



Acidic Oxidative Depolymerization Towards Functionalized Low-Molecular-Weight Lignin and High-Value-Added Aliphatic Monomers: Operating Conditions, Scale-Up, and Crosslinking

 , , ,
, , ,  and
and 
Abstract

1. Introduction
2. Results and Discussion
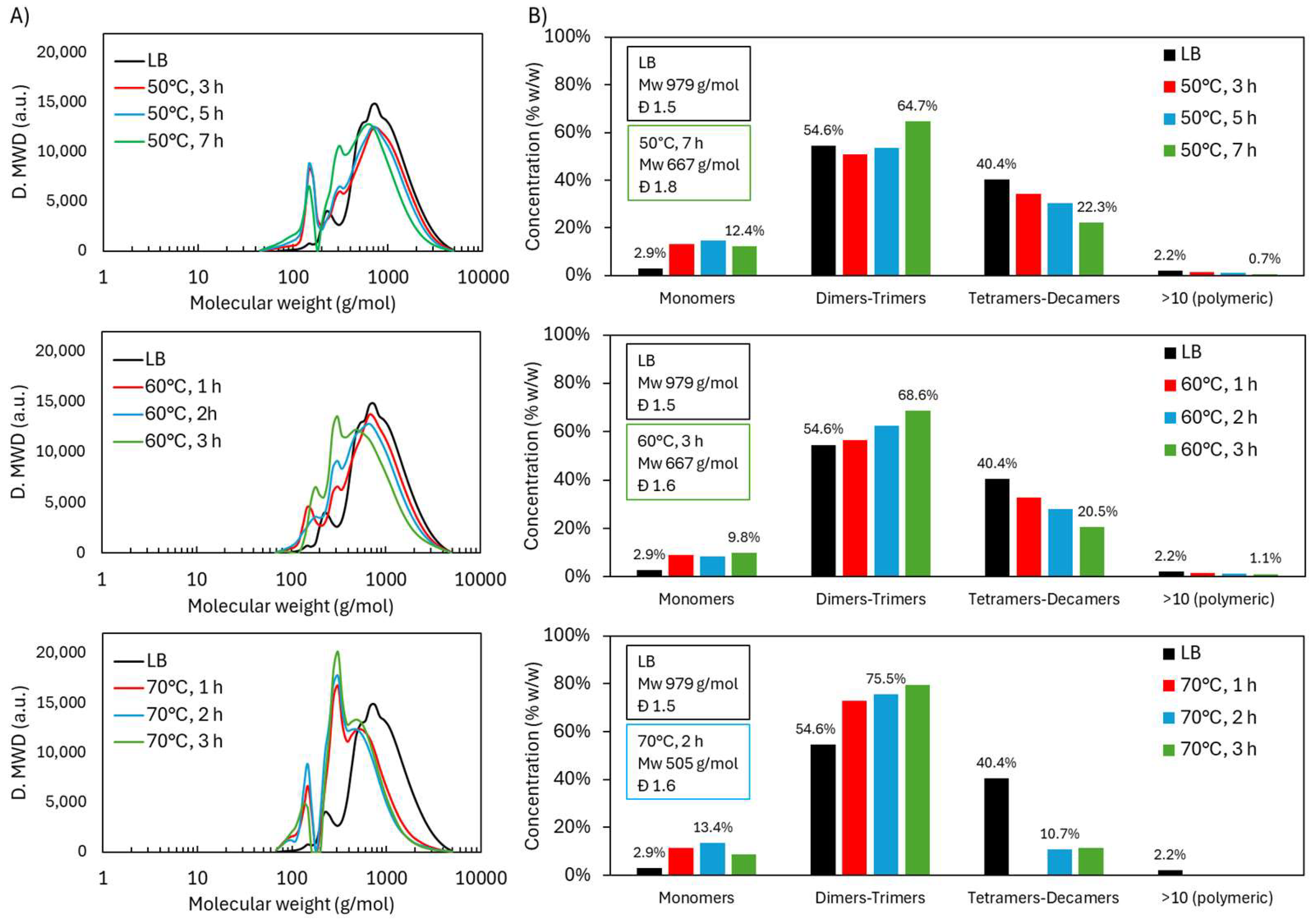
2.1. Acidic Oxidative Depolymerization Under Different Temperatures
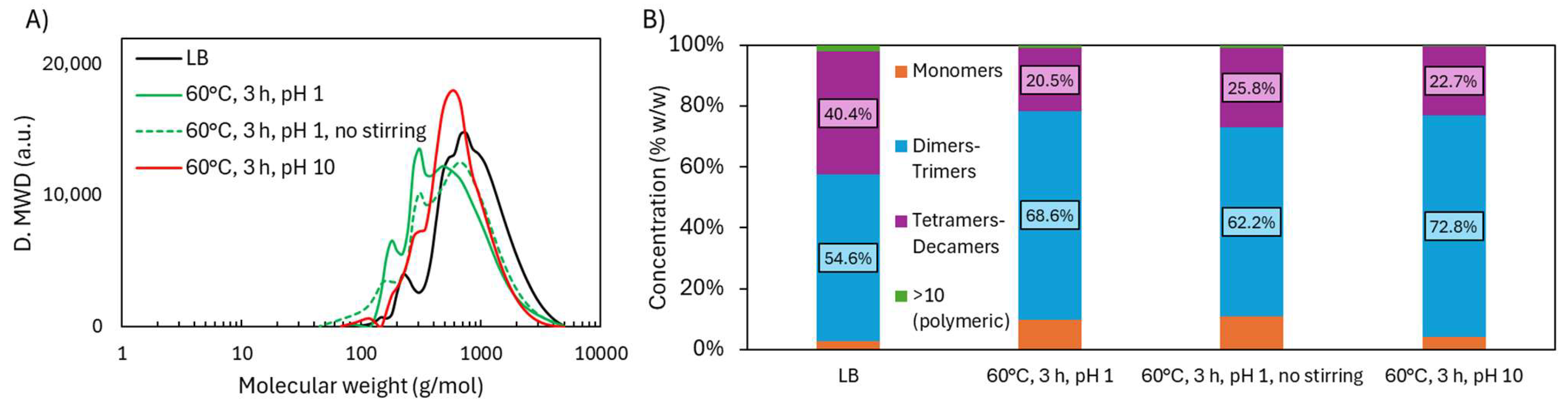
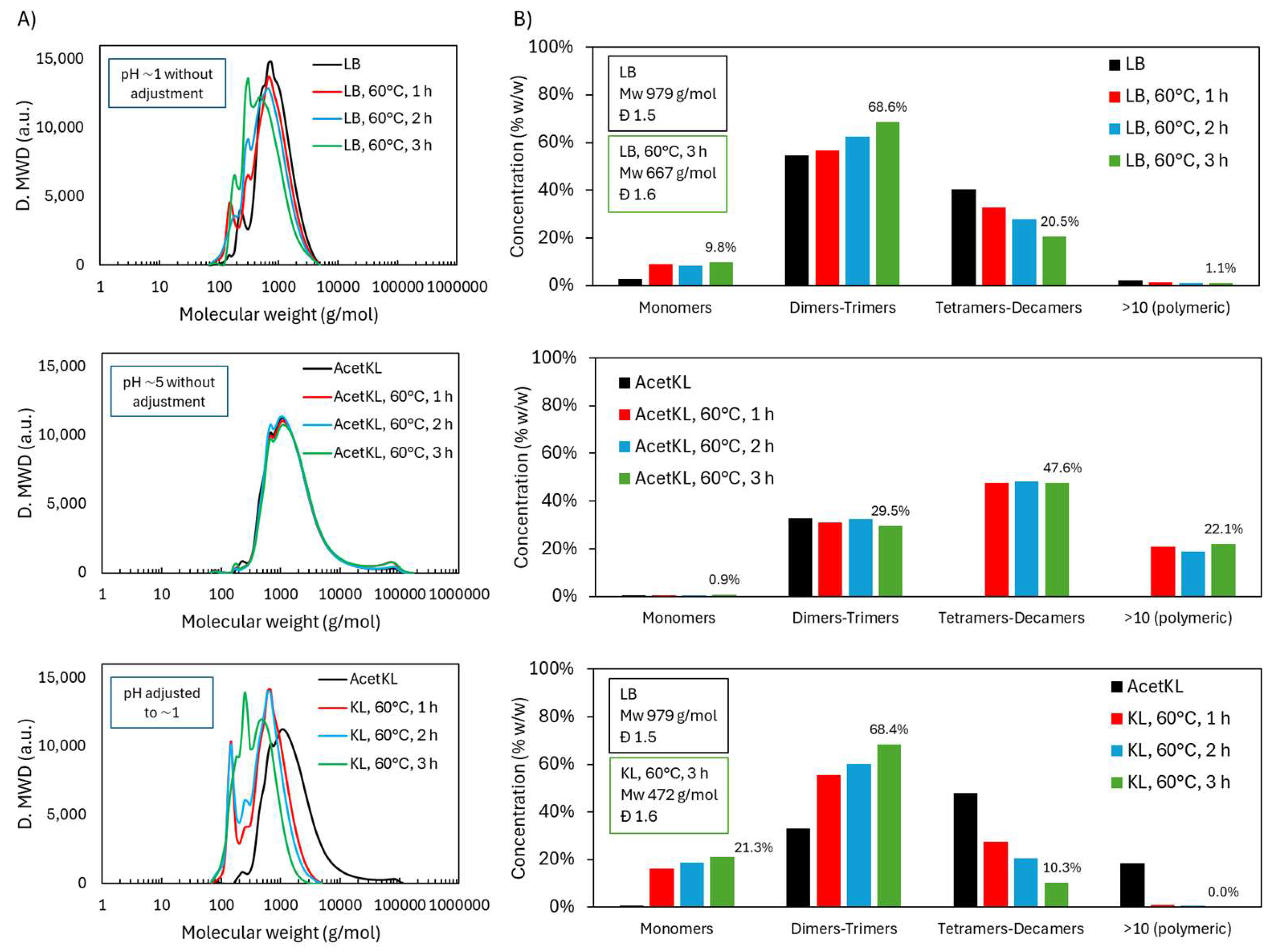
2.1.1. Molecular Weight Distribution During Acidic Oxidative Depolymerization
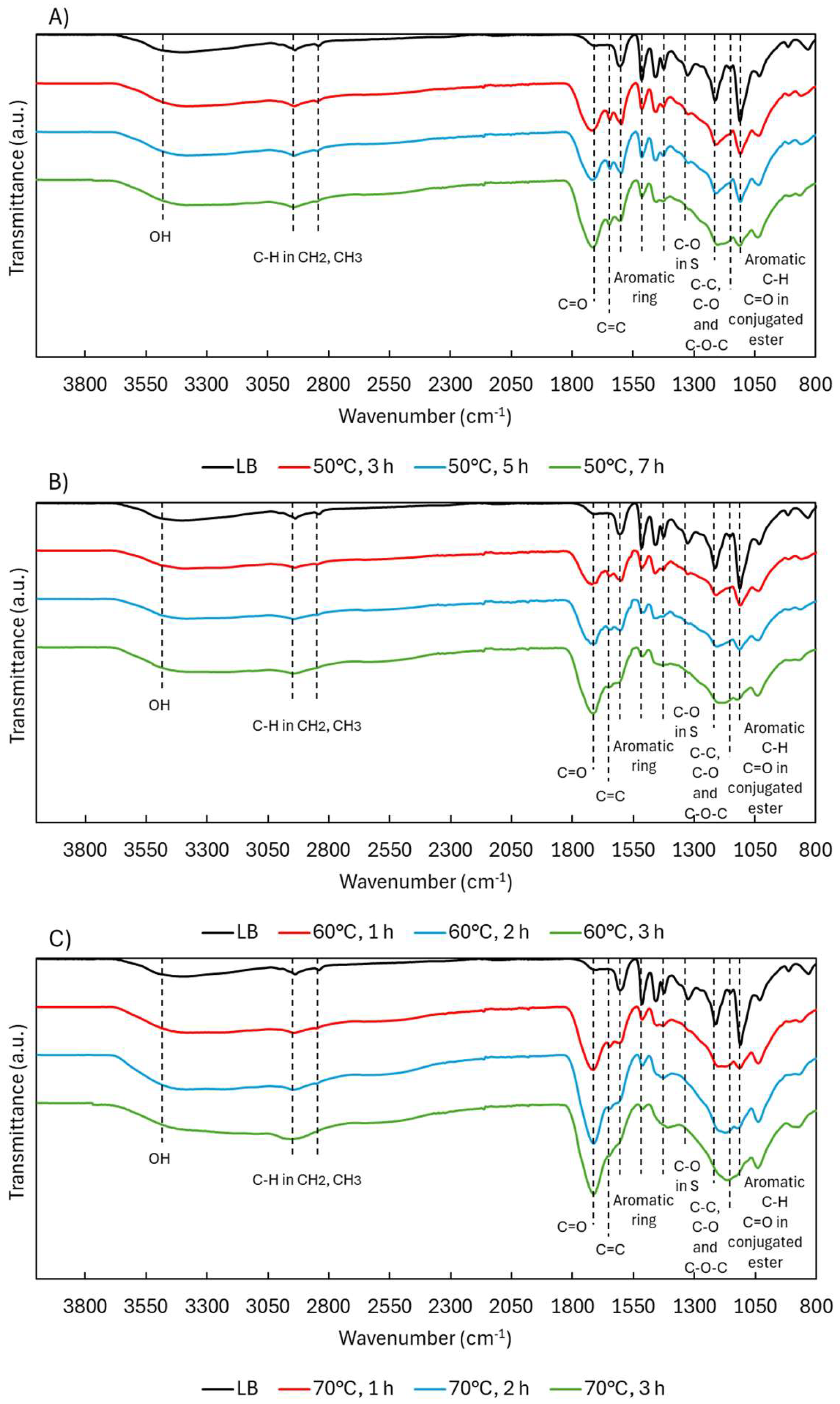
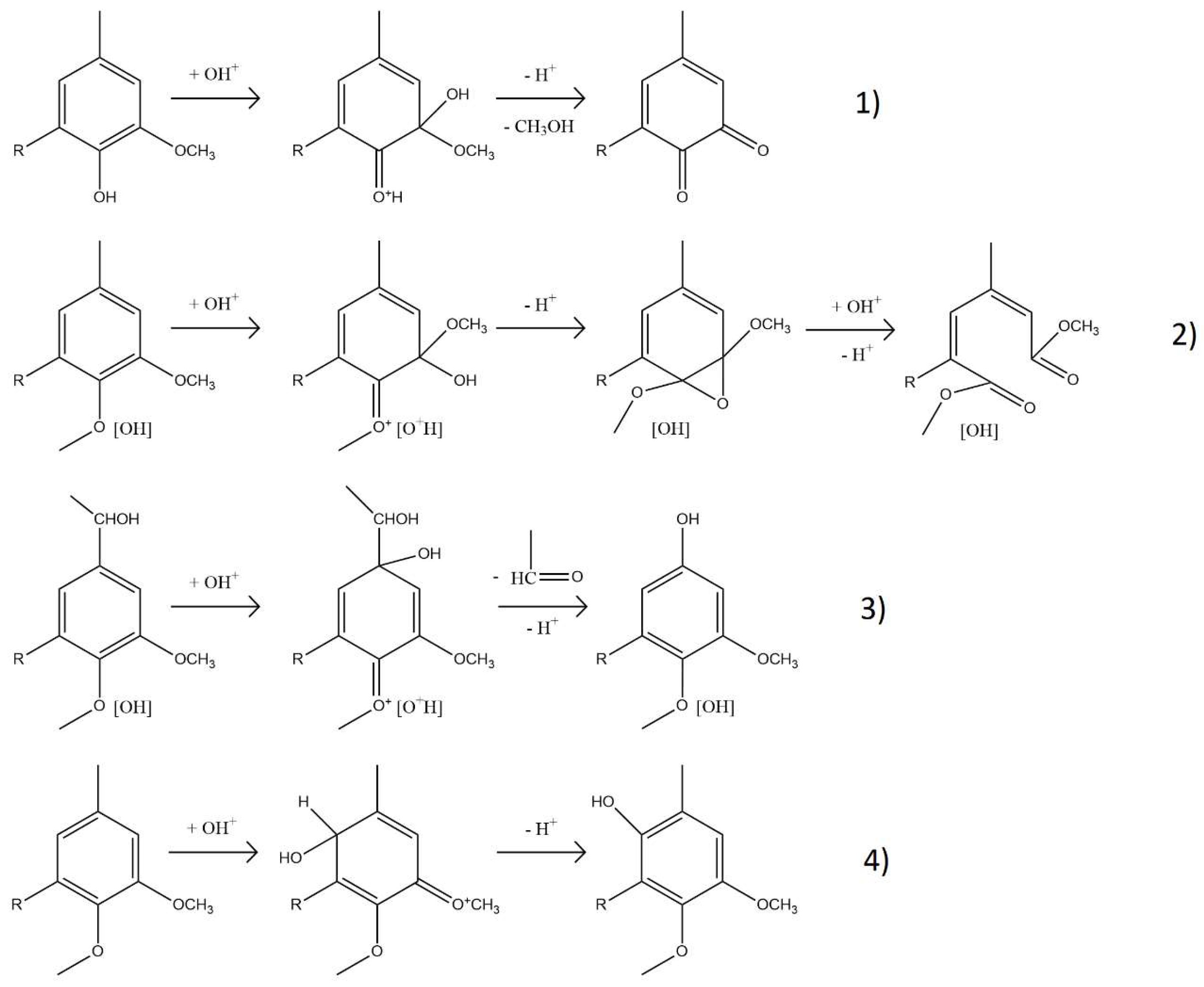
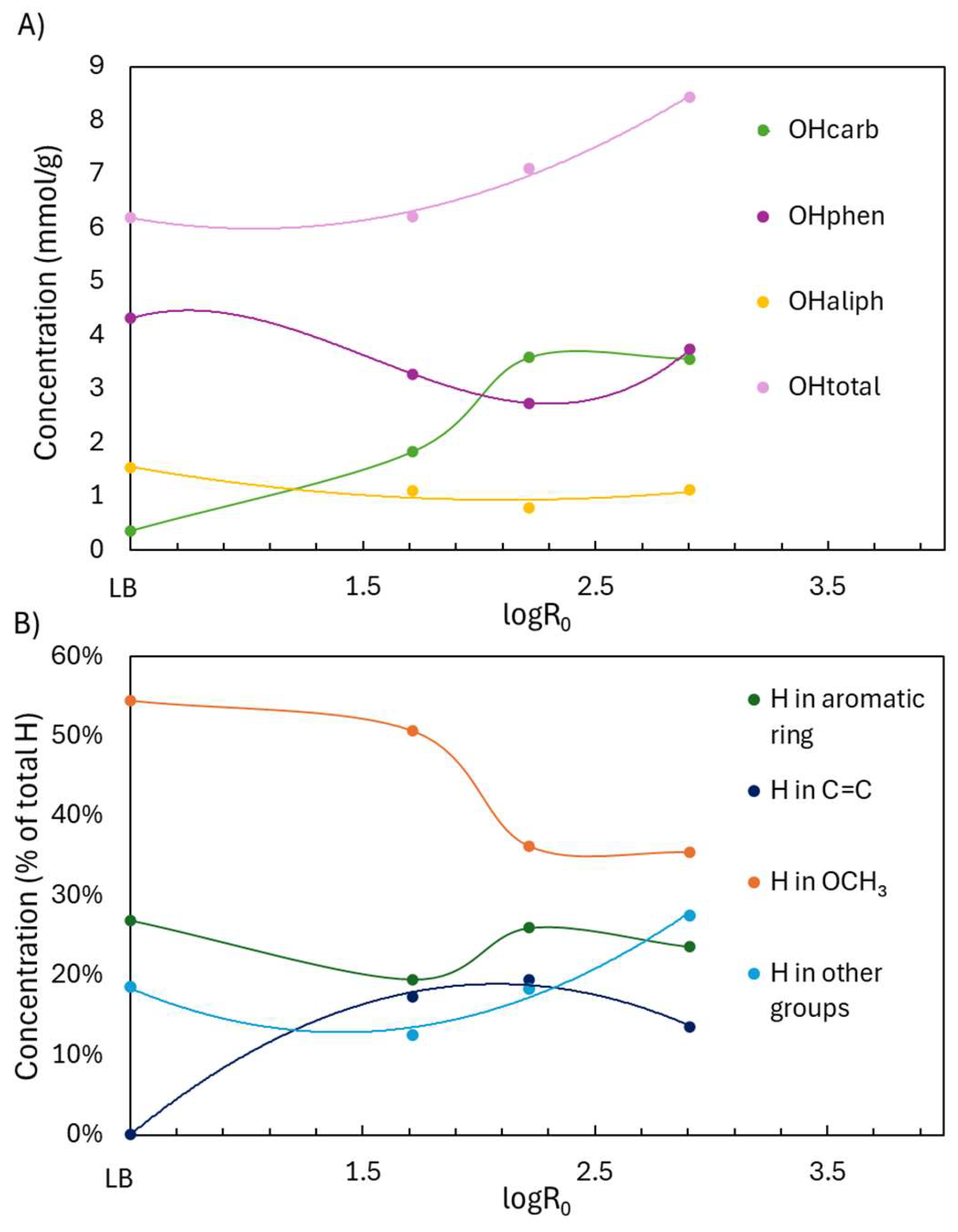
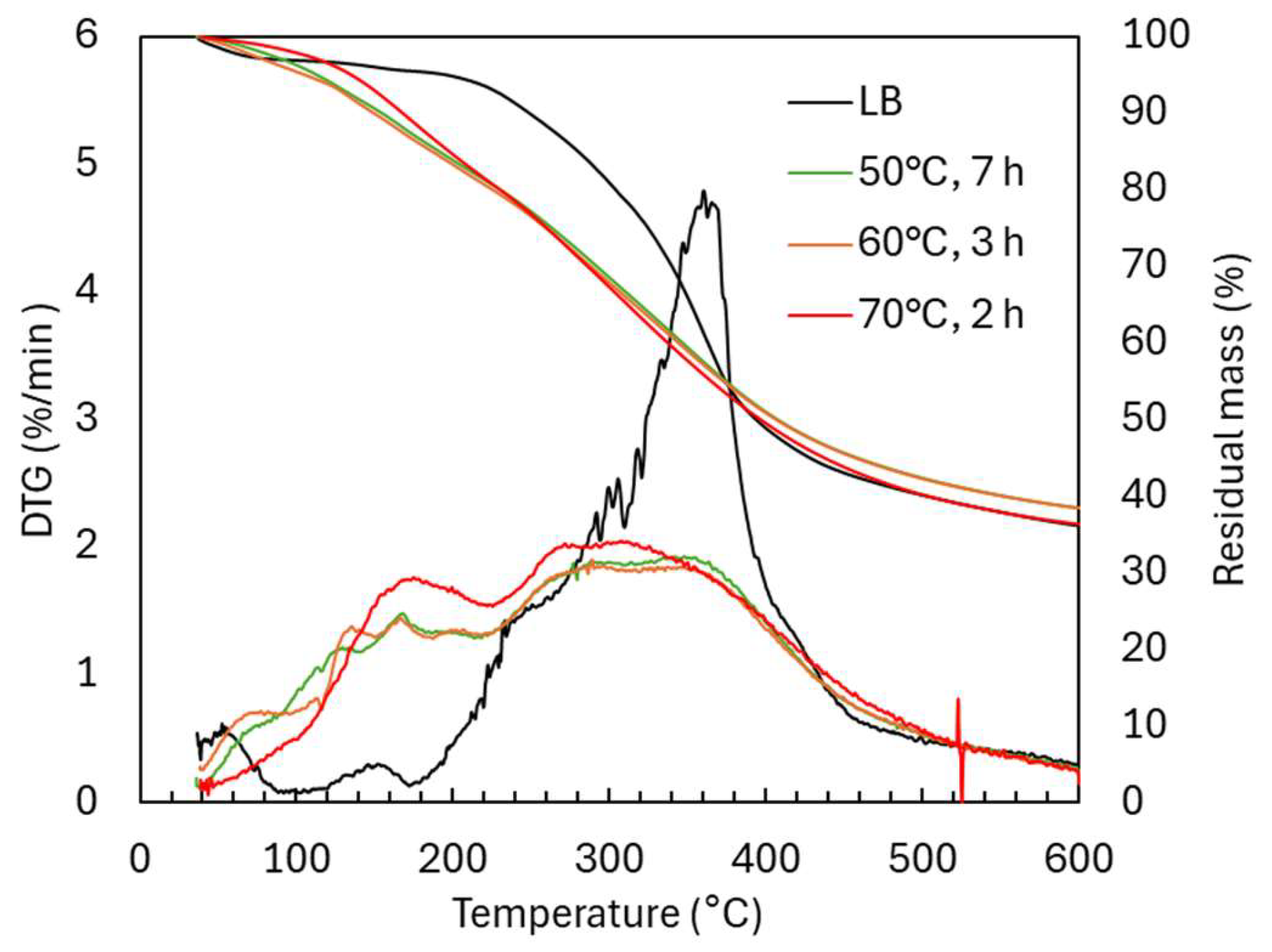
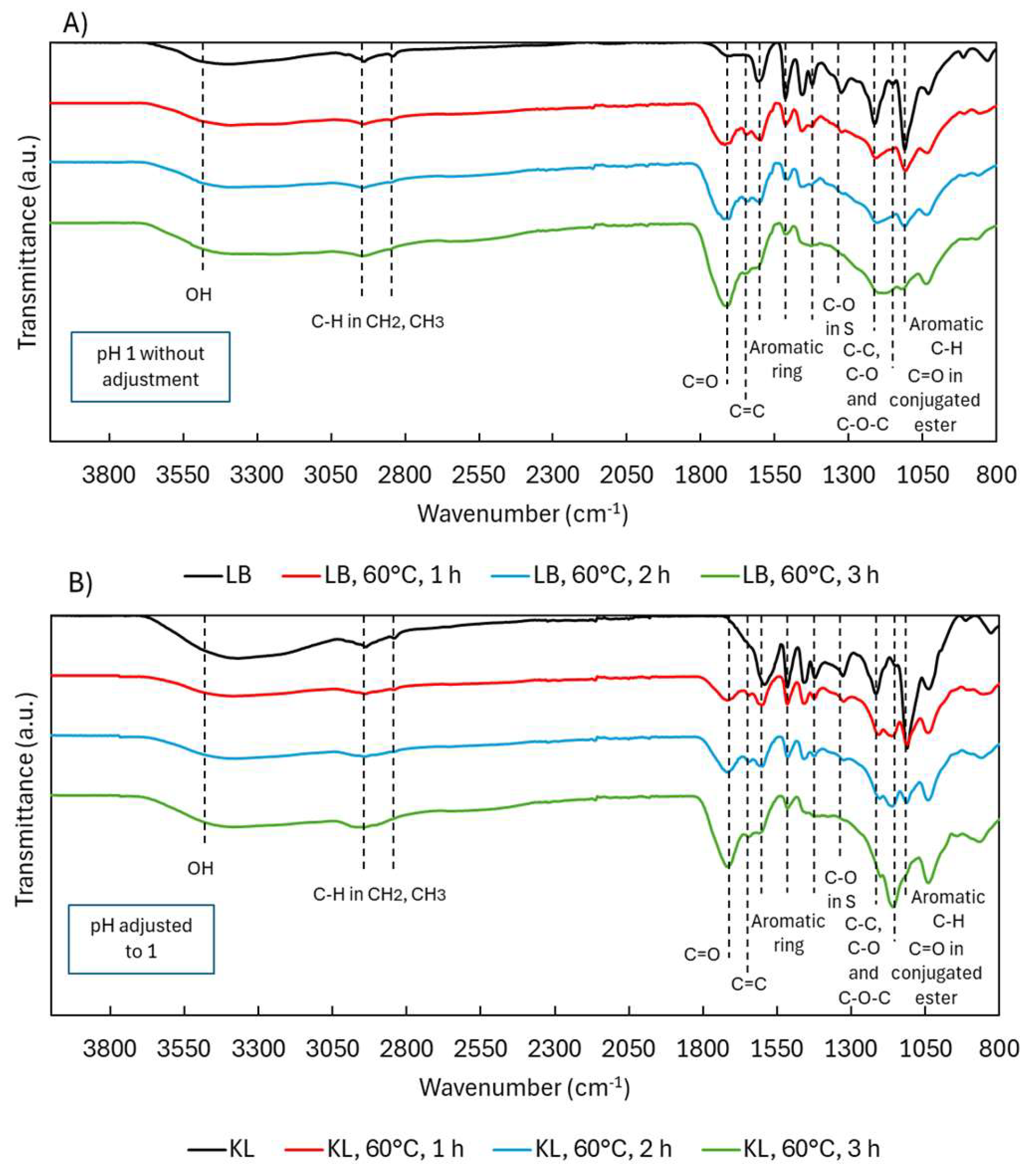
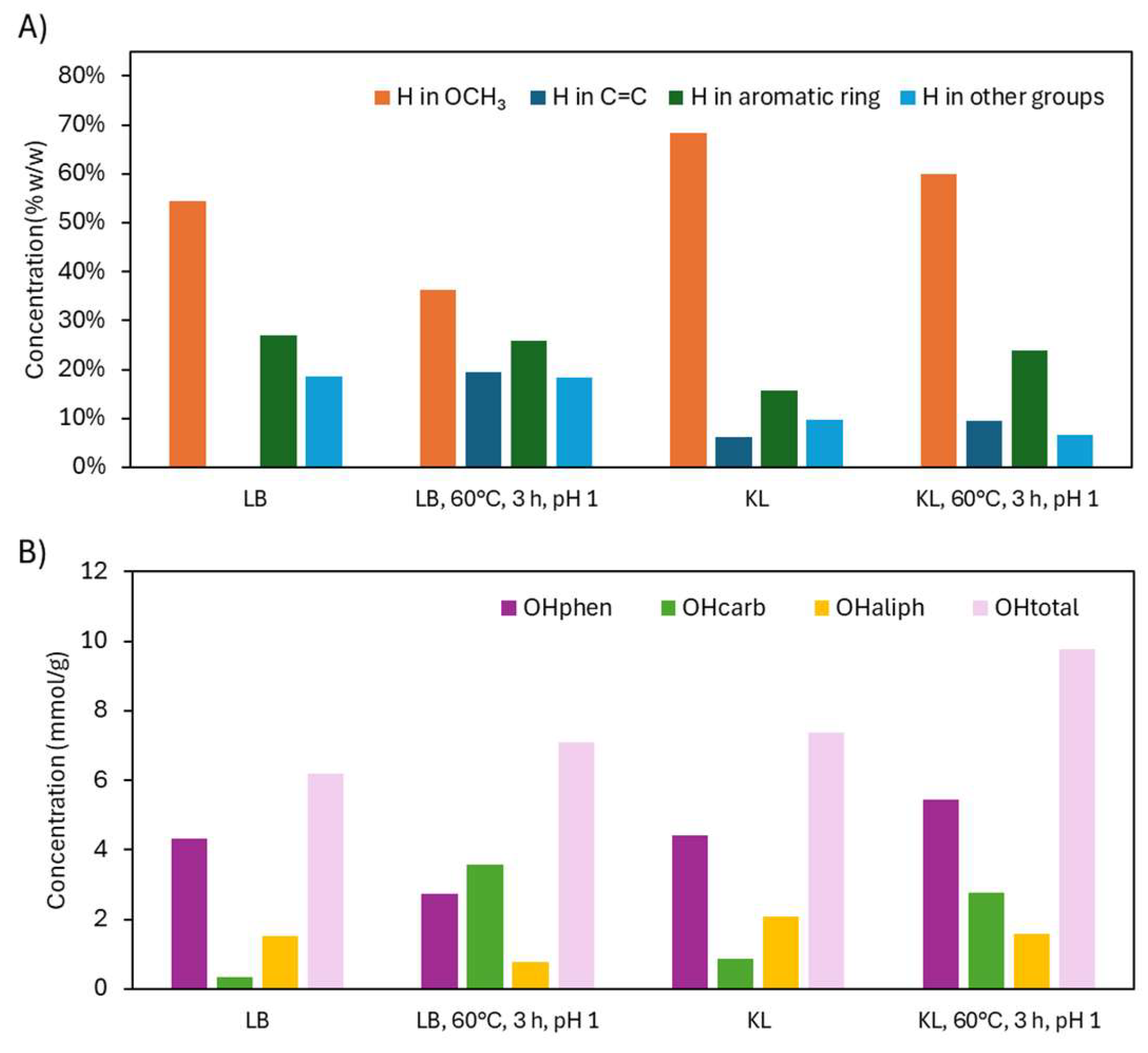
2.1.2. Structural Characterization (ATR-FTIR, 1H NMR, 31P NMR, EA, TGA, GC-FID/(TOF-MS))
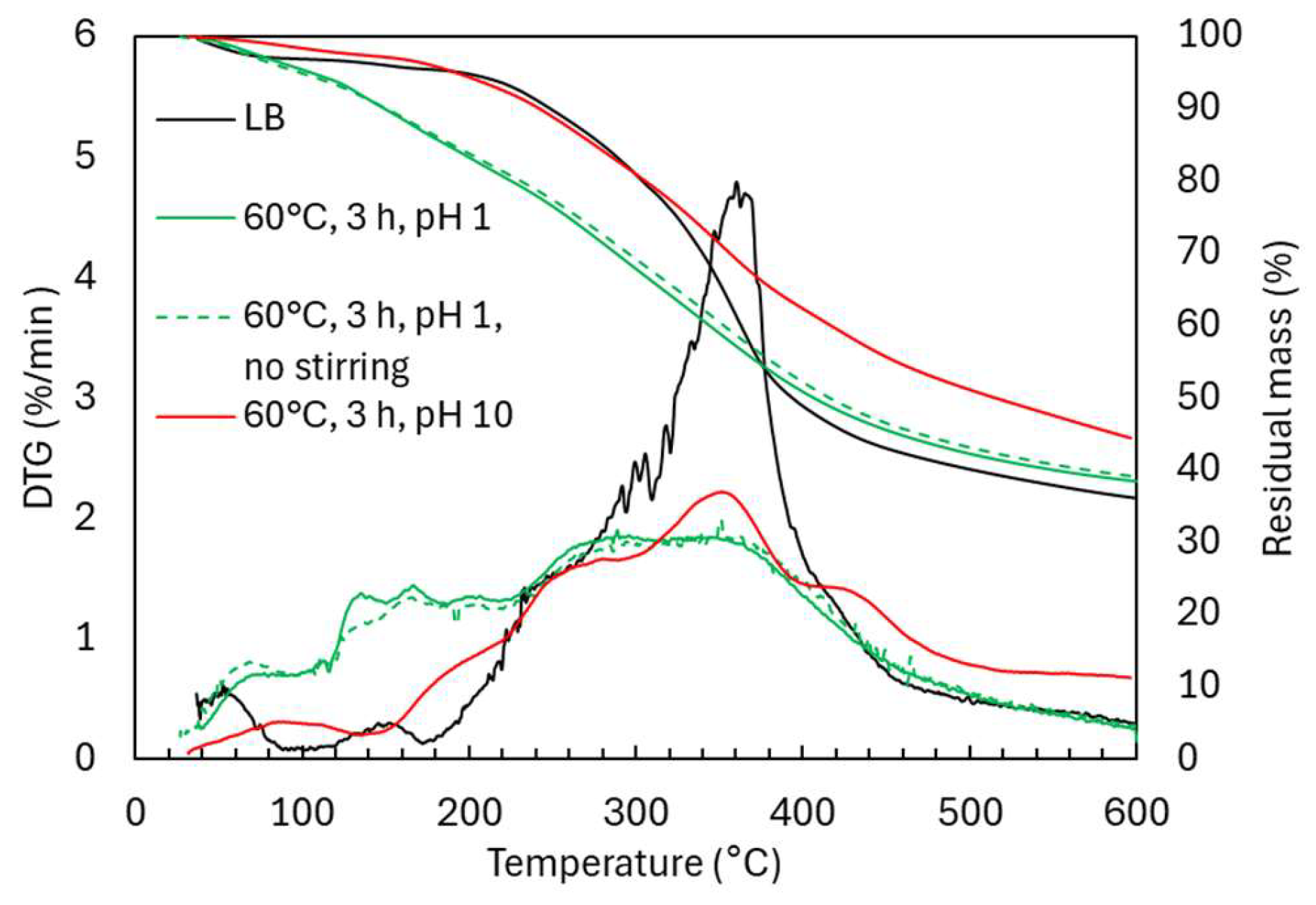
2.2. Oxidative Depolymerization: Effect of Stirring and pH
2.2.1. Molecular Weight Distribution
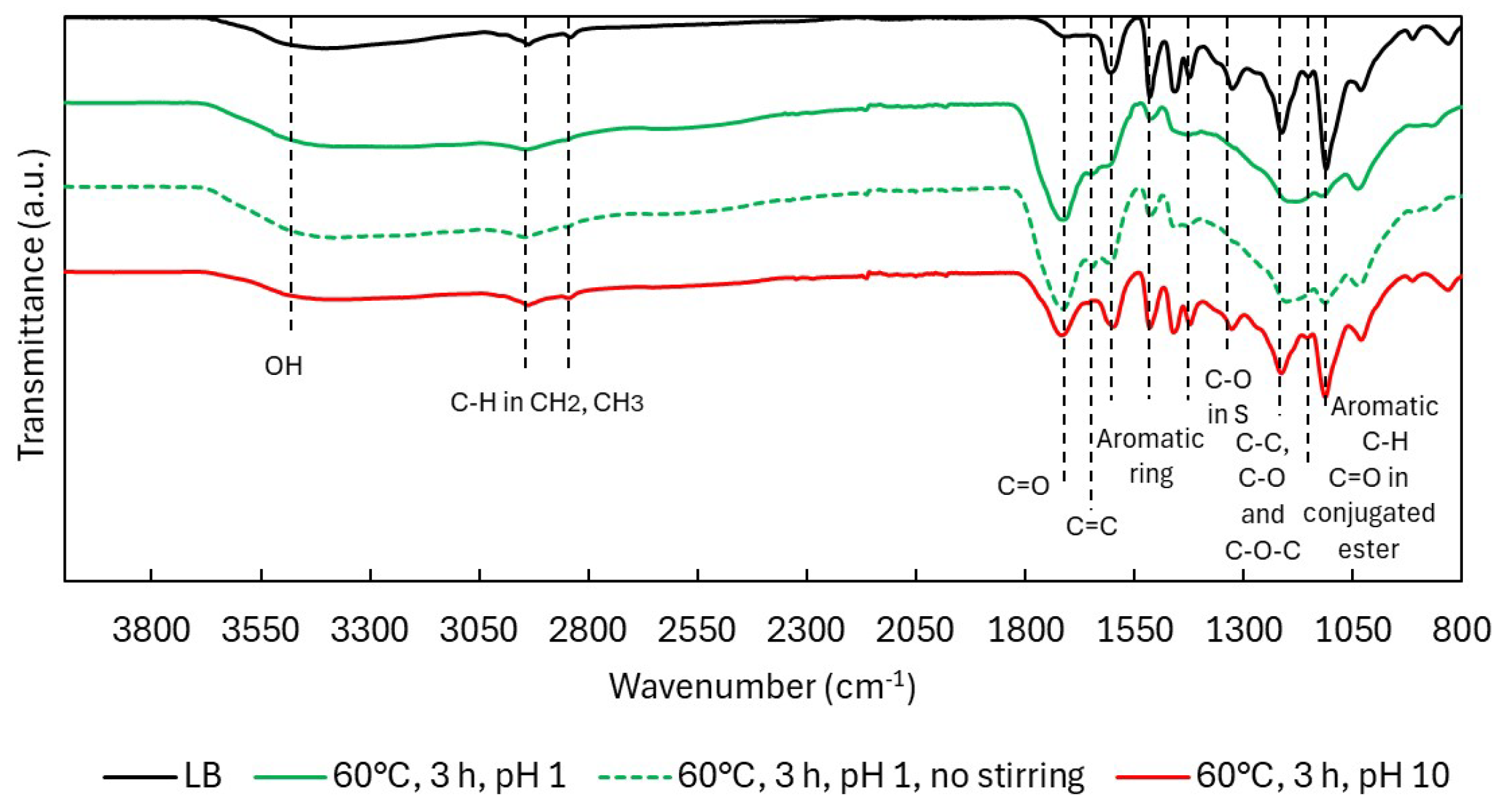
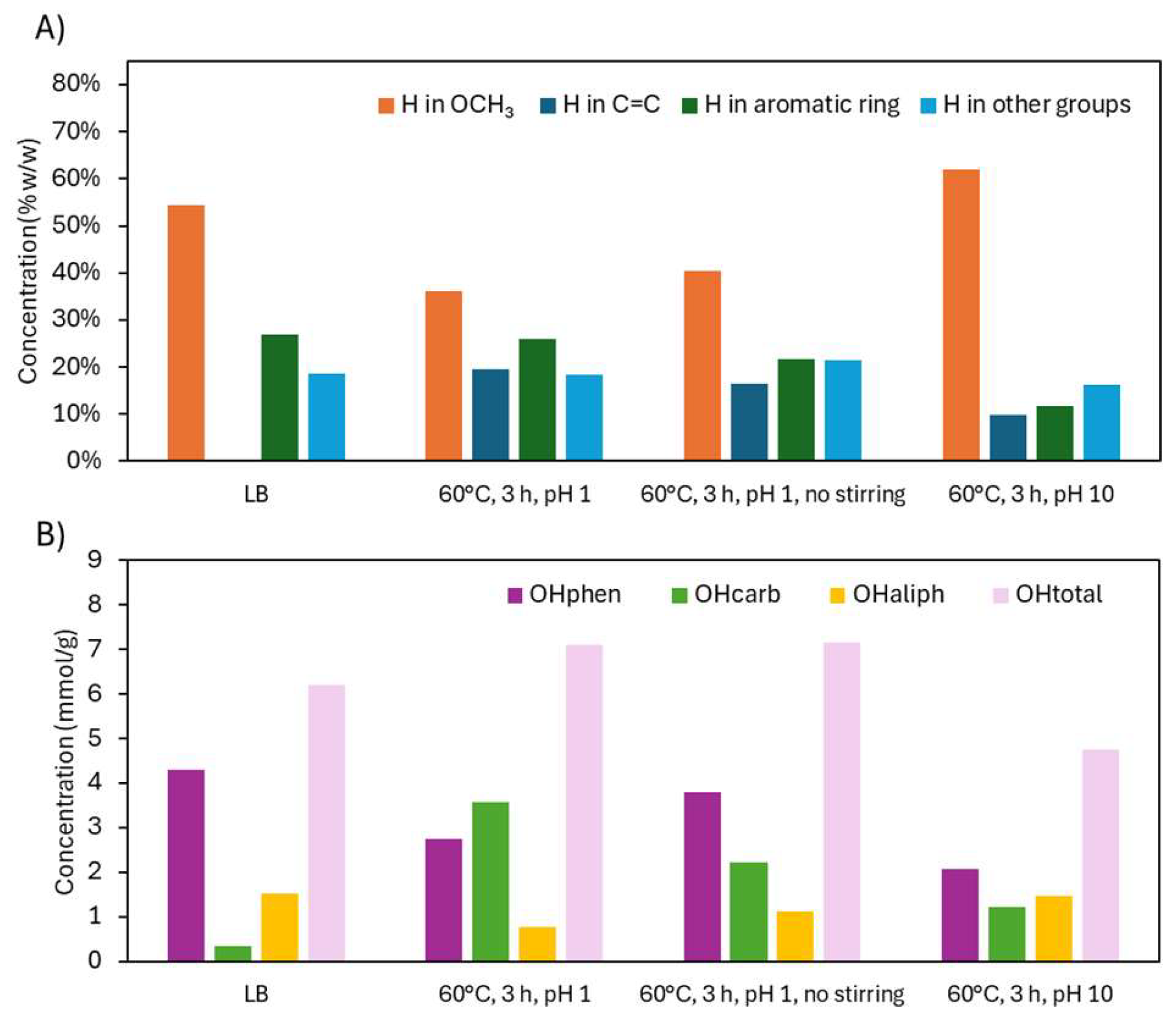
2.2.2. Structural Characterization (ATR-FTIR, 1H NMR, 31P NMR, EA, TGA)
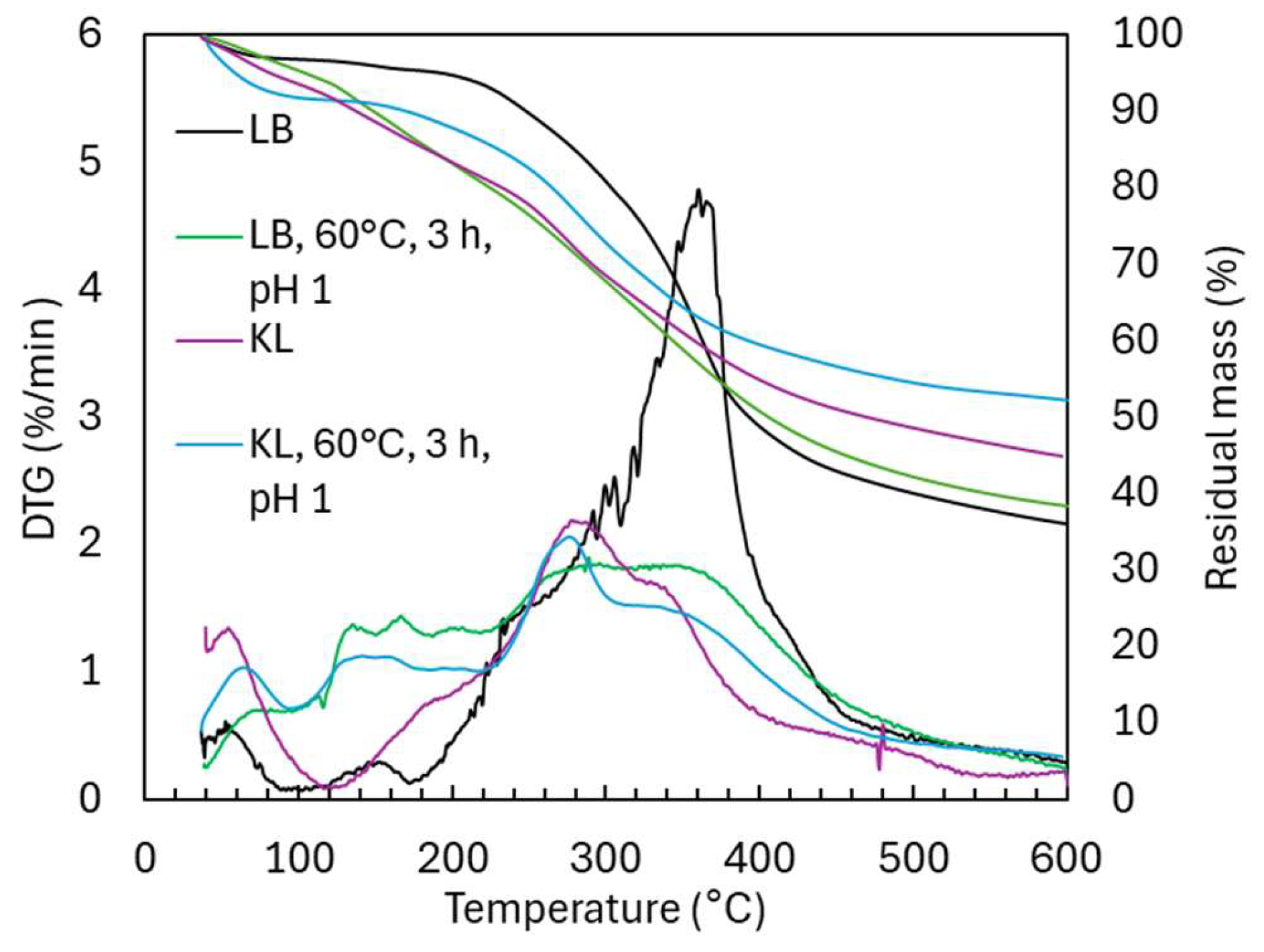
2.3. Oxidative Depolymerization of Kraft Lignin (KL) Versus Lignoboost Lignin (LB)
2.3.1. Molecular Weight Distribution During Acidic Oxidative Depolymerization
2.3.2. Structural Characterization (ATR-FTIR, 1H NMR, 31P NMR, TGA, and GC-FID/(TOF-MS))
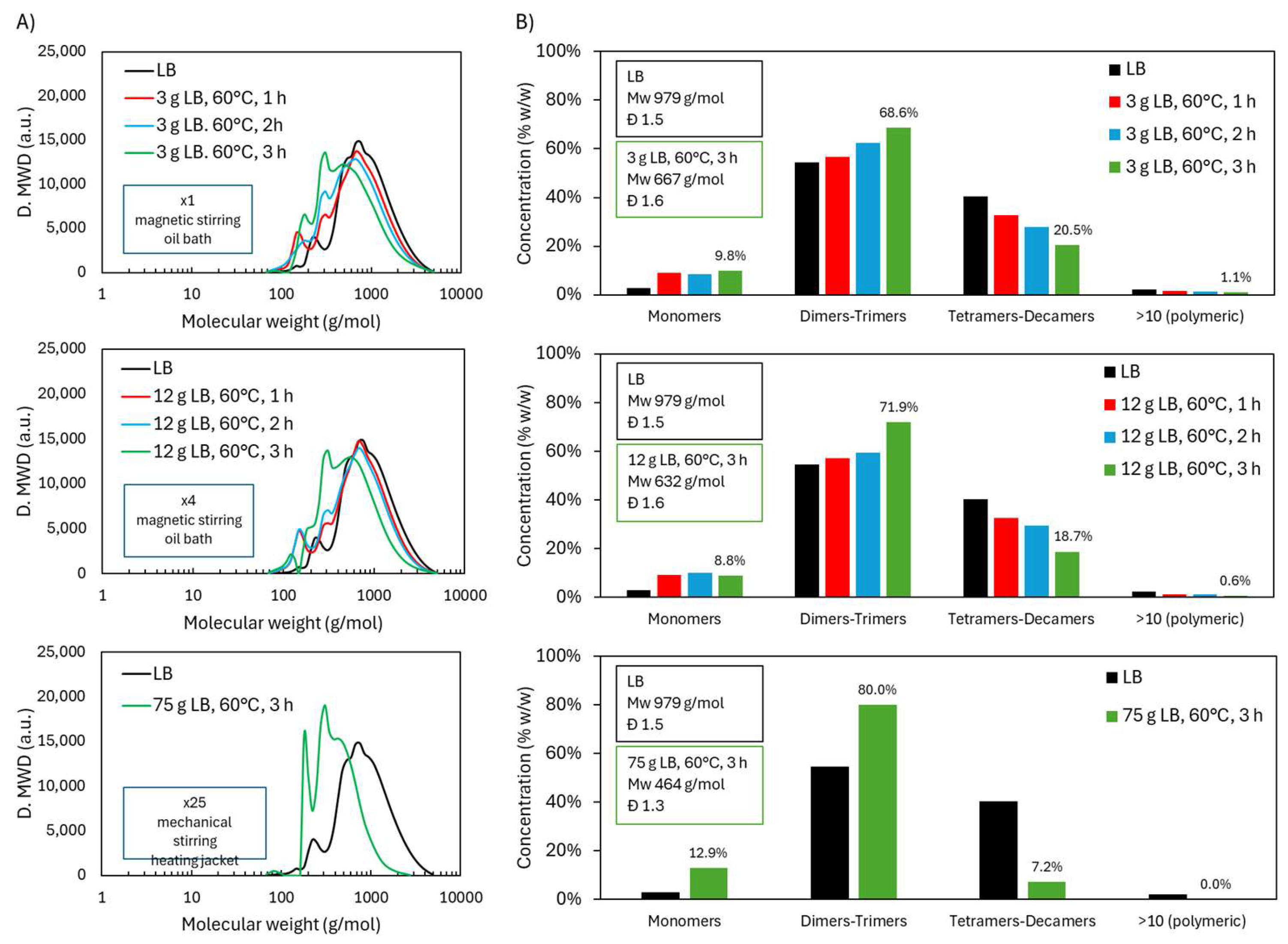
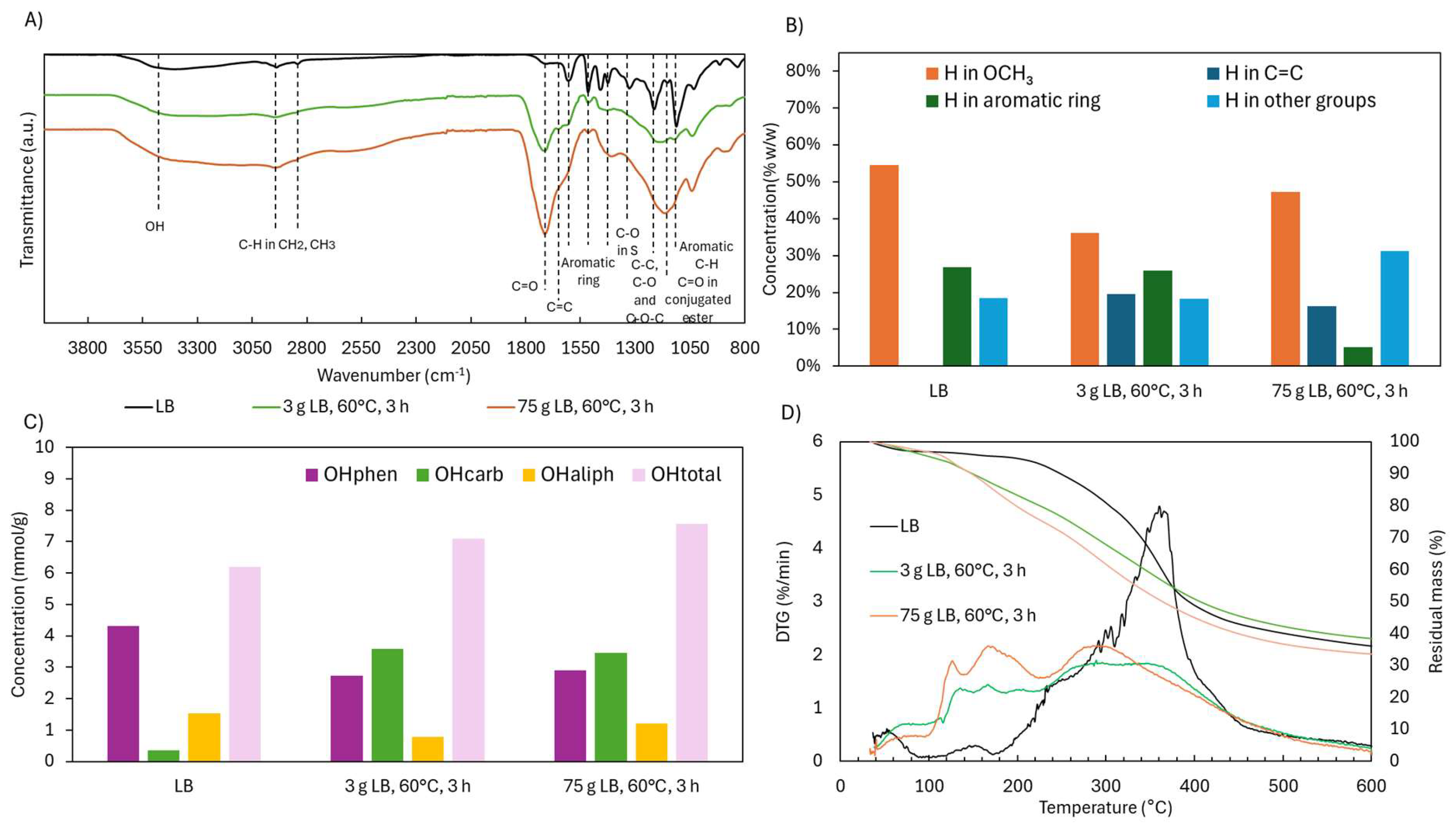
2.4. Scale-Up of Acidic Oxidative Depolymerization of Lignoboots Lignin (LB)
2.4.1. Molecular Weight Distribution During Acidic Oxidative Depolymerization
2.4.2. Structural Characterization (ATR-FTIR, 1H NMR, 31P NMR, and TGA)
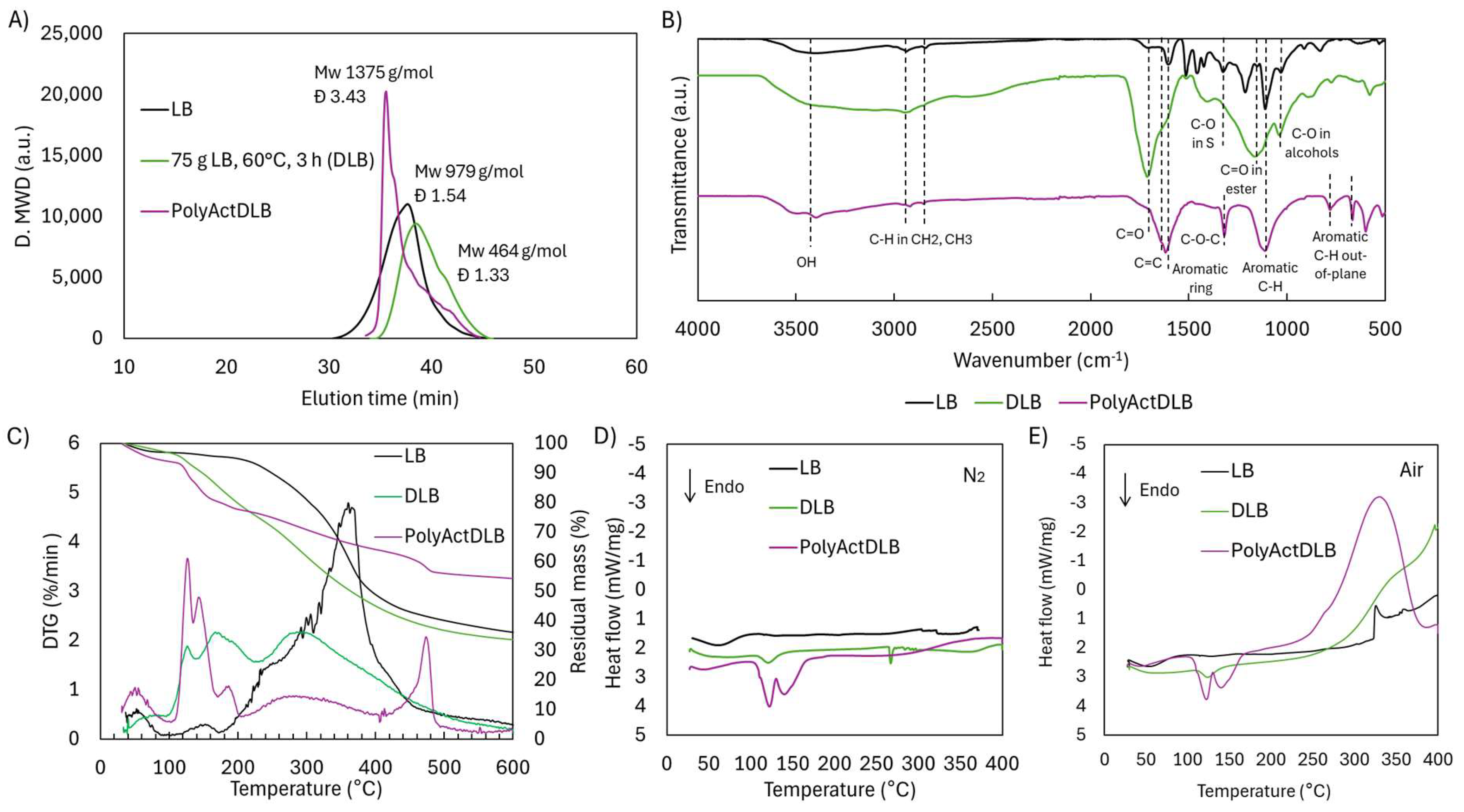
2.5. Reactivity of Depolymerized/Functionalized Lignoboost Lignin (LB) Through Radical Crosslinking
3. Materials and Methods
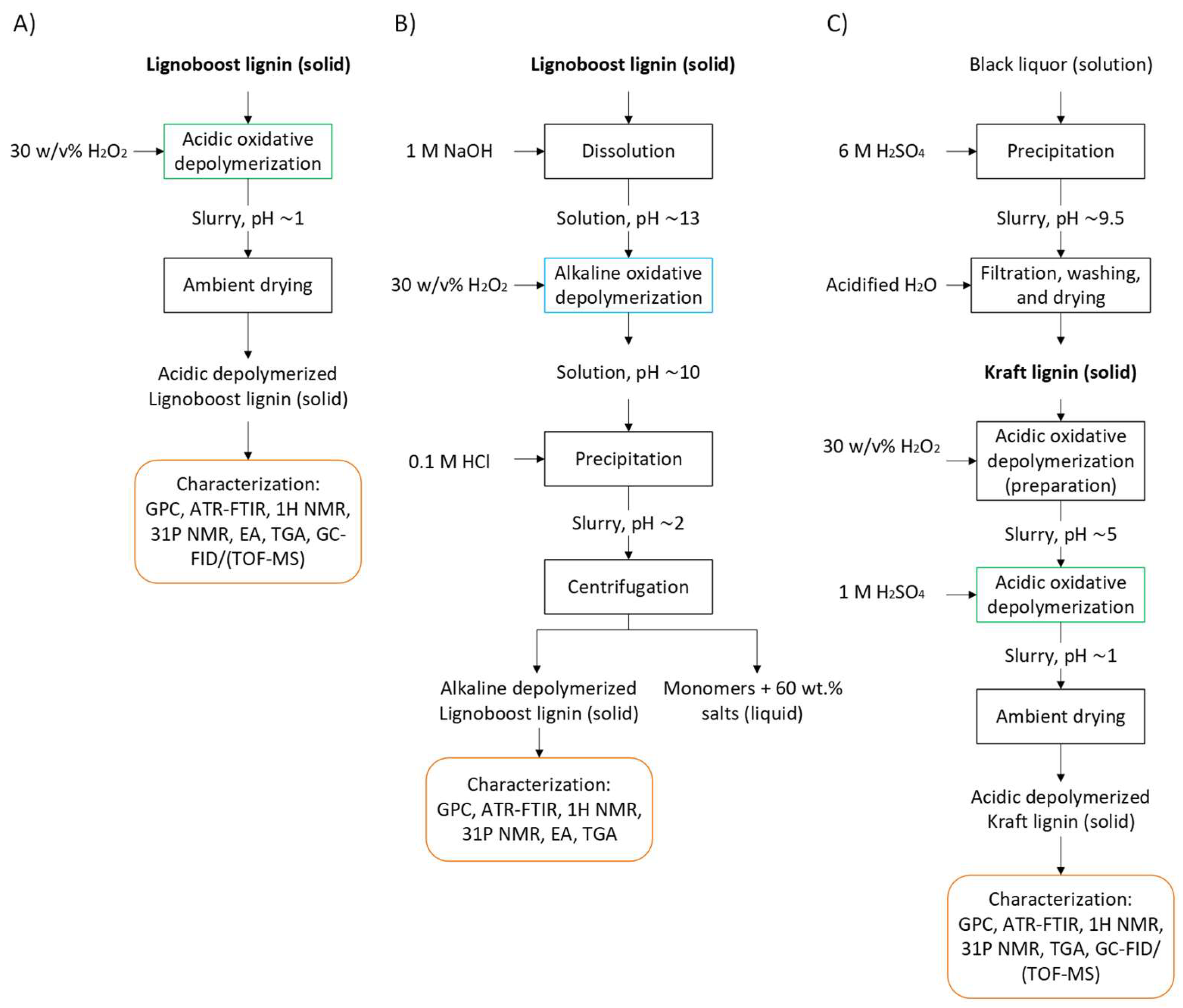
3.1. Raw Material
3.2. Oxidative Depolymerization of Lignin
3.3. Radical Crosslinking of the Depolymerized Lignin
3.4. Characterization of Lignoboost Lignin (LB), Kraft Lignin (KL), Depolymerized Lignin Samples, and Lignin-Derived Crosslinked Matrix
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| LB | Lignoboost lignin |
| KL | Kraft lignin |
| DLB | Depolymerized Lignoboost lignin |
| DP | Degree of polymerization |
| PolyActDLB | Crosslinked polymer derived from DLB |
| PPU | Phenylpropane unit |
| Mw | Weight-average molecular weight |
| Đ | Dispersity index (Mw/Mn) |
| logR0 | Logarithmic severity factor |
| OHphen | Phenolic hydroxyl group |
| OHaliph | Aliphatic hydroxyl group |
| OHcarb | Carboxylic hydroxyl group |
| OCH3 | Methoxy group |
| C=C/-CH=CH- | Aliphatic carbon-carbon double bond (alkene) |
| C=O | Carbonyl group (in ketones, acids, esters, etc.) |
| C-O-C | Ether linkage |
| THF | Tetrahydrofuran |
| DMSO-d6 | Deuterated dimethyl sulfoxide |
| MSTFA | N-Methyl-N-(trimethylsilyl)trifluoroacetamide (derivatizing agent) |
| TMDP | 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (for 31P NMR) |
| 1H NMR | Proton nuclear magnetic resonance spectroscopy |
| 31P NMR | Phosphorus-31 nuclear magnetic resonance spectroscopy |
| ATR-FTIR | Attenuated total reflectance Fourier-transform infrared spectroscopy |
| GPC | Gel permeation chromatography |
| RI | Refractive index |
| UV | Ultraviolet |
| EA | Elemental Analysis |
| TGA/DTG | Thermogravimetric analysis/Derivative thermogravimetry |
| DSC | Differential scanning calorimetry |
| GC-FID/(TOF-MS) | Gas chromatography with flame ionization detector and time-of-flight mass spectrometry |
References
- Sun, R.C. Lignin Source and Structural Characterization. ChemSusChem 2020, 13, 4385–4393. [Google Scholar] [CrossRef]
- Research and Markets Lignin Market: Analysis by Type, Form, Applications, Region Size, Trends and Forecast up to 2029. Available online: https://www.researchandmarkets.com/r/qnf8kt (accessed on 1 May 2025).
- Andriani, F.; Lawoko, M. Oxidative Carboxylation of Lignin: Exploring Reactivity of Different Lignin Types. Biomacromolecules 2024, 25, 4246–4254. [Google Scholar] [CrossRef]
- More, A.; Elder, T.; Jiang, Z. Towards a New Understanding of the Retro-Aldol Reaction for Oxidative Conversion of Lignin to Aromatic Aldehydes and Acids. Int. J. Biol. Macromol. 2021, 183, 1505–1513. [Google Scholar] [CrossRef]
- Schorr, D.; Diouf, P.N.; Stevanovic, T. Evaluation of Industrial Lignins for Biocomposites Production. Ind. Crops Prod. 2014, 52, 65–73. [Google Scholar] [CrossRef]
- Hubbe, M.A.; Alén, R.; Paleologou, M.; Kannangara, M.; Kihlman, J. Lignin Recovery from Spent Alkaline Pulping Liquors Using Acidification, Membrane Separation, and Related Processing Steps: A Review. BioResources 2019, 14, 2300–2351. [Google Scholar] [CrossRef]
- Gärtner, A.; Gellerstedt, G.; Tamminen, T. Determination of Phenolic Hydroxyl Groups in Residual Lignin Using a Modified UV-Method Anna. Nord. Pulp Pap. Res. J. 1999, 14, 163–170. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, M.; Zhang, Y.; Cao, Q.; Wang, X.; Han, Y.; Sun, G.; Li, Y.; Zhou, J. Novel Lignin–Chitosan–PVA Composite Hydrogel for Wound Dressing. Mater. Sci. Eng. C 2019, 104, 110002. [Google Scholar] [CrossRef]
- Qu, W.; Huang, Y.; Luo, Y.; Kalluru, S.; Cochran, E.; Forrester, M.; Bai, X. Controlled Radical Polymerization of Crude Lignin Bio-Oil Containing Multihydroxyl Molecules for Methacrylate Polymers and the Potential Applications. ACS Sustain. Chem. Eng. 2019, 7, 9050–9060. [Google Scholar] [CrossRef]
- Rodrigues, A.E.; Vega-Aguilar, C.A.; Filomena Barreiro, M. Effect of Methoxy Substituents on Wet Peroxide Oxidation of Lignin and Lignin Model Compounds: Understanding the Pathway to C4 Dicarboxylic Acids. Ind. Eng. Chem. Res. 2021, 60, 3543–3553. [Google Scholar] [CrossRef]
- More, A.; Elder, T.; Jiang, Z. A Review of Lignin Hydrogen Peroxide Oxidation Chemistry with Emphasis on Aromatic Aldehydes and Acids. Holzforschung 2021, 75, 806–823. [Google Scholar] [CrossRef]
- Ahmad, Z.; Dajani, W.W.A.; Paleologou, M.; Xu, C. Sustainable Process for the Depolymerization/Oxidation of Softwood and Hardwood Kraft Lignins Using Hydrogen Peroxide under Ambient Conditions. Molecules 2020, 25, 2329. [Google Scholar] [CrossRef] [PubMed]
- Junghans, U.; Bernhardt, J.J.; Wollnik, R.; Triebert, D.; Unkelbach, G.; Pufky-Heinrich, D. Valorization of Lignin via Oxidative Depolymerization with Hydrogen Peroxide: Towards Carboxyl-Rich Oligomeric Lignin Fragments. Molecules 2020, 25, 2717. [Google Scholar] [CrossRef] [PubMed]
- Ruwoldt, J.; Skunde, R.; Tanase-Opedal, M.; Syverud, K. Carboxylation of Lignin by Oxidation with Hydrogen Peroxide and Its Use as Emulsion Stabilizer. Ind. Crops Prod. 2025, 223, 120019. [Google Scholar] [CrossRef]
- Kim, J.-C.; Choi, J.-H.; Kim, J.-H.; Cho, S.-M.; Park, S.-W.; Cho, Y.-M.; Park, S.-Y.; Kwak, H.W.; Choi, I.-G. Development of Lignin-Based Polycarboxylates as a Plasticizer for Cement Paste via Peracetic Acid Oxidation. BioResources 2020, 15, 8133–8145. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y. Metal Catalyst-Free Oxidative C−C Bond Cleavage of a Lignin Model Compound by H2O2 in Formic Acid. ChemSusChem 2020, 13, 1740–1745. [Google Scholar] [CrossRef]
- Vega-Aguilar, C.A.; Barreiro, M.F.; Rodrigues, A.E. Lignin Conversion into C4 Dicarboxylic Acids by Catalytic Wet Peroxide Oxidation Using Titanium Silicalite-1. Ind. Crops Prod. 2021, 173, 114155. [Google Scholar] [CrossRef]
- Sun, S.; Qiu, X.; Hao, S.; Ravichandran, S.; Song, J.; Zhang, W. Electrochemical Conversion of Lignin to Short-Chain Carboxylic Acids. Green Chem. 2023, 25, 3127–3136. [Google Scholar] [CrossRef]
- Bi, Z.; Li, Z.; Yan, L. Catalytic Oxidation of Lignin to Dicarboxylic Acid over the CuFeS2 Nanoparticle Catalyst. Green Process. Synth. 2018, 7, 306–315. [Google Scholar] [CrossRef]
- Ding, R.; Wu, H.; Thunga, M.; Bowler, N.; Kessler, M.R. Processing and Characterization of Low-Cost Electrospun Carbon Fibers from Organosolv Lignin/Polyacrylonitrile Blends. Carbon N. Y. 2016, 100, 126–136. [Google Scholar] [CrossRef]
- Mainka, H.; Täger, O.; Körner, E.; Hilfert, L.; Busse, S.; Edelmann, F.T.; Herrmann, A.S. Lignin—An Alternative Precursor for Sustainable and Cost-Effective Automotive Carbon Fiber. J. Mater. Res. Technol. 2015, 4, 283–296. [Google Scholar] [CrossRef]
- Ouyang, Q.; Cheng, L.; Wang, H.; Li, K. Mechanism and Kinetics of the Stabilization Reactions of Itaconic Acid-Modified Polyacrylonitrile. Polym. Degrad. Stab. 2008, 93, 1415–1421. [Google Scholar] [CrossRef]
- Gierer, J. Chemistry of Delignification—Part 2: Reactions of Lignins during Bleaching. Wood Sci. Technol. 1986, 20, 1–33. [Google Scholar] [CrossRef]
- Gonçalves, S.; Martins, J.; Paiva, N.T.; Paiva, D.; Carvalho, L.H.; Magalhães, F.D. The Potential of Visible Spectroscopy as a Tool for the In-Line Monitoring of Lignin Methylolation. Polymers 2023, 15, 178. [Google Scholar] [CrossRef] [PubMed]
- Mainka, H.; Hilfert, L.; Busse, S.; Edelmann, F.; Haak, E.; Herrmann, A.S. Characterization of the Major Reactions during Conversion of Lignin to Carbon Fiber. J. Mater. Res. Technol. 2015, 4, 377–391. [Google Scholar] [CrossRef]
- Liu, X.; Xu, Y.; Yu, J.; Li, S.; Wang, J.; Wang, C.; Chu, F. Integration of Lignin and Acrylic Monomers towards Grafted Copolymers by Free Radical Polymerization. Int. J. Biol. Macromol. 2014, 67, 483–489. [Google Scholar] [CrossRef]
- Xu, X.; Xu, Y.; Deng, S.; Chen, X.; Essawy, H.; Lee, S.H.; Lum, W.C.; Zhou, X.; Zhang, J. Graft Copolymer of Tannin and Polyvinyl Alcohol with Acrylic Acid for the Preparation of Hydrophobic Biodegradable Film. Prog. Org. Coatings 2024, 186, 108090. [Google Scholar] [CrossRef]
- Falcão, L.; Araújo, M.E.M. Application of ATR-FTIR Spectroscopy to the Analysis of Tannins in Historic Leathers: The Case Study of the Upholstery from the 19th Century Portuguese Royal Train. Vib. Spectrosc. 2014, 74, 98–103. [Google Scholar] [CrossRef]
- Dutra, L.M.; Teles, P.H.V.; de Melo, N.F.; Nagata, N.; Almeida, J.R.G. da S. ATR-FTIR Spectroscopy Combined with Chemometric Tools for Rapid Distinction of Passiflora L. Species. Rev. Bras. Plantas Med. 2023, 25, 54–62. [Google Scholar] [CrossRef]
- Overend, R.P.; Chornet, E.; Gascoigne, J.A. Fractionation of Lignocellulosics by Steam-Aqueous Pretreatments. Philos. Trans. R. Soc. London. Ser. A Math. Phys. Sci. 1987, 321, 523–536. [Google Scholar] [CrossRef]
- Kim, Y.; Kreke, T.; Mosier, N.S.; Ladisch, M.R. Severity Factor Coefficients for Subcritical Liquid Hot Water Pretreatment of Hardwood Chips. Biotechnol. Bioeng. 2014, 111, 254–263. [Google Scholar] [CrossRef]
- Maitz, S.; Schlemmer, W.; Hobisch, M.A.; Hobisch, J.; Kienberger, M. Preparation and Characterization of a Water-Soluble Kraft Lignin. Adv. Sustain. Syst. 2020, 4, 2000052. [Google Scholar] [CrossRef]
- Ramos-Andrés, M.; Díaz-Cesteros, S.; Majithia, N.; García-Serna, J. Pilot-Scale Biorefinery for the Production of Purified Biopolymers Based on Hydrothermal Treatment in Flow-through Reactor Cycles. Chem. Eng. J. 2022, 437, 135123. [Google Scholar] [CrossRef]
- Sameni, J.; Krigstin, S.; Sain, M. Characterization of Lignins Isolated from Industrial Residues and Their Beneficial Uses. BioResources 2016, 11, 8435–8456. [Google Scholar] [CrossRef]
- Meng, X.; Crestini, C.; Ben, H.; Hao, N.; Pu, Y.; Ragauskas, A.J.; Argyropoulos, D.S. Determination of Hydroxyl Groups in Biorefinery Resources via Quantitative 31P NMR Spectroscopy. Nat. Protoc. 2019, 14, 2627–2647. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | logR0 | C | H | O | S | N | PPU Empirical Formula | MwPPU (g/mol) |
|---|---|---|---|---|---|---|---|---|
| LB | - | 57.90 ± 0.03 | 5.55 ± 0.02 | 32.86 ± 0.06 | 3.10 ± 0.24 | 0.52 ± 0.33 | C9H5.944O2.461S0.228N0.087(OCH3)2.372 | 235.69 |
| 50 °C, 7 h | 1.71 | 50.73 ± 0.19 | 4.71 ± 0.02 | 39.41 ± 0.34 | 4.66 ± 0.28 | 0.51 ± 0.17 | C9H6.083O4.373S0.382N0.095(OCH3)2.085 | 262.24 |
| 60 °C, 3 h | 2.21 | 48.56 ± 0.16 | 4.36 ± 0.02 | 41.90 ± 0.26 | 4.57 ± 0.21 | 0.62 ± 0.11 | C9H7.102O5.350S0.365N0.113(OCH3)1.343 | 255.60 |
| 70 °C, 2 h | 2.91 | 46.48 ± 0.09 | 4.71 ± 0.20 | 43.46 ± 0.12 | 4.84 ± 0.25 | 0.52 ± 0.12 | C9H8.254O5.860S0.410N0.101(OCH3)1.508 | 271.28 |
| Formula | Mw (g/mol) | LB 50 °C, 7 h logR0 1.71 | LB 60 °C, 3 h logR0 2.21 | LB 70 °C, 2 h logR0 2.91 | KL 60 °C, 3 h logR0 2.21 | |
|---|---|---|---|---|---|---|
| Alcohols | - | - | 2.01% | 2.04% | 1.44% | 1.33% |
| Ethylene glycol | C2H6O2 | 62.08 | 2.01% | 2.04% | 1.44% | 1.33% |
| Hydroxycarboxylic acids | - | - | 14.73% | 14.73% | 16.93% | 19.21% |
| Lactic acid | C3H6O3 | 90.09 | 0.72% | 0.74% | 0.53% | 4.24% |
| Glycolic acid | C2H4O3 | 76.06 | 9.15% | 9.27% | 11.40% | 8.94% |
| 2-Hydroxybutyric acid | C4H8O3 | 104.12 | 0.43% | 0.44% | 0.22% | 3.10% |
| Glyceric acid | C3H6O4 | 106.09 | 3.03% | 3.07% | 3.15% | 1.95% |
| Malic acid | C4H6O5 | 134.10 | 1.40% | 1.42% | 1.63% | 0.97% |
| Dicarboxylic acids | - | - | 50.28% | 53.40% | 56.21% | 29.36% |
| Oxalic acid | C2H2O4 | 90.04 | 27.14% | 27.52% | 15.72% | 8.88% |
| Propanedioic acid | C3H4O4 | 104.07 | 19.96% | 22.65% | 36.26% | 18.23% |
| Succinic acid | C4H6O4 | 118.10 | 1.76% | 1.79% | 2.19% | 1.26% |
| Ethylmalonic acid | C5H8O4 | 132.13 | 1.42% | 1.44% | 2.03% | 0.99% |
| Esters | - | - | 1.65% | 1.68% | 11.28% | 6.43% |
| Methyl 2-hydroxypropanoate | C4H8O3 | 104.12 | 0.18% | 0.18% | 1.88% | 0.99% |
| Monomethyl succinate | C5H8O4 | 132.13 | n.d. | n.d. | 3.35% | 1.28% |
| Methyl 2-hydroxyethyl malonate | C6H10O5 | 162.16 | n.d. | n.d. | 4.88% | 2.71% |
| Butyl 6-methylheptanoate | C12H24O2 | 200.36 | 1.47% | 1.49% | 1.17% | 1.45% |
| Lactones | - | - | 0.54% | 0.55% | 2.39% | 2.69% |
| 3-Hydroxy-3-hydroxymethyl-dihydro-2(3H)-furanone | C5H8O4 | 132.13 | 0.39% | 0.40% | 2.08% | 0.55% |
| 2,3,4,5-Tetrahydroxypentanoic acid-1,4-lactone | C5H8O6 | 164.11 | n.d. | n.d. | n.d. | 0.42% |
| Erythrono-1,4-lactone | C4H6O4 | 118.10 | 0.15% | 0.15% | 0.31% | 1.32% |
| D-Erythro-Pentonic Acid, γ-Lactone | C5H8O5 | 148.11 | n.d. | n.d. | n.d. | 0.41% |
| Monosaccharides | - | - | 12.16% | 10.87% | 2.10% | 29.91% |
| D-Arabinopyranose | C5H10O5 | 150.15 | 0.78% | 0.79% | 0.00% | 1.40% |
| β-Arabinopyranose | C5H10O5 | 150.15 | 0.00% | 0.00% | 0.72% | 1.58% |
| D-ribose | C5H10O5 | 150.15 | 1.44% | 0.00% | 0.55% | 0.99% |
| D-xylose | C5H10O5 | 150.15 | 4.76% | 4.83% | 0.42% | 11.95% |
| β-D(-)-Lyxopyranose | C5H10O5 | 150.15 | 5.18% | 5.25% | 0.41% | 11.56% |
| Methyl xylopyranoside | C6H12O5 | 164.18 | n.d. | n.d. | n.d. | 2.43% |
| Aromatics | - | - | 12.89% | 12.57% | 2.73% | 2.48% |
| 2,6-Dimethoxyhydroquinone | C6H4(OH)(OCH3)2 | 170.18 | 0.50% | n.d. | 0.19% | 0.34% |
| Vanillic acid | C6H4(OH)(COOH)(OCH3) | 168.16 | 1.91% | 1.93% | 0.74% | 0.89% |
| Benzoic acid, 4-hydroxy-3,5-dimethoxy- | C6H4(OH)(COOH)(OCH3)2 | 198.19 | 0.32% | 0.32% | n.d. | n.d. |
| Protocatechuic acid | C6H4(OH)(COOH)(OCH3) | 154.13 | 0.70% | 0.71% | 0.39% | n.d. |
| Syringic acid | C6H4(OH)(COOH)(OCH3)2 | 198.19 | 3.38% | 3.43% | 0.30% | n.d. |
| Acetyl syringic acid | C9H10O5 | 240.23 | 1.16% | 1.17% | 0.40% | 1.26% |
| Phthalic acid, di(2,3-dimethylphenyl) ester | C6H4(CO2R)2(OCH3)2 | 350.44 | 0.40% | 0.40% | 0.25% | n.d. |
| 4,4′-Methylenedi-2,6-xylenol | C14H14O2 | 214.28 | 3.70% | 3.75% | 0.46% | n.d. |
| 3-Hydroxy-7,8,2′,3′-tetramethoxyflavone | C19H18O7 | 358.37 | 0.83% | 0.84% | n.d. | n.d. |
| Other monomers | - | - | 5.74% | 3.97% | 6.93% | 8.58% |
| Sample | C | H | O | S | N | PPU Empirical Formula | MwPPU (g/mol) |
|---|---|---|---|---|---|---|---|
| LB | 57.9 ± 0.03 | 5.55 ± 0.02 | 32.86 ± 0.06 | 3.10 ± 0.24 | 0.52 ± 0.33 | C9H5.944O2.461S0.228N0.087(OCH3)2.372 | 235.69 |
| 60 °C, 3 h, pH 1 | 48.56 ± 0.16 | 4.36 ± 0.02 | 41.90 ± 0.26 | 4.57 ± 0.21 | 0.62 ± 0.11 | C9H7.102O5.350S0.365N0.113(OCH3)1.343 | 255.60 |
| 60 °C, 3 h, pH 1, no stirring | 49.74 ± 0.08 | 4.58 ± 0.01 | 40.97 ± 0.14 | 4.23 ± 0.12 | 0.50 ± 0.06 | C9H6.956O4.959S0.337N0.091(OCH3)1.571 | 255.06 |
| 60 °C, 3 h, pH 10 | 56.71 ± 0.22 | 5.17 ± 0.08 | 34.89 ± 0.18 | 2.62 ± 0.15 | 0.61 ± 0.26 | C9H4.837O2.732S0.202N0.107(OCH3)2.637 | 246.26 |
| Experiment | Type of Lignin | Lignin (g) | 30% (w/v) H2O2 (mL) | Stirring | pH | T (°C) | Time (h) |
|---|---|---|---|---|---|---|---|
| LB-pH1-50 °C | LB | 3 | 10 | Magnetic | 1 | 50 | 3, 5, 7 |
| LB-pH1-60 °C | 1 | 60 | 1, 2, 3 | ||||
| LB-pH1-70 °C | 1 | 70 | 1, 2, 3 | ||||
| LB-pH10-60 °C | 10 | 60 | 1, 2, 3 | ||||
| LB-pH1-0rpm-60 °C | No | 1 | 60 | 1, 2, 3 | |||
| KL-pH5-60 °C | KL | 3 | 10 | Magnetic | 5 | 60 | 1, 2, 3 |
| KL-pH1-60 °C | 1 | ||||||
| LB-pH1-60 °C-×4 | LB | 12 | 40 | Magnetic | 1 | 60 | 1, 2, 3 |
| LB-pH1-60 °C-×25 | 75 | 250 | Mechanical | 3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lourenço, M.C.; Nascimento, T.; Filho, P.J.S.; Marques, A.C.; Ramos-Andrés, M. Acidic Oxidative Depolymerization Towards Functionalized Low-Molecular-Weight Lignin and High-Value-Added Aliphatic Monomers: Operating Conditions, Scale-Up, and Crosslinking. Int. J. Mol. Sci. 2025, 26, 4872. https://doi.org/10.3390/ijms26104872
Lourenço MC, Nascimento T, Filho PJS, Marques AC, Ramos-Andrés M. Acidic Oxidative Depolymerization Towards Functionalized Low-Molecular-Weight Lignin and High-Value-Added Aliphatic Monomers: Operating Conditions, Scale-Up, and Crosslinking. International Journal of Molecular Sciences. 2025; 26(10):4872. https://doi.org/10.3390/ijms26104872
Chicago/Turabian StyleLourenço, Marta C., Talita Nascimento, Pedro José Sanches Filho, Ana C. Marques, and Marta Ramos-Andrés. 2025. "Acidic Oxidative Depolymerization Towards Functionalized Low-Molecular-Weight Lignin and High-Value-Added Aliphatic Monomers: Operating Conditions, Scale-Up, and Crosslinking" International Journal of Molecular Sciences 26, no. 10: 4872. https://doi.org/10.3390/ijms26104872
APA StyleLourenço, M. C., Nascimento, T., Filho, P. J. S., Marques, A. C., & Ramos-Andrés, M. (2025). Acidic Oxidative Depolymerization Towards Functionalized Low-Molecular-Weight Lignin and High-Value-Added Aliphatic Monomers: Operating Conditions, Scale-Up, and Crosslinking. International Journal of Molecular Sciences, 26(10), 4872. https://doi.org/10.3390/ijms26104872