
A Pilot Study: Contrasting Genomic Profiles of Lung Adenocarcinoma Between Patients of European and Latin American Ancestry

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Study Population

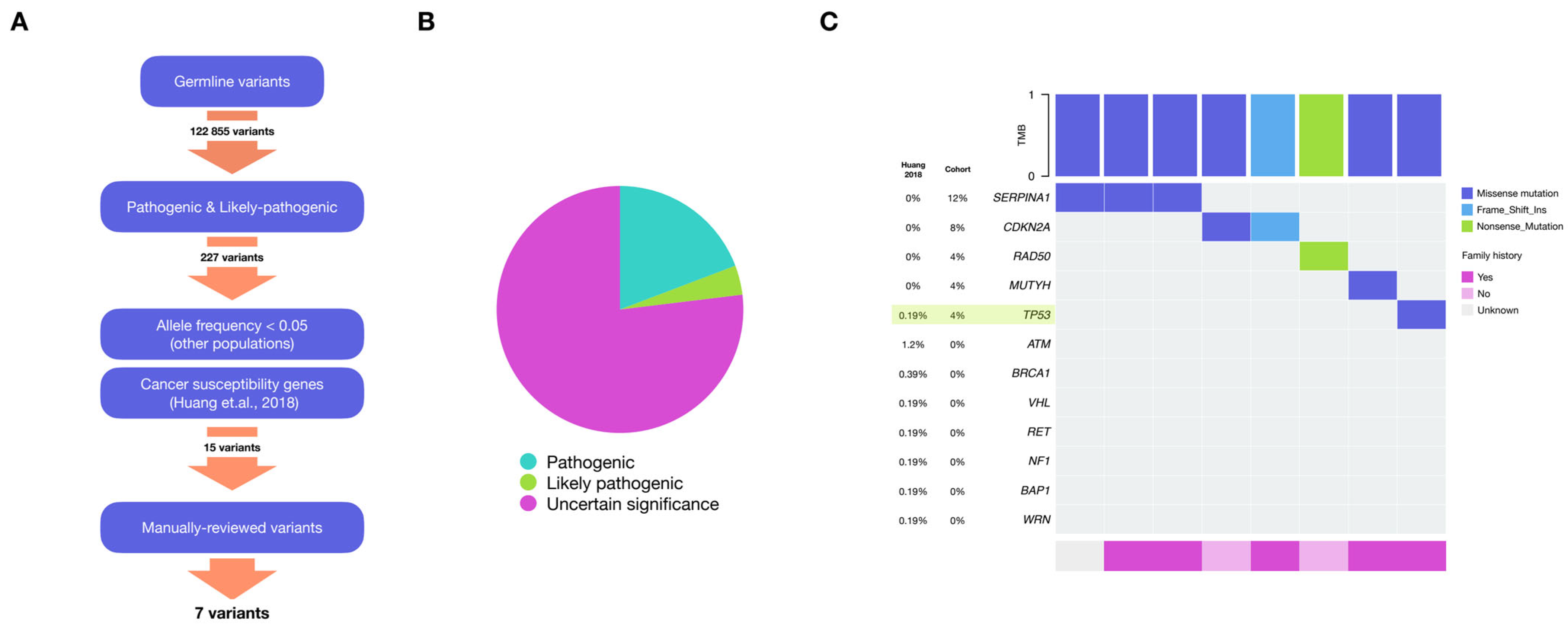
2.2. Germline Variants
2.3. Somatic Mutations
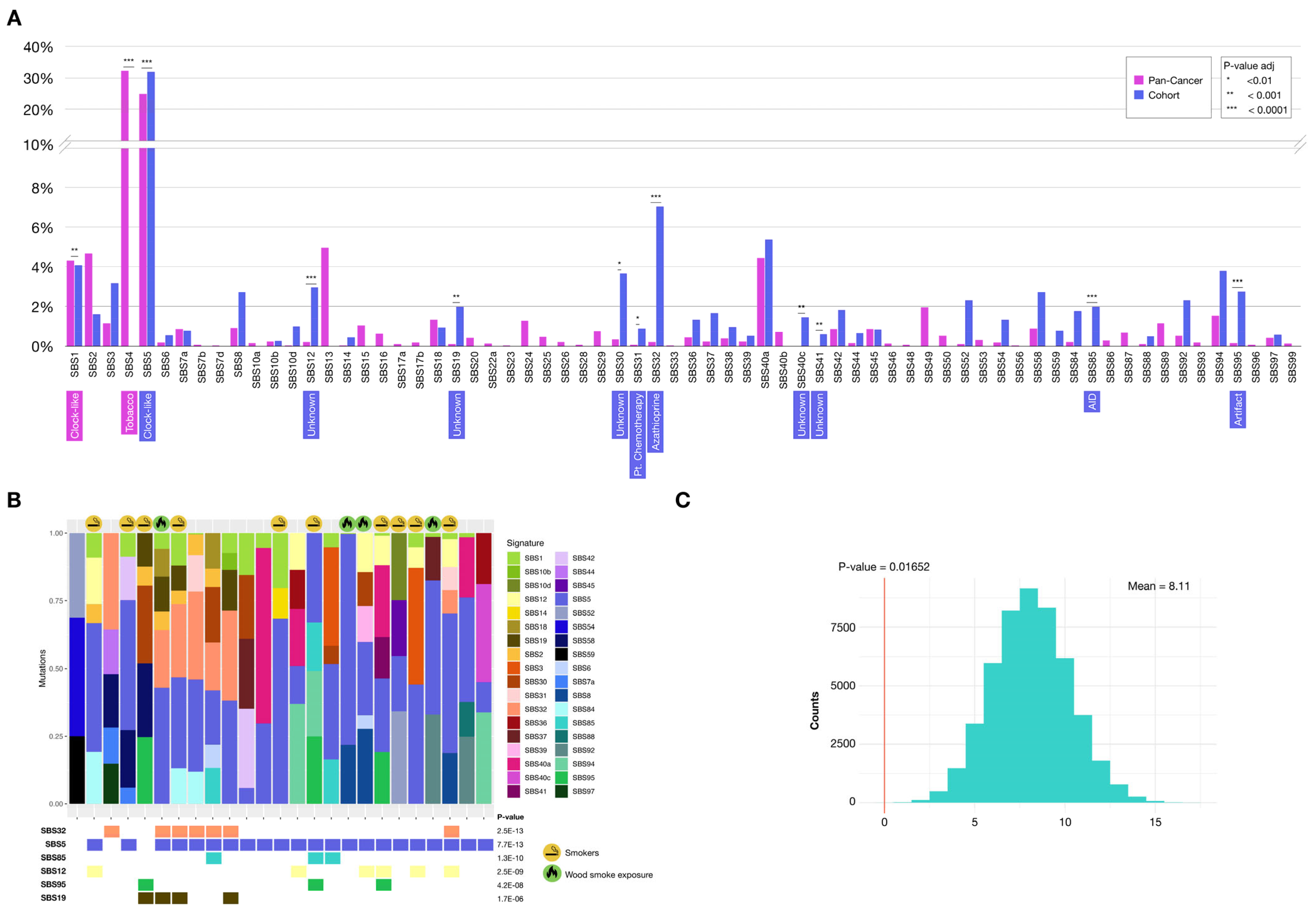
2.3.1. Mutational Signatures
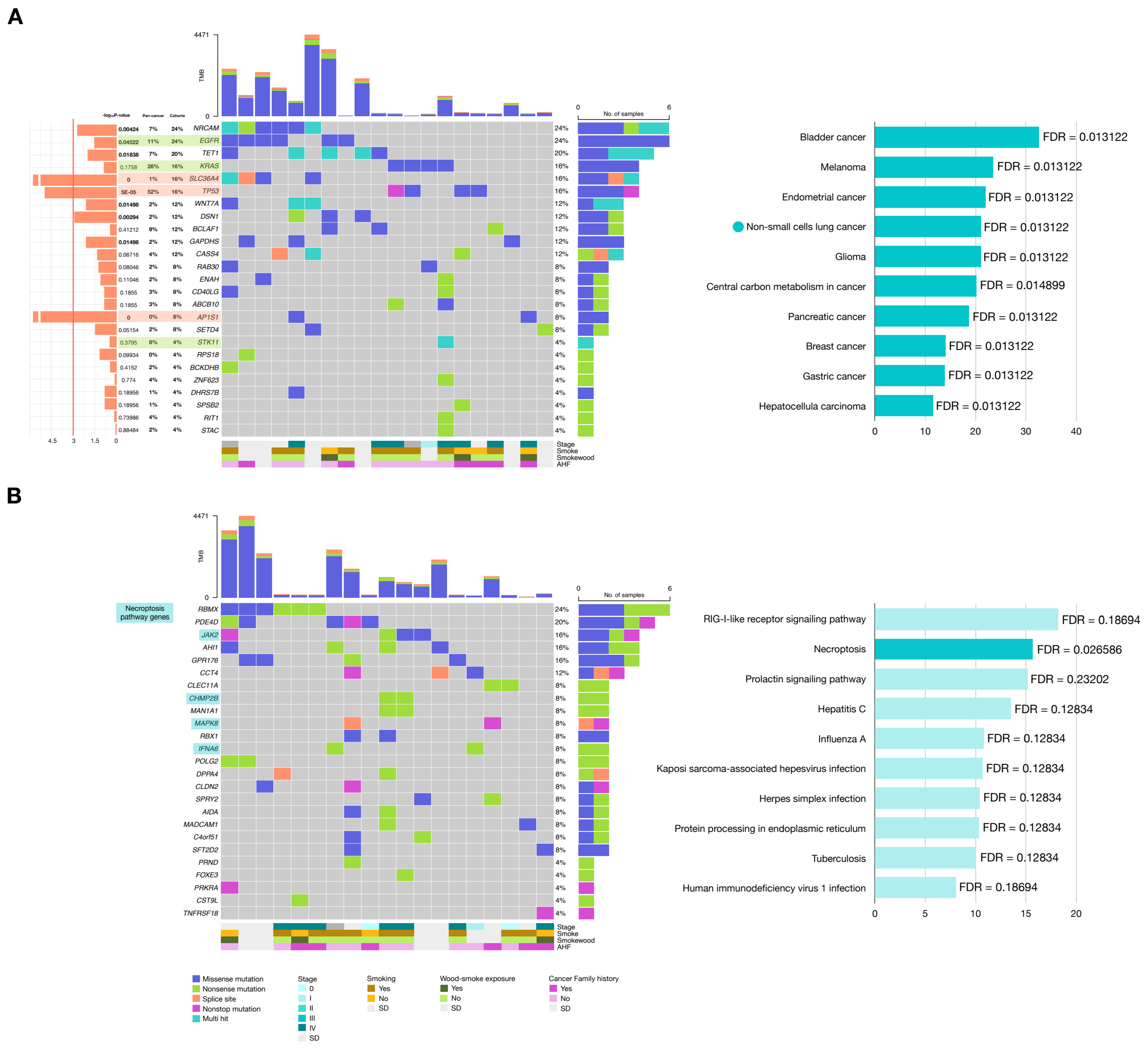
2.3.2. Genes with Potential Positive Selection (Drivers)
3. Discussion
4. Materials and Methods
4.1. Mexican Lung Adenocarcinoma Cohort
4.2. Sample Processing and Sequencing
4.3. Preprocessing
4.4. Ancestry Analysis
4.5. Germline Variants Analysis
4.6. Somatic Variants
4.6.1. Mutational Signatures Analysis
4.6.2. Positive Selection Analysis
4.7. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arrieta, O.; Zatarain-Barrón, Z.L.; Aldaco, F.; Barrón, F.; Báez-Saldaña, R.; Campos-Gómez, S.; Trejo, R.; De la Garza, J. Lung Cancer in Mexico. J. Thorac. Oncol. 2019, 14, 1695–1700. [Google Scholar] [CrossRef] [PubMed]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Prim. 2015, 1, 15009. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, H.; Teo, A.S.M.; Amer, L.B.; Sherbaf, F.G.; Tan, C.Q.; Alvarez, J.J.S.; Lu, B.; Lim, J.Q.; Takano, A.; et al. Genomic landscape of lung adenocarcinoma in East Asians. Nat. Genet. 2020, 52, 177–186. [Google Scholar] [CrossRef]
- Midha, A.; Dearden, S.; McCormack, R. EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: A systematic review and global map by ethnicity (mutMapII). Am. J. Cancer Res. 2015, 5, 2892–2911. [Google Scholar]
- Arrieta, O.; Cardona, A.F.; Martín, C.; Más-López, L.; Corrales-Rodríguez, L.; Bramuglia, G.; Castillo-Fernandez, O.; Meyerson, M.; Amieva-Rivera, E.; Campos-Parra, A.D.; et al. Updated Frequency of EGFR and KRAS Mutations in NonSmall-Cell Lung Cancer in Latin America: The Latin-American Consortium for the Investigation of Lung Cancer (CLICaP). J. Thorac. Oncol. 2015, 10, 838–843. [Google Scholar] [CrossRef]
- Zhou, W.; Christiani, D.C. East meets West: Ethnic differences in epidemiology and clinical behaviors of lung cancer between East Asians and Caucasians. Chin. J. Cancer 2011, 30, 287–292. [Google Scholar] [CrossRef] [PubMed]
- De Ver Dye, T.; Tavarez, Z.Q.; Pérez Ramos, J.G.; Fernandez, I.D.; Vega, C.V.; Vega Ocasio, D.M.; Avendaño, E.; Cardona Cordero, N.R.; Hering, C.D.; Dozier, A.M.; et al. Participation in genetic research among Latinx populations by Latin America birth-residency concordance: A global study. J. Community Genet. 2021, 12, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Molina-Aguilar, C.; Robles-Espinoza, C.D. Tackling the lack of diversity in cancer research. Dis. Models Mech. 2023, 16, dmm050275. [Google Scholar] [CrossRef]
- Park, S.L.; Cheng, I.; Haiman, C.A. Genome-Wide Association Studies of Cancer in Diverse Populations. Cancer Epidemiol. Biomark. Prev. 2018, 27, 405–417. [Google Scholar] [CrossRef]
- Arrieta, O.; Cardona, A.F.; Bramuglia, G.; Cruz-Rico, G.; Corrales, L.; Martín, C.; Imaz-Olguín, V.; Castillo, O.; Cuello, M.; Rojas-Bilbao, É.; et al. Molecular Epidemiology of ALK Rearrangements in Advanced Lung Adenocarcinoma in Latin America. Oncology 2019, 96, 207–216. [Google Scholar] [CrossRef]
- Hernández-Pedro, N.; Soca-Chafre, G.; Alaez-Versón, C.; Carrillo-Sánchez, K.; Avilés-Salas, A.; Vergara, E.; Arrieta, O. Mutational profile by targeted next generation sequencing of non-small cell lung cancer in the Mexican population. Salud Publica Mex. 2019, 61, 308. [Google Scholar] [CrossRef]
- García-Ortiz, H.; Barajas-Olmos, F.; Contreras-Cubas, C.; Cid-Soto, M.Á.; Córdova, E.J.; Centeno-Cruz, F.; Mendoza-Caamal, E.; Cicerón-Arellano, I.; Flores-Huacuja, M.; Baca, P.; et al. The genomic landscape of Mexican Indigenous populations brings insights into the peopling of the Americas. Nat. Commun. 2021, 12, 5942. [Google Scholar] [CrossRef] [PubMed]
- Ordoñez, G.; Romero, S.; Orozco, L.; Pineda, B.; Jiménez-Morales, S.; Nieto, A.; García-Ortiz, H.; Sotelo, J. Genomewide admixture study in Mexican Mestizos with multiple sclerosis. Clin. Neurol. Neurosurg. 2015, 130, 55–60. [Google Scholar] [CrossRef]
- Carrot-Zhang, J.; Chambwe, N.; Damrauer, J.S.; Knijnenburg, T.A.; Robertson, A.G.; Yau, C.; Zhou, W.; Berger, A.C.; Huang, K.L.; Newberg, J.Y.; et al. Comprehensive Analysis of Genetic Ancestry and Its Molecular Correlates in Cancer. Cancer Cell 2020, 37, 639–654.e6. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Mashl, R.J.; Wu, Y.; Ritter, D.I.; Wang, J.; Oh, C.; Paczkowska, M.; Reynolds, S.; Wyczalkowski, M.A.; Oak, N.; et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 2018, 173, 355–370.e14. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Dong, F.; Davies, K.D. Mutational Signatures in Cancer. J. Mol. Diagn. 2023, 25, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Carrot-Zhang, J.; Soca-Chafre, G.; Patterson, N.; Thorner, A.R.; Nag, A.; Watson, J.; Genovese, G.; Rodriguez, J.; Gelbard, M.K.; Corrales-Rodriguez, L.; et al. Genetic Ancestry Contributes to Somatic Mutations in Lung Cancers from Admixed Latin American Populations. Cancer Discov. 2021, 11, 591–598. [Google Scholar] [CrossRef]
- Patel, M.I.; Schupp, C.W.; Gomez, S.L.; Chang, E.T.; Wakelee, H.A. How Do Social Factors Explain Outcomes in Non–Small-Cell Lung Cancer Among Hispanics in California? Explaining the Hispanic Paradox. J. Clin. Oncol. 2013, 31, 3572–3578. [Google Scholar] [CrossRef]
- Donner, I.; Katainen, R.; Sipilä, L.J.; Aavikko, M.; Pukkala, E.; Aaltonen, L.A. Germline mutations in young non-smoking women with lung adenocarcinoma. Lung Cancer 2018, 122, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Gutiontov, S.I.; Turchan, W.T.; Spurr, L.F.; Rouhani, S.J.; Chervin, C.S.; Steinhardt, G.; Lager, A.M.; Wanjari, P.; Malik, R.; Connell, P.P.; et al. CDKN2A loss-of-function predicts immunotherapy resistance in non-small cell lung cancer. Sci. Rep. 2021, 11, 20059. [Google Scholar] [CrossRef]
- Trendowski, M.R.; Lusk, C.M.; Wenzlaff, A.S.; Neslund-Dudas, C.; Purrington, K.S.; Beebe-Dimmer, J.L.; Schwartz, A.G. Association of Germline Pathogenic Variants in MUTYH and Other DNA Damage Response Genes with Lung Cancer Risk Among Non-Hispanic Whites and African Americans. JCO Precis. Oncol. 2025, 9, e2400558. [Google Scholar] [CrossRef]
- Benusiglio, P.R.; Fallet, V.; Sanchis-Borja, M.; Coulet, F.; Cadranel, J. Lung cancer is also a hereditary disease. Eur. Respir. Rev. 2021, 30, 210045. [Google Scholar] [CrossRef]
- Liu, M.; Liu, X.; Suo, P.; Gong, Y.; Qu, B.; Peng, X.; Xiao, W.; Li, Y.; Chen, Y.; Zeng, Z.; et al. The contribution of hereditary cancer-related germline mutations to lung cancer susceptibility. Transl. Lung Cancer Res. 2020, 9, 646–658. [Google Scholar] [CrossRef]
- Peng, W.; Li, B.; Li, J.; Chang, L.; Bai, J.; Yi, Y.; Chen, R.; Zhang, Y.; Chen, C.; Pu, X.; et al. Clinical and genomic features of Chinese lung cancer patients with germline mutations. Nat. Commun. 2022, 13, 1268. [Google Scholar] [CrossRef]
- Yang, T.; Liu, H.; Li, M.; Zhang, Y.; Zhang, Y.; Liang, X.; Lou, F.; Cao, S.; Wang, H. Germline mutations among patients with non-small cell lung cancer. J. Clin. Oncol. 2021, 39 (Suppl. S15), e20527. [Google Scholar] [CrossRef]
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha1-Antitrypsin Deficiency. N. Engl. J. Med. 2020, 382, 1443–1455. [Google Scholar] [CrossRef] [PubMed]
- Torres-Durán, M.; Ruano-Ravina, A.; Parente-Lamelas, I.; Abal-Arca, J.; Leiro-Fernández, V.; Montero-Martínez, C.; Pena, C.; Castro-Añón, O.; Golpe-Gómez, A.; González-Barcala, F.J.; et al. Alpha-1 Antitrypsin Deficiency and Lung Cancer Risk. J. Thorac. Oncol. 2015, 10, 1279–1284. [Google Scholar] [CrossRef]
- Yang, P. Alpha1-Antitrypsin Deficiency Carriers, Tobacco Smoke, Chronic Obstructive Pulmonary Disease, and Lung Cancer Risk. Arch. Intern. Med. 2008, 168, 1097. [Google Scholar] [CrossRef]
- Ercetin, E.; Richtmann, S.; Delgado, B.M.; Gomez-Mariano, G.; Wrenger, S.; Korenbaum, E.; Liu, B.; DeLuca, D.; Kühnel, M.P.; Jonigk, D.; et al. Clinical Significance of SERPINA1 Gene and Its Encoded Alpha1-antitrypsin Protein in NSCLC. Cancers 2019, 11, 1306. [Google Scholar] [CrossRef] [PubMed]
- Tubío-Pérez, R.A.; Torres-Durán, M.; Fernández-Villar, A.; Ruano-Raviña, A. Alpha-1 antitrypsin deficiency and risk of lung cancer: A systematic review. Transl. Oncol. 2021, 14, 100914. [Google Scholar] [CrossRef]
- Pérez-Rubio, G.; Ambrocio-Ortiz, E.; López-Flores, L.A.; Juárez-Martín, A.I.; Jiménez-Valverde, L.O.; Zoreque-Cabrera, S.; Galicia-Negrete, G.; Ramírez-Díaz, M.E.; Cruz-Vicente, F.; Castillejos-López, M.d.J.; et al. Heterozygous Genotype rs17580 AT (PiS) in SERPINA1 is Associated with COPD Secondary to Biomass-Burning and Tobacco Smoking: A Case–Control and Populational Study. Int. J. Chron. Obs. Pulmon. Dis. 2020, 15, 1181–1190. [Google Scholar] [CrossRef]
- Sorscher, S.; LoPiccolo, J.; Heald, B.; Chen, E.; Bristow, S.L.; Michalski, S.T.; Nielsen, S.M.; Lacoste, A.; Keyder, E.; Lee, H.; et al. Rate of Pathogenic Germline Variants in Patients with Lung Cancer. JCO Precis. Oncol. 2023, 7, e2300190. [Google Scholar] [CrossRef]
- Arrieta, O.; Campos-Parra, A.D.; Zuloaga, C.; Avilés, A.; Sánchez-Reyes, R.; Manríquez, M.E.V.; Covián-Molina, E.; Martínez-Barrera, L.; Meneses, A.; Cardona, A.; et al. Clinical and Pathological Characteristics, Outcome and Mutational Profiles Regarding Non–Small-Cell Lung Cancer Related to Wood-Smoke Exposure. J. Thorac. Oncol. 2012, 7, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Rosell, R.; Cardona, A.F.; Martín, C.; Zatarain-Barrón, Z.L.; Arrieta, O. Lung cancer in never smokers: The role of different risk factors other than tobacco smoking. Crit. Rev. Oncol. Hematol. 2020, 148, 102895. [Google Scholar] [CrossRef]
- Báez-Saldaña, R.; Canseco-Raymundo, A.; Ixcot-Mejía, B.; Juárez-Verdugo, I.; Escobar-Rojas, A.; Rumbo-Nava, U.; Castillo-González, P.; León-Dueñas, S.; Arrieta, O. Case–control study about magnitude of exposure to wood smoke and risk of developing lung cancer. Eur. J. Cancer Prev. 2021, 30, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-Small Cell Lung Cancer: Epidemiology, Risk Factors, Treatment, and Survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Ernst, S.M.; Mankor, J.M.; van Riet, J.; von der Thüsen, J.H.; Dubbink, H.J.; Aerts, J.G.J.V.; de Langen, A.J.; Smit, E.F.; Dingemans, A.M.C.; Monkhorst, K. Tobacco Smoking-Related Mutational Signatures in Classifying Smoking-Associated and Nonsmoking-Associated NSCLC. J. Thorac. Oncol. 2023, 18, 487–498. [Google Scholar] [CrossRef]
- van den Heuvel, G.R.M.; Kroeze, L.I.; Ligtenberg, M.J.L.; Grünberg, K.; Jansen, E.A.M.; von Rhein, D.; de Voer, R.M.; van den Heuvel, M.M. Mutational signature analysis in non-small cell lung cancer patients with a high tumor mutational burden. Respir. Res. 2021, 22, 302. [Google Scholar] [CrossRef]
- Wang, J.; Guo, C.; Wang, J.; Zhang, X.; Qi, J.; Huang, X.; Hu, Z.; Wang, H.; Hong, B. Tumor Mutation Signature Reveals the Risk Factors of Lung Adenocarcinoma with EGFR or KRAS Mutation. Cancer Control 2025, 32, 10732748241307363. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, A.; Guo, T.; Gurevich, N.Q.; Xu, J.; Yajima, M.; Campbell, J.D. Characterization of highly active mutational signatures in tumors from a large Chinese population. medRxiv 2023. [Google Scholar] [CrossRef]
- van den Bosch, L.; Luppi, F.; Ferrara, G.; Mura, M. Immunomodulatory treatment of interstitial lung disease. Ther. Adv. Respir. Dis. 2022, 16, 175346662211170. [Google Scholar] [CrossRef] [PubMed]
- Shinmura, K.; Igarashi, H.; Goto, M.; Tao, H.; Yamada, H.; Matsuura, S.; Tajima, M.; Matsuda, T.; Yamane, A.; Funai, K.; et al. Aberrant Expression and Mutation-Inducing Activity of AID in Human Lung Cancer. Ann. Surg. Oncol. 2011, 18, 2084–2092. [Google Scholar] [CrossRef]
- Shao, M.; Xu, Y.; Zhang, J.; Mao, M.; Wang, M. Tumor mutational burden as a predictive biomarker for non-small cell lung cancer treated with immune checkpoint inhibitors of PD-1/PD-L1. Clin. Transl. Oncol. 2024, 26, 1446–1458. [Google Scholar] [CrossRef]
- Greillier, L.; Tomasini, P.; Barlesi, F. The clinical utility of tumor mutational burden in non-small cell lung cancer. Transl. Lung Cancer Res. 2018, 7, 639–646. [Google Scholar] [CrossRef]
- Vryza, P.; Fischer, T.; Mistakidi, E.; Zaravinos, A. Tumor mutation burden in the prognosis and response of lung cancer patients to immune-checkpoint inhibition therapies. Transl. Oncol. 2023, 38, 101788. [Google Scholar] [CrossRef]
- Trendowski, M.R.; Watza, D.; Lusk, C.M.; Lonardo, F.; Ratliff, V.; Wenzlaff, A.S.; Mamdani, H.; Neslund-Dudas, C.; Boerner, J.L.; Schwartz, A.G.; et al. Evaluation of the Immune Response within the Tumor Microenvironment in African American and Non-Hispanic White Patients with Non–Small Cell Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2024, 33, 1220–1228. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, L.; Thaiparambil, J.; Mai, S.; Perera, D.N.; Zhang, J.; Pan, P.Y.; Coarfa, C.; Ramos, K.; Chen, S.H.; et al. Patients with Lung Cancer of Different Racial Backgrounds Harbor Distinct Immune Cell Profiles. Cancer Res. Commun. 2022, 2, 884–893. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, H.; Huang, J.; Yan, H.; Zhao, B. The association of ROS1 mutation with cancer immunity and its impact on the efficacy of pan-cancer immunotherapy. J. Transl. Med. 2024, 22, 403. [Google Scholar] [CrossRef]
- Hernández-Verdin, I.; Akdemir, K.C.; Ramazzotti, D.; Caravagna, G.; Labreche, K.; Mokhtari, K.; Hoang-Xuan, K.; Peyre, M.; Bielle, F.; Touat, M.; et al. Pan-cancer landscape of AID-related mutations, composite mutations, and their potential role in the ICI response. npj Precis. Oncol. 2022, 6, 89. [Google Scholar] [CrossRef]
- Degasperi, A.; Zou, X.; Dias Amarante, T.; Martinez-Martinez, A.; Koh, G.C.C.; Dias, J.M.L.; Heskin, L.; Chmelova, L.; Rinaldi, G.; Wang, V.Y.W.; et al. Substitution mutational signatures in whole-genome–sequenced cancers in the UK population. Science 2022, 376, abl9283. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Biswas, A.; Sharma, A.; Sarkodie, H.; Tran, I.; Pal, I.; De, S. Mutational signatures associated with exposure to carcinogenic microplastic compounds bisphenol A and styrene oxide. NAR Cancer 2021, 3, zcab004. [Google Scholar] [CrossRef]
- Riva, L.; Pandiri, A.R.; Li, Y.R.; Droop, A.; Hewinson, J.; Quail, M.A.; Iyer, V.; Shepherd, R.; Herbert, R.A.; Campbell, P.J.; et al. The mutational signature profile of known and suspected human carcinogens in mice. Nat. Genet. 2020, 52, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Gay, M.; Zhang, T.; Hoang, P.H.; Khandekar, A.; Zhao, W.; Steele, C.D.; Otlu, B.; Nandi, S.P.; Vangara, R.; Bergstrom, E.N.; et al. The mutagenic forces shaping the genomic landscape of lung cancer in never smokers. medRxiv 2024. [Google Scholar] [CrossRef]
- Wang, G.; Liu, X.; Liu, H.; Zhang, X.; Shao, Y.; Jia, X. A novel necroptosis related gene signature and regulatory network for overall survival prediction in lung adenocarcinoma. Sci. Rep. 2023, 13, 15345. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Fan, Z.; Luo, G.; Yang, C.; Huang, Q.; Fan, K.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. The role of necroptosis in cancer biology and therapy. Mol. Cancer 2019, 18, 100. [Google Scholar] [CrossRef]
- Lee, J.H.; Park, J.E.; Hong, M.J.; Choi, J.E.; Kang, H.; Do, S.K.; Lee, S.; Jeong, J.Y.; Shin, K.M.; Do, Y.W.; et al. Genetic variants in key necroptosis regulators predict prognosis of non-small cell lung cancer after surgical resection. Thorac. Cancer 2023, 14, 2678–2686. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, L.; Zhou, Y.; Liu, L.; Jiang, W.; Zhang, H.; Liu, H. Necroptosis in Pulmonary Diseases: A New Therapeutic Target. Front. Pharmacol. 2021, 12, 737129. [Google Scholar] [CrossRef]
- Bröer, S. Amino Acid Transporters as Targets for Cancer Therapy: Why, Where, When, and How. Int. J. Mol. Sci. 2020, 21, 6156. [Google Scholar] [CrossRef]
- Zhang, N.; Yang, X.; Yuan, F.; Zhang, L.; Wang, Y.; Wang, L.; Mao, Z.; Luo, J.; Zhang, H.; Zhu, W.G.; et al. Increased Amino Acid Uptake Supports Autophagy-Deficient Cell Survival upon Glutamine Deprivation. Cell Rep. 2018, 23, 3006–3020. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Hwang, Y.E.; Lee, M.; Keum, S.; Song, S.; Kim, J.; Choi, J.; Rhee, S. Downregulation of AP1S1 causes the lysosomal degradation of EGFR in non-small cell lung cancer. J. Cell. Physiol. 2023, 238, 2335–2347. [Google Scholar] [CrossRef]
- Su, F.; Fang, Y.; Yu, J.; Jiang, T.; Lin, S.; Zhang, S.; Lv, L.; Long, T.; Pan, H.; Qi, J.; et al. The Single Nucleotide Polymorphisms of AP1S1 are Associated with Risk of Esophageal Squamous Cell Carcinoma in Chinese Population. Pharmgenom. Pers. Med. 2022, 15, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Seegobin, K.; Heng, F.; Zhou, K.; Chen, R.; Qin, H.; Manochakian, R.; Zhao, Y.; Lou, Y. Genomic landscape of lung adenocarcinomas in different races. Front. Oncol. 2022, 12, 946625. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, L.; Wang, Y.; Wang, L.; Chen, G.; Zhang, L.; Wang, D. Integrative analysis of TP53 mutations in lung adenocarcinoma for immunotherapies and prognosis. BMC Bioinform. 2023, 24, 155. [Google Scholar] [CrossRef]
- Lin, X.; Wang, L.; Xie, X.; Qin, Y.; Xie, Z.; Ouyang, M.; Zhou, C. Prognostic Biomarker TP53 Mutations for Immune Checkpoint Blockade Therapy and Its Association with Tumor Microenvironment of Lung Adenocarcinoma. Front. Mol. Biosci. 2020, 7, 602328. [Google Scholar] [CrossRef]
- Kumar, R.; Castillero, F.; Bhandari, S.; Malapati, S.; Kloecker, G. The Hispanic paradox in non-small cell lung cancer. Hematol. Oncol. Stem Cell Ther. 2021; in press. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Chen, D. NRCAM acts as a prognostic biomarker and promotes the tumor progression in gastric cancer via EMT pathway. Tissue Cell 2022, 77, 101859. [Google Scholar] [CrossRef]
- Zhou, L.; He, L.; Liu, C.-H.; Qiu, H.; Zheng, L.; Sample, K.M.; Wu, Q.; Li, J.; Xie, K.; Ampuero, J.; et al. Liver cancer stem cell dissemination and metastasis: Uncovering the role of NRCAM in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2023, 42, 311. [Google Scholar] [CrossRef]
- Liu, F.; Ma, J.; Wang, K.; Li, Z.; Jiang, Q.; Chen, H.; Li, W.; Xia, J. High expression of PDE4D correlates with poor prognosis and clinical progression in pancreaticductal adenocarcinoma. J. Cancer 2019, 10, 6252–6260. [Google Scholar] [CrossRef]
- Xu, Y.; Jin, J.; Xu, J.; Shao, Y.W.; Fan, Y. JAK2 variations and functions in lung adenocarcinoma. Tumor Biol. 2017, 39, 101042831771114. [Google Scholar] [CrossRef]
- Zhang, Y.; Gu, X.; Zhu, F.; Li, Y.; Huang, Y.; Ju, S. High expression of GPR176 predicts poor prognosis of gastric cancer patients and promotes the proliferation, migration, and invasion of gastric cancer cells. Sci. Rep. 2023, 13, 9360. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Chen, S.; Liu, J.; Li, H.; Zhao, H.; Zheng, C.; Zhang, Y.; Huang, H.; Huang, J.; Wang, B.; et al. GPR176 Is a Biomarker for Predicting Prognosis and Immune Infiltration in Stomach Adenocarcinoma. Mediat. Inflamm. 2023, 2023, 7123568. [Google Scholar] [CrossRef]
- Liu, X.; Rothe, K.; Yen, R.; Fruhstorfer, C.; Maetzig, T.; Chen, M.; Forrest, D.L.; Humphries, R.K.; Jiang, X. A novel AHI-1–BCR-ABL–DNM2 complex regulates leukemic properties of primitive CML cells through enhanced cellular endocytosis and ROS-mediated autophagy. Leukemia 2017, 31, 2376–2387. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, Y.; Hu, Q.; Liu, Y.; Qi, X.; Tang, Z.; Hu, H.; Lin, N.; Zeng, S.; Yu, L. Unveiling tumor immune evasion mechanisms: Abnormal expression of transporters on immune cells in the tumor microenvironment. Front. Immunol. 2023, 14, 1225948. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.-J.; Snell, C.; Turley, H.; Li, J.-L.; McCormick, R.; Perera, S.M.W.; Heublein, S.; Kazi, S.; Azad, A.; Wilson, C.; et al. PAT4 levels control amino-acid sensitivity of rapamycin-resistant mTORC1 from the Golgi and affect clinical outcome in colorectal cancer. Oncogene 2016, 35, 3004–3015. [Google Scholar] [CrossRef]
- Goberdhan, D.C.I. Intracellular amino acid sensing and mTORC1-regulated growth: New ways to block an old target? Curr. Opin. Investig. Drugs 2010, 11, 1360–1367. [Google Scholar]
- Yang, Q.; Dang, H.; Liu, J.; Wang, X.; Wang, J.; Lan, X.; Ji, M.; Xing, M.; Hou, P. Hypoxia switches TET1 from being tumor-suppressive to oncogenic. Oncogene 2023, 42, 1634–1648. [Google Scholar] [CrossRef]
- Tang, J.; Peng, W.; Ji, J.; Peng, C.; Wang, T.; Yang, P.; Gu, J.; Feng, Y.; Jin, K.; Wang, X.; et al. GPR176 Promotes Cancer Progression by Interacting with G Protein GNAS to Restrain Cell Mitophagy in Colorectal Cancer. Adv. Sci. 2023, 10, 2205627. [Google Scholar] [CrossRef]
- Lucchetta, M.; da Piedade, I.; Mounir, M.; Vabistsevits, M.; Terkelsen, T.; Papaleo, E. Distinct signatures of lung cancer types: Aberrant mucin O-glycosylation and compromised immune response. BMC Cancer 2019, 19, 824. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [PubMed]
- Ansari, J.; Shackelford, R.E.; El-Osta, H. Epigenetics in non-small cell lung cancer: From basics to therapeutics. Transl. Lung Cancer Res. 2016, 5, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Ghazimoradi, M.H.; Pakravan, K.; Khalafizadeh, A.; Babashah, S. TET1 regulates stem cell properties and cell cycle of Cancer stem cells in triple-negative breast cancer via DNA demethylation. Biochem. Pharmacol. 2024, 219, 115913. [Google Scholar] [CrossRef]
- Filipczak, P.T.; Leng, S.; Tellez, C.S.; Do, K.C.; Grimes, M.J.; Thomas, C.L.; Walton-Filipczak, S.R.; Picchi, M.A.; Belinsky, S.A. p53-Suppressed Oncogene TET1 Prevents Cellular Aging in Lung Cancer. Cancer Res. 2019, 79, 1758–1768. [Google Scholar] [CrossRef]
- Alrehaili, A.A.; Gharib, A.F.; Alghamdi, S.A.; Alhazmi, A.; Al-Shehri, S.S.; Hagag, H.M.; Alsaeedi, F.A.; Alhuthali, H.M.; Raafat, N.; Etewa, R.L.; et al. Evaluation of TET Family Gene Expression and 5-Hydroxymethylcytosine as Potential Epigenetic Markers in Non-small Cell Lung Cancer. In Vivo 2023, 37, 445–453. [Google Scholar] [CrossRef]
- Tuersun, H.; Liu, L.; Zhang, J.; Maimaitizunong, R.; Tang, X.; Li, H. m6A reading protein RBMX as a biomarker for prognosis and tumor progression in esophageal cancer. Transl. Cancer Res. 2023, 12, 2319–2335. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, T.; Chen, D.; Huang, J.; Zhao, Y.; Ma, W.; Liu, H. RBMX involves in telomere stability maintenance by regulating TERRA expression. PLoS Genet. 2023, 19, e1010937. [Google Scholar] [CrossRef]
- Sheng, Y.; Lei, K.; Sun, C.; Liu, J.; Tu, Z.; Zhu, X.; Huang, K. Aberrant RBMX expression is relevant for cancer prognosis and immunotherapy response. Aging 2024, 16, 226. [Google Scholar] [CrossRef]
- Wang, Z.; Xing, Y.; Li, B.; Li, X.; Liu, B.; Wang, Y. Molecular pathways, resistance mechanisms and targeted interventions in non-small-cell lung cancer. Mol. Biomed. 2022, 3, 42. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Yang, J.C.-H.; Lee, C.K.; Kurata, T.; Kim, D.-W.; John, T.; Nogami, N.; Ohe, Y.; Mann, H.; Rukazenkov, Y.; et al. Osimertinib As First-Line Treatment of EGFR Mutation–Positive Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 841–849. [Google Scholar] [CrossRef]
- Cannon, M.; Stevenson, J.; Stahl, K.; Basu, R.; Coffman, A.; Kiwala, S.; McMichael, J.F.; Kuzma, K.; Morrissey, D.; Cotto, K.; et al. DGIdb 5.0: Rebuilding the drug–gene interaction database for precision medicine and drug discovery platforms. Nucleic Acids Res. 2024, 52, D1227–D1235. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Zhao, D.; Yu, X.; Shen, X.; Zhou, Y.; Wang, S.; Qiu, Y.; Chen, Y.; Zhu, F. TTD: Therapeutic Target Database describing target druggability information. Nucleic Acids Res. 2024, 52, D1465–D1477. [Google Scholar] [CrossRef] [PubMed]
- Forloni, M.; Gupta, R.; Nagarajan, A.; Sun, L.-S.; Dong, Y.; Pirazzoli, V.; Toki, M.; Wurtz, A.; Melnick, M.A.; Kobayashi, S.; et al. Oncogenic EGFR Represses the TET1 DNA Demethylase to Induce Silencing of Tumor Suppressors in Cancer Cells. Cell Rep. 2016, 16, 457–471. [Google Scholar] [CrossRef]
- Wu, H.-X.; Chen, Y.-X.; Wang, Z.-X.; Zhao, Q.; He, M.-M.; Wang, Y.-N.; Wang, F.; Xu, R.H. Alteration in TET1 as potential biomarker for immune checkpoint blockade in multiple cancers. J. Immunother. Cancer 2019, 7, 264. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Wang, C.; Zhou, J.-X.; Xu, Z.-M.; Gao, J.; Sui, P.; Walsh, C.P.; Ji, H.; Xu, G.L. Loss of TET reprograms Wnt signaling through impaired demethylation to promote lung cancer development. Proc. Natl. Acad. Sci. USA 2022, 119, e2107599119. [Google Scholar] [CrossRef]
- Yan, Q.; Zeng, P.; Zhou, X.; Zhao, X.; Chen, R.; Qiao, J.; Feng, L.; Zhu, Z.; Zhang, G.; Chen, C. RBMX suppresses tumorigenicity and progression of bladder cancer by interacting with the hnRNP A1 protein to regulate PKM alternative splicing. Oncogene 2021, 40, 2635–2650. [Google Scholar] [CrossRef] [PubMed]
- Renieri, A.; Mencarelli, M.A.; Cetta, F.; Baldassarri, M.; Mari, F.; Furini, S.; Piu, P.; Ariani, F.; Dragani, T.A.; Frullanti, E. Oligogenic germline mutations identified in early non-smokers lung adenocarcinoma patients. Lung Cancer 2014, 85, 168–174. [Google Scholar] [CrossRef]
- Feng, B.; Pan, B.; Huang, J.; Du, Y.; Wang, X.; Wu, J.; Ma, R.; Shen, B.; Huang, G.; Feng, J. PDE4D/cAMP/IL-23 axis determines the immunotherapy efficacy of lung adenocarcinoma via activating the IL-9 autocrine loop of cytotoxic T lymphocytes. Cancer Lett. 2023, 565, 216224. [Google Scholar] [CrossRef]
- Pullamsetti, S.S.; Banat, G.A.; Schmall, A.; Szibor, M.; Pomagruk, D.; Hänze, J.; Kolosionek, E.; Wilhelm, J.; Braun, T.; Grimminger, F.; et al. Phosphodiesterase-4 promotes proliferation and angiogenesis of lung cancer by crosstalk with HIF. Oncogene 2013, 32, 1121–1134. [Google Scholar] [CrossRef]
- Hsien Lai, S.; Zervoudakis, G.; Chou, J.; Gurney, M.E.; Quesnelle, K.M. PDE4 subtypes in cancer. Oncogene 2020, 39, 3791–3802. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef]
- Sanchez, P.; Espinosa, M.; Maldonado, V.; Barquera, R.; Belem-Gabiño, N.; Torres, J.; Cravioto, A.; Melendez-Zajgla, J. Pancreatic ductal adenocarcinomas from Mexican patients present a distinct genomic mutational pattern. Mol. Biol. Rep. 2020, 47, 5175–5184. [Google Scholar] [CrossRef]
- Bergstrom, E.N.; Huang, M.N.; Mahto, U.; Barnes, M.; Stratton, M.R.; Rozen, S.G.; Alexandrov, L.B. SigProfilerMatrixGenerator: A tool for visualizing and exploring patterns of small mutational events. BMC Genomics 2019, 20, 685. [Google Scholar] [CrossRef]
- Díaz-Gay, M.; Vangara, R.; Barnes, M.; Wang, X.; Islam, S.M.A.; Vermes, I.; Duke, S.; Narasimman, N.B.; Yang, T.; Jiang, Z.; et al. Assigning mutational signatures to individual samples and individual somatic mutations with SigProfilerAssignment. Bioinformatics 2023, 39, btad756. [Google Scholar] [CrossRef]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041.e21. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Mularoni, L.; Sabarinathan, R.; Deu-Pons, J.; Gonzalez-Perez, A.; López-Bigas, N. OncodriveFML: A general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 2016, 17, 128. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Vasaikar, S.; Shi, Z.; Greer, M.; Zhang, B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017, 45, W130–W137. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rueda-Zarazua, B.; Gutiérrez, H.; García-Ortiz, H.; Orozco, L.; Ramírez-Martínez, G.; Jiménez-Alvarez, L.; Bolaños-Morales, F.V.; Zuñiga, J.; Ávila-Moreno, F.; Melendez-Zajgla, J. A Pilot Study: Contrasting Genomic Profiles of Lung Adenocarcinoma Between Patients of European and Latin American Ancestry. Int. J. Mol. Sci. 2025, 26, 4865. https://doi.org/10.3390/ijms26104865
Rueda-Zarazua B, Gutiérrez H, García-Ortiz H, Orozco L, Ramírez-Martínez G, Jiménez-Alvarez L, Bolaños-Morales FV, Zuñiga J, Ávila-Moreno F, Melendez-Zajgla J. A Pilot Study: Contrasting Genomic Profiles of Lung Adenocarcinoma Between Patients of European and Latin American Ancestry. International Journal of Molecular Sciences. 2025; 26(10):4865. https://doi.org/10.3390/ijms26104865
Chicago/Turabian StyleRueda-Zarazua, Bertha, Humberto Gutiérrez, Humberto García-Ortiz, Lorena Orozco, Gustavo Ramírez-Martínez, Luis Jiménez-Alvarez, Francina V. Bolaños-Morales, Joaquín Zuñiga, Federico Ávila-Moreno, and Jorge Melendez-Zajgla. 2025. "A Pilot Study: Contrasting Genomic Profiles of Lung Adenocarcinoma Between Patients of European and Latin American Ancestry" International Journal of Molecular Sciences 26, no. 10: 4865. https://doi.org/10.3390/ijms26104865
APA StyleRueda-Zarazua, B., Gutiérrez, H., García-Ortiz, H., Orozco, L., Ramírez-Martínez, G., Jiménez-Alvarez, L., Bolaños-Morales, F. V., Zuñiga, J., Ávila-Moreno, F., & Melendez-Zajgla, J. (2025). A Pilot Study: Contrasting Genomic Profiles of Lung Adenocarcinoma Between Patients of European and Latin American Ancestry. International Journal of Molecular Sciences, 26(10), 4865. https://doi.org/10.3390/ijms26104865