
Liquid Biopsy in B and T Cell Lymphomas: From Bench to Bedside

 ,
,  , , ,
, , , 
Abstract
1. Introduction
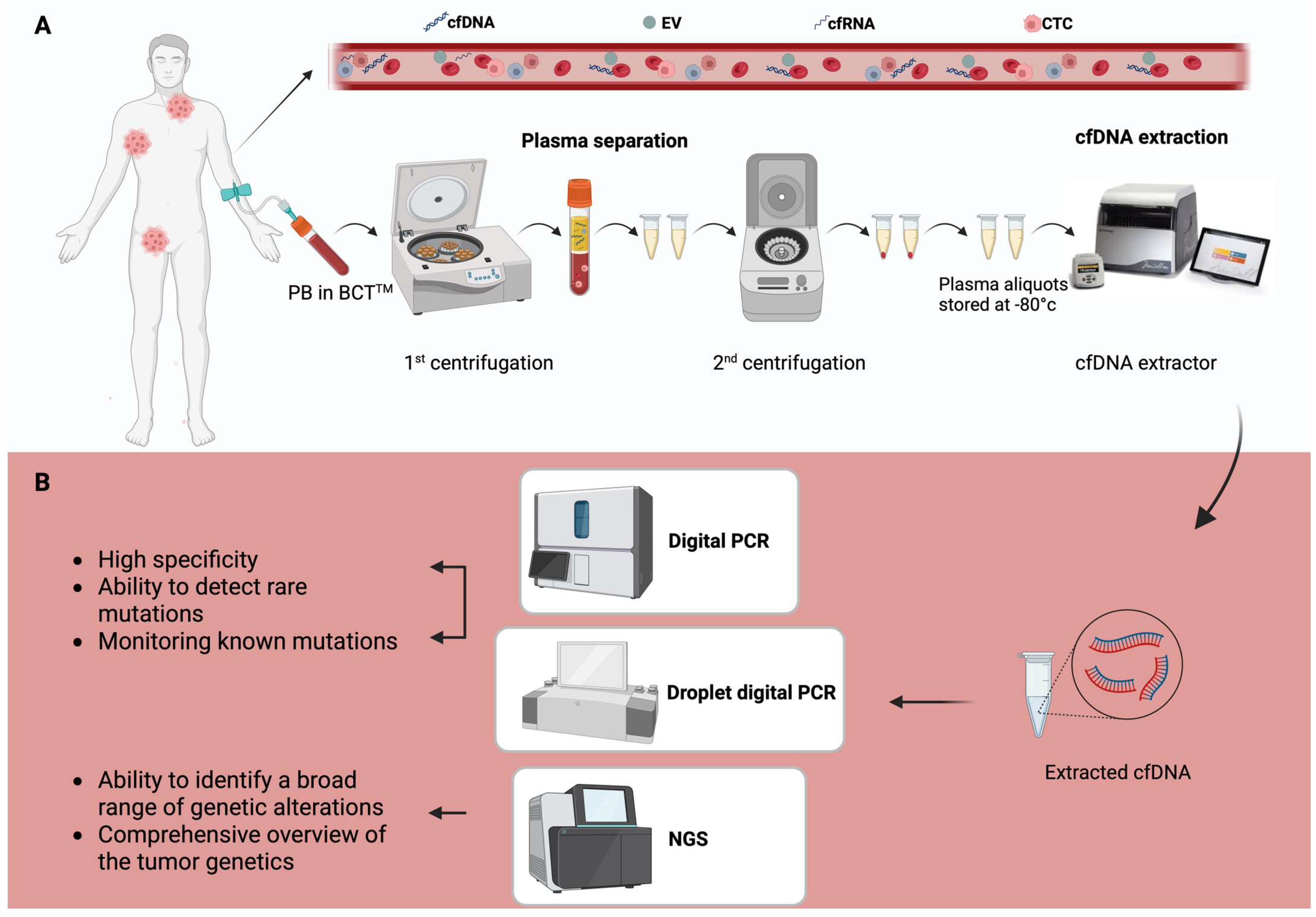
1.1. Biological Basis of ctDNA
1.2. Detection and Analytical Techniques
2. ctDNA in B-Cell Lymphoma
2.1. Diffuse Large-B-Cell Lymphoma
2.2. Hodgkin’s Lymphoma
2.3. Central Nervous System Lymphomas
2.4. Follicular Lymphoma
2.5. Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
3. T-Cell Lymphomas
3.1. ENKTL
3.2. PTCL
4. ctDNA in Lymphoma Clinical Trials
5. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| GENE | Full Name |
| BCL2 | B-cell Lymphoma 2 |
| CD19 | Cluster of Differentiation 19 |
| CD274 | Cluster of Differentiation 274 (also known as PD-L1, Programmed Death-Ligand 1) |
| CD5 | Cluster of Differentiation 5 |
| CD79 | Cluster of Differentiation 79 |
| CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A |
| CXCR4 | C-X-C Chemokine Receptor Type 4 |
| DDX3X | DEAD-Box Helicase 3 X-Linked |
| DNMT3A | DNA Methyltransferase 3 Alpha |
| EZH2 | Enhancer of Zeste Homolog 2 |
| IDH2 | Isocitrate Dehydrogenase 2 |
| IRF8 | Interferon Regulatory Factor 8 |
| JAK | Janus Kinase |
| KMT2D | Lysine Methyltransferase 2D (also known as MLL2) |
| MYD88 | Myeloid Differentiation Primary Response 88 |
| NFKB | Nuclear Factor Kappa-light-chain-enhancer of Activated B cells |
| NOTCH1 | Notch Receptor 1 |
| PAX5 | Paired Box 5 |
| PI3K | Phosphoinositide 3-Kinases |
| PIM1 | Pim-1 Proto-Oncogene, Serine/Threonine Kinase |
| POD24 | Progression of Disease within 24 months |
| RHOA | Ras Homolog Family Member A |
| SOCS1 | Suppressor of Cytokine Signaling 1 |
| STAT | Signal Transducer and Activator of Transcription |
| TET2 | Tet Methylcytosine Dioxygenase 2 |
| TMEM30A | Transmembrane Protein 30A |
| TP53 | Tumor Protein p53 |
| XPO1 | Exportin 1 |
References
- Mugnaini, E.N.; Ghosh, N. Lymphoma. Prim. Care 2016, 43, 661–675. [Google Scholar] [CrossRef] [PubMed]
- Malone, E.R.; Oliva, M.; Sabatini, P.J.B.; Stockley, T.L.; Siu, L.L. Molecular profiling for precision cancer therapies. Genome Med. 2020, 12, 8. [Google Scholar] [CrossRef]
- Cirillo, M.; Craig, A.F.M.; Borchmann, S.; Kurtz, D.M. Liquid biopsy in lymphoma: Molecular methods and clinical applications. Cancer Treat. Rev. 2020, 91, 102106. [Google Scholar] [CrossRef]
- Chan, H.T.; Chin, Y.M.; Low, S.K. Circulating Tumor DNA-Based Genomic Profiling Assays in Adult Solid Tumors for Precision Oncology: Recent Advancements and Future Challenges. Cancers 2022, 14, 3275. [Google Scholar] [CrossRef]
- Talotta, D.; Almasri, M.; Cosentino, C.; Gaidano, G.; Moia, R. Liquid biopsy in hematological malignancies: Current and future applications. Front. Oncol. 2023, 13, 1164517. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Diop, F.; Spaccarotella, E.; Monti, S.; Zanni, M.; Rasi, S.; Deambrogi, C.; Spina, V.; Bruscaggin, A.; Favini, C.; et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood 2017, 129, 1947–1957. [Google Scholar] [CrossRef]
- Ma, L.; Guo, H.; Zhao, Y.; Liu, Z.; Wang, C.; Bu, J.; Sun, T.; Wei, J. Liquid biopsy in cancer: Current status, challenges and future prospects. Signal Transduct. Target. Ther. 2024, 9, 336. [Google Scholar] [CrossRef]
- Matuszczak, M.; Schalken, J.A.; Salagierski, M. Prostate Cancer Liquid Biopsy Biomarkers’ Clinical Utility in Diagnosis and Prognosis. Cancers 2021, 13, 3373. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88. [Google Scholar] [CrossRef]
- Liu, S.; Wang, J. Current and Future Perspectives of Cell-Free DNA in Liquid Biopsy. Curr. Issues Mol. Biol. 2022, 44, 2695–2709. [Google Scholar] [CrossRef]
- Moia, R.; Favini, C.; Ferri, V.; Forestieri, G.; Terzi Di Bergamo, L.; Schipani, M.; Sagiraju, S.; Andorno, A.; Rasi, S.; Adhinaveni, R.; et al. Multiregional sequencing and circulating tumour DNA analysis provide complementary approaches for comprehensive disease profiling of small lymphocytic lymphoma. Br. J. Haematol. 2021, 195, 108–112. [Google Scholar] [CrossRef]
- Savino, F.D.; Rigali, F.; Giustini, V.; D’Aliberti, D.; Spinelli, S.; Piazza, R.; Sacco, A.; Roccaro, A.M. Liquid Biopsy in Cancer: Focus on Lymphoproliferative Disorders. Cancers 2022, 14, 5378. [Google Scholar] [CrossRef] [PubMed]
- Jamal, E.; Poynton, E.; Elbogdady, M.; Shamaa, S.; Okosun, J. Prospects for liquid biopsy approaches in lymphomas. Leuk. Lymphoma 2024, 65, 1923–1933. [Google Scholar] [CrossRef]
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA apoptosis and active DNA release. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Khier, S.; Lohan, L. Kinetics of circulating cell-free DNA for biomedical applications: Critical appraisal of the literature. Future Sci. OA 2018, 4, FSO295. [Google Scholar] [CrossRef] [PubMed]
- Lo, Y.M.; Corbetta, N.; Chamberlain, P.F.; Rai, V.; Sargent, I.L.; Redman, C.W.; Wainscoat, J.S. Presence of fetal DNA in maternal plasma and serum. Lancet 1997, 350, 485–487. [Google Scholar] [CrossRef]
- Chan, K.C.; Jiang, P.; Sun, K.; Cheng, Y.K.; Tong, Y.K.; Cheng, S.H.; Wong, A.I.; Hudecova, I.; Leung, T.Y.; Chiu, R.W.; et al. Second generation noninvasive fetal genome analysis reveals de novo mutations, single-base parental inheritance, and preferred DNA ends. Proc. Natl. Acad. Sci. USA 2016, 113, E8159–E8168. [Google Scholar] [CrossRef]
- Fan, H.C.; Blumenfeld, Y.J.; Chitkara, U.; Hudgins, L.; Quake, S.R. Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood. Proc. Natl. Acad. Sci. USA 2008, 105, 16266–16271. [Google Scholar] [CrossRef]
- Li, J.Y.; Zuo, L.P.; Xu, J.; Sun, C.Y. Clinical applications of circulating tumor DNA in hematological malignancies: From past to the future. Blood Rev. 2024, 68, 101237. [Google Scholar] [CrossRef]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, M.; Yokoyama, K.; Imoto, S.; Tojo, A. Role of Circulating Tumor DNA in Hematological Malignancy. Cancers 2021, 13, 2078. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Spina, V.; Bruscaggin, A.; Gaidano, G. Liquid biopsy in lymphoma. Haematologica 2019, 104, 648–652. [Google Scholar] [CrossRef]
- Zhu, D.; Wang, H.; Wu, W.; Geng, S.; Zhong, G.; Li, Y.; Guo, H.; Long, G.; Ren, Q.; Luan, Y.; et al. Circulating cell-free DNA fragmentation is a stepwise and conserved process linked to apoptosis. BMC Biol. 2023, 21, 253. [Google Scholar] [CrossRef]
- Schroers-Martin, J.G.; Alig, S.; Garofalo, A.; Tessoulin, B.; Sugio, T.; Alizadeh, A.A. Molecular Monitoring of Lymphomas. Annu. Rev. Pathol. 2023, 18, 149–180. [Google Scholar] [CrossRef]
- Tivey, A.; Church, M.; Rothwell, D.; Dive, C.; Cook, N. Circulating tumour DNA—Looking beyond the blood. Nat. Rev. Clin. Oncol. 2022, 19, 600–612. [Google Scholar] [CrossRef] [PubMed]
- Malentacchi, F.; Pizzamiglio, S.; Verderio, P.; Pazzagli, M.; Orlando, C.; Ciniselli, C.M.; Günther, K.; Gelmini, S. Influence of storage conditions and extraction methods on the quantity and quality of circulating cell-free DNA (ccfDNA): The SPIDIA-DNAplas External Quality Assessment experience. Clin. Chem. Lab. Med. 2015, 53, 1935–1942. [Google Scholar] [CrossRef]
- Lee, J.S.; Kim, M.; Seong, M.W.; Kim, H.S.; Lee, Y.K.; Kang, H.J. Plasma vs. serum in circulating tumor DNA measurement: Characterization by DNA fragment sizing and digital droplet polymerase chain reaction. Clin. Chem. Lab. Med. 2020, 58, 527–532. [Google Scholar] [CrossRef]
- Toro, P.V.; Erlanger, B.; Beaver, J.A.; Cochran, R.L.; VanDenBerg, D.A.; Yakim, E.; Cravero, K.; Chu, D.; Zabransky, D.J.; Wong, H.Y.; et al. Comparison of cell stabilizing blood collection tubes for circulating plasma tumor DNA. Clin. Biochem. 2015, 48, 993–998. [Google Scholar] [CrossRef]
- Kang, Q.; Henry, N.L.; Paoletti, C.; Jiang, H.; Vats, P.; Chinnaiyan, A.M.; Hayes, D.F.; Merajver, S.D.; Rae, J.M.; Tewari, M. Comparative analysis of circulating tumor DNA stability In K(3)EDTA, Streck, and CellSave blood collection tubes. Clin. Biochem. 2016, 49, 1354–1360. [Google Scholar] [CrossRef]
- Donaldson, J.; Park, B.H. Circulating Tumor DNA: Measurement and Clinical Utility. Annu. Rev. Med. 2018, 69, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Page, K.; Powles, T.; Slade, M.J.; MT, D.E.B.; Walker, R.A.; Coombes, R.C.; Shaw, J.A. The importance of careful blood processing in isolation of cell-free DNA. Ann. N. Y. Acad. Sci. 2006, 1075, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Pascual, J.; Attard, G.; Bidard, F.C.; Curigliano, G.; De Mattos-Arruda, L.; Diehn, M.; Italiano, A.; Lindberg, J.; Merker, J.D.; Montagut, C.; et al. ESMO recommendations on the use of circulating tumour DNA assays for patients with cancer: A report from the ESMO Precision Medicine Working Group. Ann. Oncol. 2022, 33, 750–768. [Google Scholar] [CrossRef]
- Johansson, G.; Andersson, D.; Filges, S.; Li, J.; Muth, A.; Godfrey, T.E.; Ståhlberg, A. Considerations and quality controls when analyzing cell-free tumor DNA. Biomol. Detect. Quantif. 2019, 17, 100078. [Google Scholar] [CrossRef]
- Roschewski, M.; Rossi, D.; Kurtz, D.M.; Alizadeh, A.A.; Wilson, W.H. Circulating Tumor DNA in Lymphoma: Principles and Future Directions. Blood Cancer Discov. 2022, 3, 5–15. [Google Scholar] [CrossRef]
- Postel, M.; Roosen, A.; Laurent-Puig, P.; Taly, V.; Wang-Renault, S.F. Droplet-based digital PCR and next generation sequencing for monitoring circulating tumor DNA: A cancer diagnostic perspective. Expert. Rev. Mol. Diagn. 2018, 18, 7–17. [Google Scholar] [CrossRef]
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Dang, D.K.; Park, B.H. Circulating tumor DNA: Current challenges for clinical utility. J. Clin. Investig. 2022, 132, e154941. [Google Scholar] [CrossRef]
- Sater, V.; Viailly, P.J.; Lecroq, T.; Ruminy, P.; Bérard, C.; Prieur-Gaston, É.; Jardin, F. UMI-Gen: A UMI-based read simulator for variant calling evaluation in paired-end sequencing NGS libraries. Comput. Struct. Biotechnol. J. 2020, 18, 2270–2280. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Dalla-Favera, R. The genetic landscape of diffuse large B-cell lymphoma. Semin. Hematol. 2015, 52, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Sehn, L.H.; Gascoyne, R.D. Diffuse large B-cell lymphoma: Optimizing outcome in the context of clinical and biologic heterogeneity. Blood 2015, 125, 22–32. [Google Scholar] [CrossRef]
- Tilly, H.; Morschhauser, F.; Sehn, L.H.; Friedberg, J.W.; Trněný, M.; Sharman, J.P.; Herbaux, C.; Burke, J.M.; Matasar, M.; Rai, S.; et al. Polatuzumab Vedotin in Previously Untreated Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2022, 386, 351–363. [Google Scholar] [CrossRef]
- Xu-Monette, Z.Y.; Wu, L.; Visco, C.; Tai, Y.C.; Tzankov, A.; Liu, W.M.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A.; Orazi, A.; et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: Report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012, 120, 3986–3996. [Google Scholar] [CrossRef] [PubMed]
- Moia, R.; Talotta, D.; Terzi di Bergamo, L.; Almasri, M.; Dondolin, R.; Salehi, M.; Cosentino, C.; Soscia, R.; Della Starza, I.; Bruscaggin, A.; et al. Molecular clustering on ctDNA improves the prognostic stratification of DLBCL patients compared to ctDNA levels. Blood Adv. 2025, 9, 1692–1701. [Google Scholar] [CrossRef]
- Scherer, F.; Kurtz, D.M.; Newman, A.M.; Stehr, H.; Craig, A.F.; Esfahani, M.S.; Lovejoy, A.F.; Chabon, J.J.; Klass, D.M.; Liu, C.L.; et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transl. Med. 2016, 8, 364ra155. [Google Scholar] [CrossRef]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating tumor DNA reveals genetics, clonal evolution, and residual disease in classical Hodgkin lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef]
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568.e514. [Google Scholar] [CrossRef]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Scherer, F.; Jin, M.C.; Soo, J.; Craig, A.F.M.; Esfahani, M.S.; Chabon, J.J.; Stehr, H.; Liu, C.L.; Tibshirani, R.; et al. Circulating Tumor DNA Measurements As Early Outcome Predictors in Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2018, 36, 2845–2853. [Google Scholar] [CrossRef]
- Camus, V.; Viennot, M.; Lequesne, J.; Viailly, P.J.; Bohers, E.; Bessi, L.; Marcq, B.; Etancelin, P.; Dubois, S.; Picquenot, J.M.; et al. Targeted genotyping of circulating tumor DNA for classical Hodgkin lymphoma monitoring: A prospective study. Haematologica 2021, 106, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Bobillo, S.; Crespo, M.; Escudero, L.; Mayor, R.; Raheja, P.; Carpio, C.; Rubio-Perez, C.; Tazón-Vega, B.; Palacio, C.; Carabia, J.; et al. Cell free circulating tumor DNA in cerebrospinal fluid detects and monitors central nervous system involvement of B-cell lymphomas. Haematologica 2021, 106, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Mutter, J.A.; Alig, S.K.; Esfahani, M.S.; Lauer, E.M.; Mitschke, J.; Kurtz, D.M.; Kühn, J.; Bleul, S.; Olsen, M.; Liu, C.L.; et al. Circulating Tumor DNA Profiling for Detection, Risk Stratification, and Classification of Brain Lymphomas. J. Clin. Oncol. 2023, 41, 1684–1694. [Google Scholar] [CrossRef] [PubMed]
- Sarkozy, C.; Huet, S.; Carlton, V.E.; Fabiani, B.; Delmer, A.; Jardin, F.; Delfau-Larue, M.H.; Hacini, M.; Ribrag, V.; Guidez, S.; et al. The prognostic value of clonal heterogeneity and quantitative assessment of plasma circulating clonal IG-VDJ sequences at diagnosis in patients with follicular lymphoma. Oncotarget 2017, 8, 8765–8774. [Google Scholar] [CrossRef]
- Jiménez-Ubieto, A.; Poza, M.; Martin-Muñoz, A.; Ruiz-Heredia, Y.; Dorado, S.; Figaredo, G.; Rosa-Rosa, J.M.; Rodriguez, A.; Barcena, C.; Navamuel, L.P.; et al. Real-life disease monitoring in follicular lymphoma patients using liquid biopsy ultra-deep sequencing and PET/CT. Leukemia 2023, 37, 659–669. [Google Scholar] [CrossRef]
- Yeh, P.; Hunter, T.; Sinha, D.; Ftouni, S.; Wallach, E.; Jiang, D.; Chan, Y.C.; Wong, S.Q.; Silva, M.J.; Vedururu, R.; et al. Circulating tumour DNA reflects treatment response and clonal evolution in chronic lymphocytic leukaemia. Nat. Commun. 2017, 8, 14756. [Google Scholar] [CrossRef]
- Fürstenau, M.; Weiss, J.; Giza, A.; Franzen, F.; Robrecht, S.; Fink, A.M.; Fischer, K.; Schneider, C.; Tausch, E.; Stilgenbauer, S.; et al. Circulating Tumor DNA-Based MRD Assessment in Patients with CLL Treated with Obinutuzumab, Acalabrutinib, and Venetoclax. Clin. Cancer Res. 2022, 28, 4203–4211. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, W.; Li, J.; Xiong, J.; Liu, J.; Chen, T.; Wen, Q.; Zeng, Y.; Gao, L.; Gao, L.; et al. Plasma circulating tumor DNA assessment reveals KMT2D as a potential poor prognostic factor in extranodal NK/T-cell lymphoma. Biomark. Res. 2020, 8, 27. [Google Scholar] [CrossRef]
- Wei, C.; Wang, W.; Zhang, Y.; Zhao, D.; Zhang, W.; Zhou, D. Mutation profiling, tumour burden assessment, outcome prediction and disease monitoring by circulating tumour DNA in peripheral T-cell lymphoma. Br. J. Haematol. 2023, 202, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Meriranta, L.; Alkodsi, A.; Pasanen, A.; Lepistö, M.; Mapar, P.; Blaker, Y.N.; Jørgensen, J.; Karjalainen-Lindsberg, M.L.; Fiskvik, I.; Mikalsen, L.T.G.; et al. Molecular features encoded in the ctDNA reveal heterogeneity and predict outcome in high-risk aggressive B-cell lymphoma. Blood 2022, 139, 1863–1877. [Google Scholar] [CrossRef] [PubMed]
- Rivas-Delgado, A.; Nadeu, F.; Enjuanes, A.; Casanueva-Eliceiry, S.; Mozas, P.; Magnano, L.; Castrejón de Anta, N.; Rovira, J.; Dlouhy, I.; Martín, S.; et al. Mutational Landscape and Tumor Burden Assessed by Cell-free DNA in Diffuse Large B-Cell Lymphoma in a Population-Based Study. Clin. Cancer Res. 2021, 27, 513–521. [Google Scholar] [CrossRef]
- Alig, S.; Macaulay, C.W.; Kurtz, D.M.; Dührsen, U.; Hüttmann, A.; Schmitz, C.; Jin, M.C.; Sworder, B.J.; Garofalo, A.; Shahrokh Esfahani, M.; et al. Short Diagnosis-to-Treatment Interval Is Associated With Higher Circulating Tumor DNA Levels in Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2021, 39, 2605–2616. [Google Scholar] [CrossRef]
- Lauer, E.M.; Mutter, J.; Scherer, F. Circulating tumor DNA in B-cell lymphoma: Technical advances, clinical applications, and perspectives for translational research. Leukemia 2022, 36, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Bruscaggin, A.; di Bergamo, L.T.; Spina, V.; Hodkinson, B.; Forestieri, G.; Bonfiglio, F.; Condoluci, A.; Wu, W.; Pirosa, M.C.; Faderl, M.R.; et al. Circulating tumor DNA for comprehensive noninvasive monitoring of lymphoma treated with ibrutinib plus nivolumab. Blood Adv. 2021, 5, 4674–4685. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Esfahani, M.S.; Scherer, F.; Soo, J.; Jin, M.C.; Liu, C.L.; Newman, A.M.; Dührsen, U.; Hüttmann, A.; Casasnovas, O.; et al. Dynamic Risk Profiling Using Serial Tumor Biomarkers for Personalized Outcome Prediction. Cell 2019, 178, 699–713.e619. [Google Scholar] [CrossRef]
- Roschewski, M.; Lindenberg, L.; Mena, E.; Lakhotia, R.; Melani, C.; Steinberg, S.M.; Schultz, A.; Hogan, G.; Chabon, J.J.; Close, S.; et al. End-of-Treatment Response Assessment after Frontline Therapy for Aggressive B-Cell Lymphoma: Landmark Comparison of a Singular PET/CT Scan Versus Ultrasensitive Circulating Tumor DNA. Blood 2023, 142, 192. [Google Scholar] [CrossRef]
- Zou, H.; Liu, W.; Wang, X.; Wang, Y.; Wang, C.; Qiu, C.; Liu, H.; Shan, D.; Xie, T.; Huang, W.; et al. Dynamic monitoring of circulating tumor DNA reveals outcomes and genomic alterations in patients with relapsed or refractory large B-cell lymphoma undergoing CAR T-cell therapy. J. Immunother. Cancer 2024, 12, e008450. [Google Scholar] [CrossRef]
- Sworder, B.J.; Kurtz, D.M.; Alig, S.K.; Frank, M.J.; Shukla, N.; Garofalo, A.; Macaulay, C.W.; Shahrokh Esfahani, M.; Olsen, M.N.; Hamilton, J.; et al. Determinants of resistance to engineered T cell therapies targeting CD19 in large B cell lymphomas. Cancer Cell 2023, 41, 210–225.e215. [Google Scholar] [CrossRef]
- Mouhssine, S.; Maher, N.; Gaidano, G. A STEP ahead for CAR-T cell therapy of large B cell lymphoma: Understanding the molecular determinants of resistance. Transl. Cancer Res. 2023, 12, 2970–2975. [Google Scholar] [CrossRef] [PubMed]
- Herrera, A.F.; Kim, H.T.; Kong, K.A.; Faham, M.; Sun, H.; Sohani, A.R.; Alyea, E.P.; Carlton, V.E.; Chen, Y.B.; Cutler, C.S.; et al. Next-generation sequencing-based detection of circulating tumour DNA After allogeneic stem cell transplantation for lymphoma. Br. J. Haematol. 2016, 175, 841–850. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Diehn, M.; Craig, A.F.M.; Chen, B.; Liu, C.L.; Hamilton, E.G.; Scherer, F.; Jin, M.C.; Macaulay, C.; Keh, L.C.T.; et al. Phased Variant Enrichment for Enhanced Minimal Residual Disease Detection from Cell-Free DNA. Blood 2019, 134, 552. [Google Scholar] [CrossRef]
- Kurtz, D.M.; Soo, J.; Co Ting Keh, L.; Alig, S.; Chabon, J.J.; Sworder, B.J.; Schultz, A.; Jin, M.C.; Scherer, F.; Garofalo, A.; et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat. Biotechnol. 2021, 39, 1537–1547. [Google Scholar] [CrossRef]
- Scherer, F.; Navarrete, M.A.; Bertinetti-Lapatki, C.; Boehm, J.; Schmitt-Graeff, A.; Veelken, H. Isotype-switched follicular lymphoma displays dissociation between activation-induced cytidine deaminase expression and somatic hypermutation. Leuk. Lymphoma 2016, 57, 151–160. [Google Scholar] [CrossRef]
- Ansell, S.M. Hodgkin lymphoma: 2025 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2024, 99, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Oki, Y.; Neelapu, S.S.; Fanale, M.; Kwak, L.W.; Fayad, L.; Rodriguez, M.A.; Wallace, M.; Klinger, M.; Carlton, V.; Kong, K.; et al. Detection of classical Hodgkin lymphoma specific sequence in peripheral blood using a next-generation sequencing approach. Br. J. Haematol. 2015, 169, 689–693. [Google Scholar] [CrossRef]
- Desch, A.K.; Hartung, K.; Botzen, A.; Brobeil, A.; Rummel, M.; Kurch, L.; Georgi, T.; Jox, T.; Bielack, S.; Burdach, S.; et al. Genotyping circulating tumor DNA of pediatric Hodgkin lymphoma. Leukemia 2020, 34, 151–166. [Google Scholar] [CrossRef]
- Georgiadi, E.C.; Sachinis, N.; Dimtsas, G.; Vassilakopoulos, T.P.; Kittas, C.; Doussis-Anagnostopoulou, I.A. Evaluation of apoptosis in classical Hodgkin’s lymphoma comparing different methods. J. Balk. Union Oncol. 2012, 17, 746–752. [Google Scholar]
- Hu, Z.; Chen, H.; Long, Y.; Li, P.; Gu, Y. The main sources of circulating cell-free DNA: Apoptosis, necrosis and active secretion. Crit. Rev. Oncol. Hematol. 2021, 157, 103166. [Google Scholar] [CrossRef]
- Camus, V.; Stamatoullas, A.; Mareschal, S.; Viailly, P.J.; Sarafan-Vasseur, N.; Bohers, E.; Dubois, S.; Picquenot, J.M.; Ruminy, P.; Maingonnat, C.; et al. Detection and prognostic value of recurrent exportin 1 mutations in tumor and cell-free circulating DNA of patients with classical Hodgkin lymphoma. Haematologica 2016, 101, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Jardin, F.; Pujals, A.; Pelletier, L.; Bohers, E.; Camus, V.; Mareschal, S.; Dubois, S.; Sola, B.; Ochmann, M.; Lemonnier, F.; et al. Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B-cell lymphoma. Am. J. Hematol. 2016, 91, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Van Slambrouck, C.; Huh, J.; Suh, C.; Song, J.Y.; Menon, M.P.; Sohani, A.R.; Duffield, A.S.; Goldberg, R.C.; Dama, P.; Kiyotani, K.; et al. Diagnostic utility of STAT6(YE361) expression in classical Hodgkin lymphoma and related entities. Mod. Pathol. 2020, 33, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Buedts, L.; Wlodarska, I.; Finalet-Ferreiro, J.; Gheysens, O.; Dehaspe, L.; Tousseyn, T.; Fornecker, L.M.; Lazarovici, J.; Casasnovas, R.O.; Gac, A.C.; et al. The landscape of copy number variations in classical Hodgkin lymphoma: A joint KU Leuven and LYSA study on cell-free DNA. Blood Adv. 2021, 5, 1991–2002. [Google Scholar] [CrossRef]
- Alig, S.K.; Shahrokh Esfahani, M.; Garofalo, A.; Li, M.Y.; Rossi, C.; Flerlage, T.; Flerlage, J.E.; Adams, R.; Binkley, M.S.; Shukla, N.; et al. Distinct Hodgkin lymphoma subtypes defined by noninvasive genomic profiling. Nature 2024, 625, 778–787. [Google Scholar] [CrossRef]
- Sobesky, S.; Mammadova, L.; Cirillo, M.; Drees, E.E.E.; Mattlener, J.; Dörr, H.; Altmüller, J.; Shi, Z.; Bröckelmann, P.J.; Weiss, J.; et al. In-depth cell-free DNA sequencing reveals genomic landscape of Hodgkin’s lymphoma and facilitates ultrasensitive residual disease detection. Med 2021, 2, 1171–1193.e11. [Google Scholar] [CrossRef]
- Lynch, R.C.; Ujjani, C.S.; Poh, C.; Warren, E.H.; Smith, S.D.; Shadman, M.; Till, B.; Raghunathan, V.M.; Alig, S.; Alizadeh, A.A.; et al. Concurrent pembrolizumab with AVD for untreated classic Hodgkin lymphoma. Blood 2023, 141, 2576–2586. [Google Scholar] [CrossRef] [PubMed]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Grommes, C.; DeAngelis, L.M. Primary CNS Lymphoma. J. Clin. Oncol. 2017, 35, 2410–2418. [Google Scholar] [CrossRef]
- Bobillo, S.; Khwaja, J.; Ferreri, A.J.M.; Cwynarski, K. Prevention and management of secondary central nervous system lymphoma. Haematologica 2023, 108, 673–689. [Google Scholar] [CrossRef]
- Ferreri, A.J.M.; Calimeri, T.; Cwynarski, K.; Dietrich, J.; Grommes, C.; Hoang-Xuan, K.; Hu, L.S.; Illerhaus, G.; Nayak, L.; Ponzoni, M.; et al. Primary central nervous system lymphoma. Nat. Rev. Dis. Primers 2023, 9, 29. [Google Scholar] [CrossRef]
- Nishihara, M.; Sasayama, T.; Kudo, H.; Kohmura, E. Morbidity of stereotactic biopsy for intracranial lesions. Kobe J. Med. Sci. 2011, 56, E148–E153. [Google Scholar] [PubMed]
- Hickmann, A.K.; Frick, M.; Hadaschik, D.; Battke, F.; Bittl, M.; Ganslandt, O.; Biskup, S.; Döcker, D. Molecular tumor analysis and liquid biopsy: A feasibility investigation analyzing circulating tumor DNA in patients with central nervous system lymphomas. BMC Cancer 2019, 19, 192. [Google Scholar] [CrossRef]
- Nayak, L.; Bettegowda, C.; Scherer, F.; Galldiks, N.; Ahluwalia, M.; Baraniskin, A.; von Baumgarten, L.; Bromberg, J.E.C.; Ferreri, A.J.M.; Grommes, C.; et al. Liquid biopsy for improving diagnosis and monitoring of CNS lymphomas: A RANO review. Neuro Oncol. 2024, 26, 993–1011. [Google Scholar] [CrossRef] [PubMed]
- Fukumura, K.; Kawazu, M.; Kojima, S.; Ueno, T.; Sai, E.; Soda, M.; Ueda, H.; Yasuda, T.; Yamaguchi, H.; Lee, J.; et al. Genomic characterization of primary central nervous system lymphoma. Acta Neuropathol. 2016, 131, 865–875. [Google Scholar] [CrossRef]
- Nayyar, N.; White, M.D.; Gill, C.M.; Lastrapes, M.; Bertalan, M.; Kaplan, A.; D’Andrea, M.R.; Bihun, I.; Kaneb, A.; Dietrich, J.; et al. MYD88 L265P mutation and CDKN2A loss are early mutational events in primary central nervous system diffuse large B-cell lymphomas. Blood Adv. 2019, 3, 375–383. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Godlewska, E.; Brunn, A.; Wiestler, O.D.; Siebert, R.; Deckert, M. Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol. 2011, 122, 791–792. [Google Scholar] [CrossRef] [PubMed]
- Poulain, S.; Boyle, E.M.; Tricot, S.; Demarquette, H.; Doye, E.; Roumier, C.; Duthilleul, P.; Preudhomme, C.; Maurage, C.A.; Morschhauser, F. Absence of CXCR4 mutations but high incidence of double mutant in CD79A/B and MYD88 in primary central nervous system lymphoma. Br. J. Haematol. 2015, 170, 285–287. [Google Scholar] [CrossRef]
- Ferreri, A.J.M.; Calimeri, T.; Lopedote, P.; Francaviglia, I.; Daverio, R.; Iacona, C.; Belloni, C.; Steffanoni, S.; Gulino, A.; Anghileri, E.; et al. MYD88 L265P mutation and interleukin-10 detection in cerebrospinal fluid are highly specific discriminating markers in patients with primary central nervous system lymphoma: Results from a prospective study. Br. J. Haematol. 2021, 193, 497–505. [Google Scholar] [CrossRef]
- Rimelen, V.; Ahle, G.; Pencreach, E.; Zinniger, N.; Debliquis, A.; Zalmaï, L.; Harzallah, I.; Hurstel, R.; Alamome, I.; Lamy, F.; et al. Tumor cell-free DNA detection in CSF for primary CNS lymphoma diagnosis. Acta Neuropathol. Commun. 2019, 7, 43. [Google Scholar] [CrossRef]
- Watanabe, J.; Natsumeda, M.; Okada, M.; Kobayashi, D.; Kanemaru, Y.; Tsukamoto, Y.; Oishi, M.; Kakita, A.; Fujii, Y. High Detection Rate of MYD88 Mutations in Cerebrospinal Fluid From Patients With CNS Lymphomas. JCO Precis. Oncol. 2019, 3, 1–13. [Google Scholar] [CrossRef]
- Montesinos-Rongen, M.; Brunn, A.; Tuchscherer, A.; Borchmann, P.; Schorb, E.; Kasenda, B.; Altmüller, J.; Illerhaus, G.; Ruge, M.I.; Maarouf, M.; et al. Analysis of Driver Mutational Hot Spots in Blood-Derived Cell-Free DNA of Patients with Primary Central Nervous System Lymphoma Obtained before Intracerebral Biopsy. J. Mol. Diagn. 2020, 22, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Burns, E.J.; Georgantas, N.Z.; Thierauf, J.; Nayyar, N.; Gordon, A.; Jones, S.S.; Pisapia, M.; Sun, Y.; Burns, R.P.; et al. A rapid genotyping panel for detection of primary central nervous system lymphoma. Blood 2021, 138, 382–386. [Google Scholar] [CrossRef]
- Yamagishi, Y.; Sasaki, N.; Nakano, Y.; Matushita, Y.; Omura, T.; Shimizu, S.; Saito, K.; Kobayashi, K.; Narita, Y.; Kondo, A.; et al. Liquid biopsy of cerebrospinal fluid for MYD88 L265P mutation is useful for diagnosis of central nervous system lymphoma. Cancer Sci. 2021, 112, 4702–4710. [Google Scholar] [CrossRef] [PubMed]
- Bonzheim, I.; Giese, S.; Deuter, C.; Süsskind, D.; Zierhut, M.; Waizel, M.; Szurman, P.; Federmann, B.; Schmidt, J.; Quintanilla-Martinez, L.; et al. High frequency of MYD88 mutations in vitreoretinal B-cell lymphoma: A valuable tool to improve diagnostic yield of vitreous aspirates. Blood 2015, 126, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Miserocchi, E.; Ferreri, A.J.M.; Giuffrè, C.; Cangi, M.G.; Francaviglia, I.; Calimeri, T.; Ponzoni, M.; Pecciarini, L.; Bandello, F.M.; Modorati, G.M. MYD88 L265P Mutation Detection in the Aqueous Humor of Patients with Vitreoretinal Lymphoma. Retina 2019, 39, 679–684. [Google Scholar] [CrossRef]
- Fontanilles, M.; Marguet, F.; Bohers, É.; Viailly, P.J.; Dubois, S.; Bertrand, P.; Camus, V.; Mareschal, S.; Ruminy, P.; Maingonnat, C.; et al. Non-invasive detection of somatic mutations using next-generation sequencing in primary central nervous system lymphoma. Oncotarget 2017, 8, 48157–48168. [Google Scholar] [CrossRef]
- Hattori, K.; Sakata-Yanagimoto, M.; Suehara, Y.; Yokoyama, Y.; Kato, T.; Kurita, N.; Nishikii, H.; Obara, N.; Takano, S.; Ishikawa, E.; et al. Clinical significance of disease-specific MYD88 mutations in circulating DNA in primary central nervous system lymphoma. Cancer Sci. 2018, 109, 225–230. [Google Scholar] [CrossRef]
- Zorofchian, S.; Lu, G.; Zhu, J.J.; Duose, D.Y.; Windham, J.; Esquenazi, Y.; Ballester, L.Y. Detection of the MYD88 p.L265P Mutation in the CSF of a Patient With Secondary Central Nervous System Lymphoma. Front. Oncol. 2018, 8, 382. [Google Scholar] [CrossRef]
- Liang, J.H.; Wu, Y.F.; Shen, H.R.; Li, Y.; Liang, J.H.; Gao, R.; Hua, W.; Shang, C.Y.; Du, K.X.; Xing, T.Y.; et al. Clinical implications of CSF-ctDNA positivity in newly diagnosed diffuse large B cell lymphoma. Leukemia 2024, 38, 1541–1552. [Google Scholar] [CrossRef]
- Heger, J.M.; Mattlener, J.; Schneider, J.; Gödel, P.; Sieg, N.; Ullrich, F.; Lewis, R.; Bucaciuc-Mracica, T.; Schwarz, R.F.; Rueß, D.; et al. Entirely noninvasive outcome prediction in central nervous system lymphomas using circulating tumor DNA. Blood 2024, 143, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Roulland, S.; Gloghini, A.; Younes, A.; von Keudell, G.; López-Guillermo, A.; Fitzgibbon, J. Follicular lymphoma. Nat. Rev. Dis. Primers 2019, 5, 83. [Google Scholar] [CrossRef]
- Delfau-Larue, M.H.; van der Gucht, A.; Dupuis, J.; Jais, J.P.; Nel, I.; Beldi-Ferchiou, A.; Hamdane, S.; Benmaad, I.; Laboure, G.; Verret, B.; et al. Total metabolic tumor volume, circulating tumor cells, cell-free DNA: Distinct prognostic value in follicular lymphoma. Blood Adv. 2018, 2, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Miranda, I.; Pedrosa, L.; Llanos, M.; Franco, F.F.; Gómez, S.; Martín-Acosta, P.; García-Arroyo, F.R.; Gumá, J.; Horcajo, B.; Ballesteros, A.K.; et al. Monitoring of Circulating Tumor DNA Predicts Response to Treatment and Early Progression in Follicular Lymphoma: Results of a Prospective Pilot Study. Clin. Cancer Res. 2023, 29, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, S.; Genuardi, E.; Mazziotta, F.; Iovino, L.; Morabito, F.; Grassi, S.; Ciabatti, E.; Guerrini, F.; Petrini, M. The Minimal Residual Disease in Non-Hodgkin’s Lymphomas: From the Laboratory to the Clinical Practice. Front. Oncol. 2019, 9, 528. [Google Scholar] [CrossRef]
- Maher, N.; Mouhssine, S.; Matti, B.F.; Alwan, A.F.; Gaidano, G. Molecular Mechanisms in the Transformation from Indolent to Aggressive B Cell Malignancies. Cancers 2025, 17, 907. [Google Scholar] [CrossRef]
- Quintanilla-Martinez, L. The 2016 updated WHO classification of lymphoid neoplasias. Hematol. Oncol. 2017, 35 (Suppl. 1), 37–45. [Google Scholar] [CrossRef]
- Sander, B.; Campo, E.; Hsi, E.D. Chronic lymphocytic leukaemia/small lymphocytic lymphoma and mantle cell lymphoma: From early lesions to transformation. Virchows Arch. 2023, 482, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Gaidano, G.; Rossi, D. The mutational landscape of chronic lymphocytic leukemia and its impact on prognosis and treatment. Hematol. Am. Soc. Hematol. Educ. Program. 2017, 2017, 329–337. [Google Scholar] [CrossRef]
- Nabki, J.; Al Deeban, B.; Sium, A.M.; Cosentino, C.; Almasri, M.; Awikeh, B.; Maher, N.; Bellia, M.; Dondolin, R.; Mouhssine, S.; et al. Immunoglobulin light chain mutational status refines IGHV prognostic value in identifying chronic lymphocytic leukemia patients with early treatment requirement. Leukemia 2025, 39, 643–649. [Google Scholar] [CrossRef]
- McKeague, S.; Tam, C. Prognostic factors in chronic lymphocytic leukaemia—The old, the new and the future. Leuk. Lymphoma 2025, 66, 847–857. [Google Scholar] [CrossRef]
- Mouhssine, S.; Maher, N.; Kogila, S.; Cerchione, C.; Martinelli, G.; Gaidano, G. Current Therapeutic Sequencing in Chronic Lymphocytic Leukemia. Hematol. Rep. 2024, 16, 270–282. [Google Scholar] [CrossRef] [PubMed]
- Fürstenau, M.; De Silva, N.; Eichhorst, B.; Hallek, M. Minimal Residual Disease Assessment in CLL: Ready for Use in Clinical Routine? Hemasphere 2019, 3, e287. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M.; Fischer, K.; Fingerle-Rowson, G.; Fink, A.M.; Busch, R.; Mayer, J.; Hensel, M.; Hopfinger, G.; Hess, G.; von Grünhagen, U.; et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: A randomised, open-label, phase 3 trial. Lancet 2010, 376, 1164–1174. [Google Scholar] [CrossRef]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Gerecitano, J.; Robak, T.; De la Serna, J.; et al. Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2018, 378, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Lew, T.E.; Anderson, M.A.; Lin, V.S.; Handunnetti, S.M.; Came, N.A.; Blombery, P.; Westerman, D.A.; Wall, M.; Tam, C.S.; Roberts, A.W.; et al. Undetectable peripheral blood MRD should be the goal of venetoclax in CLL, but attainment plateaus after 24 months. Blood Adv. 2020, 4, 165–173. [Google Scholar] [CrossRef]
- Thompson, P.A.; Srivastava, J.; Peterson, C.; Strati, P.; Jorgensen, J.L.; Hether, T.; Keating, M.J.; O’Brien, S.M.; Ferrajoli, A.; Burger, J.A.; et al. Minimal residual disease undetectable by next-generation sequencing predicts improved outcome in CLL after chemoimmunotherapy. Blood 2019, 134, 1951–1959. [Google Scholar] [CrossRef]
- Rawstron, A.C.; Villamor, N.; Ritgen, M.; Böttcher, S.; Ghia, P.; Zehnder, J.L.; Lozanski, G.; Colomer, D.; Moreno, C.; Geuna, M.; et al. International standardized approach for flow cytometric residual disease monitoring in chronic lymphocytic leukaemia. Leukemia 2007, 21, 956–964. [Google Scholar] [CrossRef]
- Kovacs, G.; Robrecht, S.; Fink, A.M.; Bahlo, J.; Cramer, P.; von Tresckow, J.; Maurer, C.; Langerbeins, P.; Fingerle-Rowson, G.; Ritgen, M.; et al. Minimal Residual Disease Assessment Improves Prediction of Outcome in Patients With Chronic Lymphocytic Leukemia (CLL) Who Achieve Partial Response: Comprehensive Analysis of Two Phase III Studies of the German CLL Study Group. J. Clin. Oncol. 2016, 34, 3758–3765. [Google Scholar] [CrossRef]
- Condoluci, A.; Romano, I.; Dietrich, D.; Pini, K.; Stussi, G.; Müller, G.; Cantoni, N.; Cathomas, R.; Mey, U.J.M.; Widmer, A.A.; et al. Ibrutinib Lead-in followed by Venetoclax Plus Ibrutinib in Relapsed/Refractory Chronic Lymphocytic Leukemia—SAKK 34/17. Blood 2025, in press. [Google Scholar] [CrossRef]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef]
- Ong, S.Y.; Zain, J.M. Aggressive T-cell lymphomas: 2024: Updates on diagnosis, risk stratification, and management. Am. J. Hematol. 2024, 99, 439–456. [Google Scholar] [CrossRef]
- Huang, Z.; Fu, Y.; Yang, H.; Zhou, Y.; Shi, M.; Li, Q.; Liu, W.; Liang, J.; Zhu, L.; Qin, S.; et al. Liquid biopsy in T-cell lymphoma: Biomarker detection techniques and clinical application. Mol. Cancer 2024, 23, 36. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Chen, C.; Xia, Y.; Wang, Y.; Liu, P.P.; Bi, X.W.; Shao, Y.W.; Ou, Q.X.; Wu, X.; Yang, H.; et al. Mutation Profiling of Malignant Lymphoma by Next-Generation Sequencing of Circulating Cell-Free DNA. J. Cancer 2019, 10, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Cao, Z.; Chen, B.; Chai, Y.; Lin, J.; Ye, J.; Wei, Y.; Liu, H.; Han-Zhang, H.; Mao, X.; et al. Liquid biopsy in extranodal NK/T-cell lymphoma: A prospective analysis of cell-free DNA genotyping and monitoring. Blood Adv. 2021, 5, 2505–2514. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Shen, J.; Zhou, H.; Zhang, F.; Li, H.; Ma, Z.; Huang, W.; Chen, L.; Chen, Y.; Liu, T. Mutation profiling of circulating tumor DNA identifies distinct mutation patterns in non-Hodgkin lymphoma. Eur. J. Haematol. 2022, 108, 298–309. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, Y.J.; Yoon, S.E.; Ryu, K.J.; Park, B.; Park, D.; Cho, D.; Kim, H.Y.; Cho, J.; Ko, Y.H.; et al. Circulating Tumor DNA-Based Genotyping and Monitoring for Predicting Disease Relapses of Patients with Peripheral T-Cell Lymphomas. Cancer Res. Treat. 2023, 55, 291–303. [Google Scholar] [CrossRef]
- Miljkovic, M.D.; Melani, C.; Pittaluga, S.; Lakhotia, R.; Lucas, N.; Jacob, A.; Yusko, E.; Jaffe, E.S.; Wilson, W.H.; Roschewski, M. Next-generation sequencing-based monitoring of circulating tumor DNA reveals clonotypic heterogeneity in untreated PTCL. Blood Adv. 2021, 5, 4198–4210. [Google Scholar] [CrossRef]
- Kharuzhyk, S.; Zhavrid, E.; Dziuban, A.; Sukolinskaja, E.; Kalenik, O. Comparison of whole-body MRI with diffusion-weighted imaging and PET/CT in lymphoma staging. Eur. Radiol. 2020, 30, 3915–3923. [Google Scholar] [CrossRef]
- Salehi, M.; Pirosa, M.C.; Bruscaggin, A.; Terzi Di Bergamo, L.; Jauk, F.; Forestieri, G.; Bocchetta, S.; Piffaretti, D.; Moia, R.; Cristaldi, V.; et al. A Comprehensive Genetic Study of Classical Hodgkin Lymphoma Using Ctdna. Blood 2024, 144, 854. [Google Scholar] [CrossRef]
- Sheng, L.; Ouyang, G.; Lai, Y.; Nie, S.; Wang, D.; Tang, S.; Pan, Y.; Xu, K.; Sun, Y.; Zhang, P.; et al. Preliminary Data on the Efficacy and Safety of Orelabrutinib Combined with Rituximab and Methotrexate (ORM Regimen) As First-Line Treatment for Primary Central Nervous System Lymphoma, and the Exploration of Cerebrospinal Fluid Ctdna Dynamic Monitoring. Blood 2024, 144, 4500. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Lymphoma Subtype | References | Origin of ctDNA | Patient Count | Technologies | Sensitivity | Clinical Application |
|---|---|---|---|---|---|---|
| DLBCL | Rossi et al., 2017 [6] | Plasma | 30 | CAPP-Seq | ~10−3 ctDNA detected in 66.6% | Disease genotyping |
| Kurtz et al., 2018 [52] | Plasma | 217 | CAPP-Seq | Detected in 98% | Early relapse prediction | |
| Moia et al., 2025 [46] | Plasma | 166 | CAPP-Seq | 95.8% concordance with tissue biopsy | Molecular clustering | |
| HL | Spina et al., 2018 [48] | Plasma | 112 | CAPP-Seq | ~10−3 Detected in 81.2% | Monitoring of clonal evolution and treatment response assessment |
| Camus et al., 2021 [53] | Plasma | 94 | dPCR, NGS | - | MRD monitoring and risk stratification | |
| CNSL | Bobillo et al., 2021 [54] | CSF | 19 | Targeted NGS, ddPCR | - | Disease genotyping |
| Mutter et al., 2023 [55] | CSF, plasma | 92 | CAPPSeq, PhasED-seq | Detectable in 78% of plasma and 100% of CSF | Treatment response assessment and risk stratification | |
| FL | Sarkozy et al., 2017 [56] | Plasma | 34 | NGS-based immunosequencing | Detected in 86% | Prognostic biomarker |
| Jiménez-Ubieto et al., 2023 [57] | Plasma | 84 | LiqBio-MRD | Detected in 80% | Treatment monitoring | |
| CLL | Yeh et al., 2019 [58] | Plasma | 32 | dPCR, TS | Detected in 80% | MRD assessment |
| Fürstenau et al., 2022 [59] | Plasma | 46 | dPCR | - | MRD assessment | |
| T-cell lymphomas | Li et al., 2020 [60] | Plasma | 65 | Targeted sequencing | 93.8% concordance with tissue biopsy | Guiding therapeutic decision-making |
| Wei et al., 2023 [61] | Plasma | 47 | NGS | 73.9% concordance with tissue biopsy | Mutation profiling and disease monitoring |
| Disease | Methods | Endpoints | Study Design | Identifier |
|---|---|---|---|---|
| PCNSL | ctDNA | Evaluate ctDNA conversion rate in CSF | Interventional (Phase II) | NCT04401774 |
| DLBCL | ctDNA and PET | Evaluate a PET/CT- and ctDNA-oriented therapy in DLBCL to test treatment response | Interventional (Phase II) | NCT04604067 |
| MCL | Flow cytometry, PCR and ctDNA | Estimate MRD negative response rates by aggregate measure of peripheral blood/or bone marrow flow cytometry, PCR, and ctDNA | Interventional (Phase II) | NCT04234061 |
| PCNSL | ctDNA | Characterizing ctDNA for early response assessment | Observational | NCT06755619 |
| DLBCL | ctDNA | Measure ctDNA in real time during standard treatment | Interventional | NCT06693830 |
| HL | ctDNA | MRD assessment by ctDNA | Observational | NCT05254821 |
| PTCL | ctDNA | Evaluate the feasibility of ctDNA measurement in blood plasma for treatment evaluation and MRD surveillance | Observational | NCT06362148 |
| DLBCL, MCL, and FL | ctDNA | To determine the clinical utility of ctDNA as well as CTC-based MRD assessment in CAR therapy patients | Observational | NCT05255354 |
| LBCL | ctDNA | Early assessment of lymphoma treatment response using ctDNA analyzed by phased variant analysis | Observational | NCT06550427 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almasri, M.; Maher, N.; Al Deeban, B.; Diop, N.M.; Moia, R.; Gaidano, G. Liquid Biopsy in B and T Cell Lymphomas: From Bench to Bedside. Int. J. Mol. Sci. 2025, 26, 4869. https://doi.org/10.3390/ijms26104869
Almasri M, Maher N, Al Deeban B, Diop NM, Moia R, Gaidano G. Liquid Biopsy in B and T Cell Lymphomas: From Bench to Bedside. International Journal of Molecular Sciences. 2025; 26(10):4869. https://doi.org/10.3390/ijms26104869
Chicago/Turabian StyleAlmasri, Mohammad, Nawar Maher, Bashar Al Deeban, Ndeye Marie Diop, Riccardo Moia, and Gianluca Gaidano. 2025. "Liquid Biopsy in B and T Cell Lymphomas: From Bench to Bedside" International Journal of Molecular Sciences 26, no. 10: 4869. https://doi.org/10.3390/ijms26104869
APA StyleAlmasri, M., Maher, N., Al Deeban, B., Diop, N. M., Moia, R., & Gaidano, G. (2025). Liquid Biopsy in B and T Cell Lymphomas: From Bench to Bedside. International Journal of Molecular Sciences, 26(10), 4869. https://doi.org/10.3390/ijms26104869