive Androstane Receptor, Aryl Hydrocarbon Receptor and Peroxisome Proliferator-Activated Receptor alpha at the Crossroads of Toxicokinetics and Toxicodynamics. Basic. Clin. Pharmacol. Toxicol. 2018, 123 (Suppl. S5), 42–50. [Google Scholar] [CrossRef]
- Botts, S.; Ennulat, D.; Francke-Carroll, S.; Graham, M.; Maronpot, R.R.; Mohutsky, M. Introduction to hepatic drug metabolizing enzyme induction in drug safety evaluation studies. Toxicol. Pathol. 2010, 38, 796–798. [Google Scholar] [CrossRef]
- Oladimeji, P.; Cui, H.; Zhang, C.; Chen, T. Regulation of PXR and CAR by protein-protein interaction and signaling crosstalk. Expert Opin. Drug Metab. Toxicol. 2016, 12, 997–1010. [Google Scholar] [CrossRef]
- Xiao, L.; Nickbarg, E.; Wang, W.; Thomas, A.; Ziebell, M.; Prosise, W.W.; Lesburg, C.A.; Taremi, S.S.; Gerlach, V.L.; Le, H.V.; et al. Evaluation of in vitro PXR-based assays and in silico modeling approaches for understanding the binding of a structurally diverse set of drugs to PXR. Biochem. Pharmacol. 2011, 81, 669–679. [Google Scholar] [CrossRef]
- Konno, Y.; Degawa, M. Gene activations of CYP2B1 and CYP3A1 by dihydropyridine calcium channel antagonists in the rat liver: The structure-activity relationship. Biol. Pharm. Bull. 2004, 27, 903–905. [Google Scholar] [CrossRef]
- Lanz, M.C.; Zatulovskiy, E.; Swaffer, M.P.; Zhang, L.; Ilerten, I.; Zhang, S.; You, D.S.; Marinov, G.; McAlpine, P.; Elias, J.E.; et al. Increasing cell size remodels the proteome and promotes senescence. Mol. Cell 2022, 82, 3255–3269.e8. [Google Scholar] [CrossRef]
- Rhind, N. Cell-size control. Curr. Biol. 2021, 31, R1414–R1420. [Google Scholar] [CrossRef]
- Kempe, H.; Schwabe, A.; Cremazy, F.; Verschure, P.J.; Bruggeman, F.J. The volumes and transcript counts of single cells reveal concentration homeostasis and capture biological noise. Mol. Biol. Cell 2015, 26, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Mucs, D.; Norinder, U.; Svensson, F. LightGBM: An Effective and Scalable Algorithm for Prediction of Chemical Toxicity-Application to the Tox21 and Mutagenicity Data Sets. J. Chem. Inf. Model. 2019, 59, 4150–4158. [Google Scholar] [CrossRef] [PubMed]
- Jaganathan, K.; Geethika, P.R.; Ramakrishnan, S.; Sundaram, D. Explainable AI and tree-based ensemble models: A comparative study in predicting chemical pulmonary toxicity. Eur. Phys. J. Spec. Top. 2024. [Google Scholar] [CrossRef]
- Seto, H.; Oyama, A.; Kitora, S.; Toki, H.; Yamamoto, R.; Kotoku, J.; Haga, A.; Shinzawa, M.; Yamakawa, M.; Fukui, S.; et al. Gradient boosting decision tree becomes more reliable than logistic regression in predicting probability for diabetes with big data. Sci. Rep. 2022, 12, 15889. [Google Scholar] [CrossRef]
- David, W.; Hosmer, J.; Lemeshow, S.; Sturdivant, R.X. Applied Logistic Regression, 3rd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Van Messem, A. Chapter 10—Support vector machines: A robust prediction method with applications in bioinformatics. In Handbook of Statistics; Srinivasa Rao, A.S.R., Rao, C.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; Volume 43, pp. 391–466. [Google Scholar]
- Genuer, R.; Poggi, J.-M.; Tuleau-Malot, C.; Villa-Vialaneix, N. Random Forests for Big Data. Big Data Res. 2017, 9, 28–46. [Google Scholar] [CrossRef]
- Cover, T.; Hart, P. Nearest neighbor pattern classification. IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Scardino, V. Machine Learning Toxicity Prediction: Latest Advances by Toxicity End Point. ACS Omega 2022, 7, 47536–47546. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gong, Y. The Comparison of LightGBM and XGBoost Coupling Factor Analysis and Prediagnosis of Acute Liver Failure. IEEE Access 2020, 8, 220990–221003. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Y.; Zhao, Y. LightGBM: An Effective miRNA Classification Method in Breast Cancer Patients. In Proceedings of the 2017 International Conference on Computational Biology and Bioinformatics, Newark, NJ, USA, 18–20 October 2017; Association for Computing Machinery: Newark, NJ, USA, 2017; pp. 7–11. [Google Scholar]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dose Level | Treatment Duration (d) | Total Samples | Positive Samples | Positive Rates (%) |
|---|---|---|---|---|
| High | 28 | 393 | 110 | 28.0 |
| 14 | 420 | 97 | 23.1 | |
| 7 | 429 | 67 | 15.6 | |
| 3 | 429 | 40 | 9.3 | |
| Middle | 28 | 423 | 73 | 17.3 |
| 14 | 423 | 44 | 10.4 | |
| 7 | 423 | 26 | 6.1 | |
| 3 | 423 | 15 | 3.5 | |
| Low | 28 | 423 | 24 | 5.7 |
| 14 | 423 | 9 | 2.1 | |
| 7 | 423 | 3 | 7.0 | |
| 3 | 423 | 0 | 0 | |
| All | - | 5055 | 508 | 10.1 |
| Fold | Total Samples | Positive Samples | Positive Rates (%) | Number of Unique Compounds |
|---|---|---|---|---|
| 0 | 1004 | 104 | 10.4 | 28 |
| 1 | 1023 | 114 | 11.1 | 30 |
| 2 | 1039 | 108 | 10.4 | 29 |
| 3 | 991 | 79 | 8.0 | 28 |
| 4 | 998 | 103 | 10.3 | 28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikoma, K.; Hosaka, T.; Ooka, A.; Shizu, R.; Yoshinari, K. Machine Learning on Toxicogenomic Data Reveals a Strong Association Between the Induction of Drug-Metabolizing Enzymes and Centrilobular Hepatocyte Hypertrophy in Rats. Int. J. Mol. Sci. 2025, 26, 4886. https://doi.org/10.3390/ijms26104886
Ikoma K, Hosaka T, Ooka A, Shizu R, Yoshinari K. Machine Learning on Toxicogenomic Data Reveals a Strong Association Between the Induction of Drug-Metabolizing Enzymes and Centrilobular Hepatocyte Hypertrophy in Rats. International Journal of Molecular Sciences. 2025; 26(10):4886. https://doi.org/10.3390/ijms26104886
Chicago/Turabian StyleIkoma, Kazuki, Takuomi Hosaka, Akira Ooka, Ryota Shizu, and Kouichi Yoshinari. 2025. "Machine Learning on Toxicogenomic Data Reveals a Strong Association Between the Induction of Drug-Metabolizing Enzymes and Centrilobular Hepatocyte Hypertrophy in Rats" International Journal of Molecular Sciences 26, no. 10: 4886. https://doi.org/10.3390/ijms26104886
APA StyleIkoma, K., Hosaka, T., Ooka, A., Shizu, R., & Yoshinari, K. (2025). Machine Learning on Toxicogenomic Data Reveals a Strong Association Between the Induction of Drug-Metabolizing Enzymes and Centrilobular Hepatocyte Hypertrophy in Rats. International Journal of Molecular Sciences, 26(10), 4886. https://doi.org/10.3390/ijms26104886