


DNA Methylation and Alternative Splicing Safeguard Genome and Transcriptome After a Retrotransposition Burst in Arabidopsis thaliana

 , , ,
, , , 
Abstract
1. Introduction
2. Results
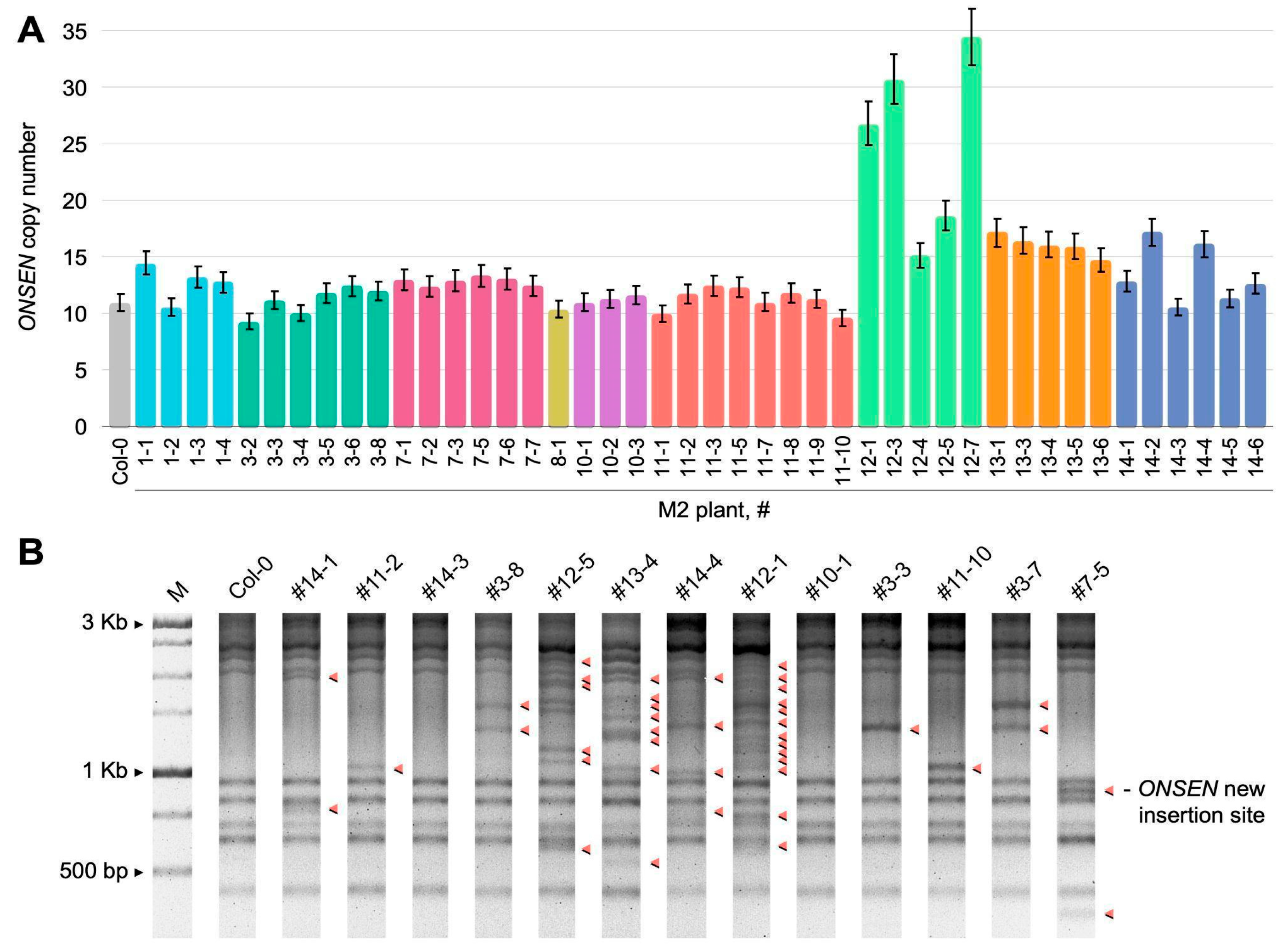
2.1. Obtaining A. thaliana Plants with an Increased Number of ONSEN Insertions
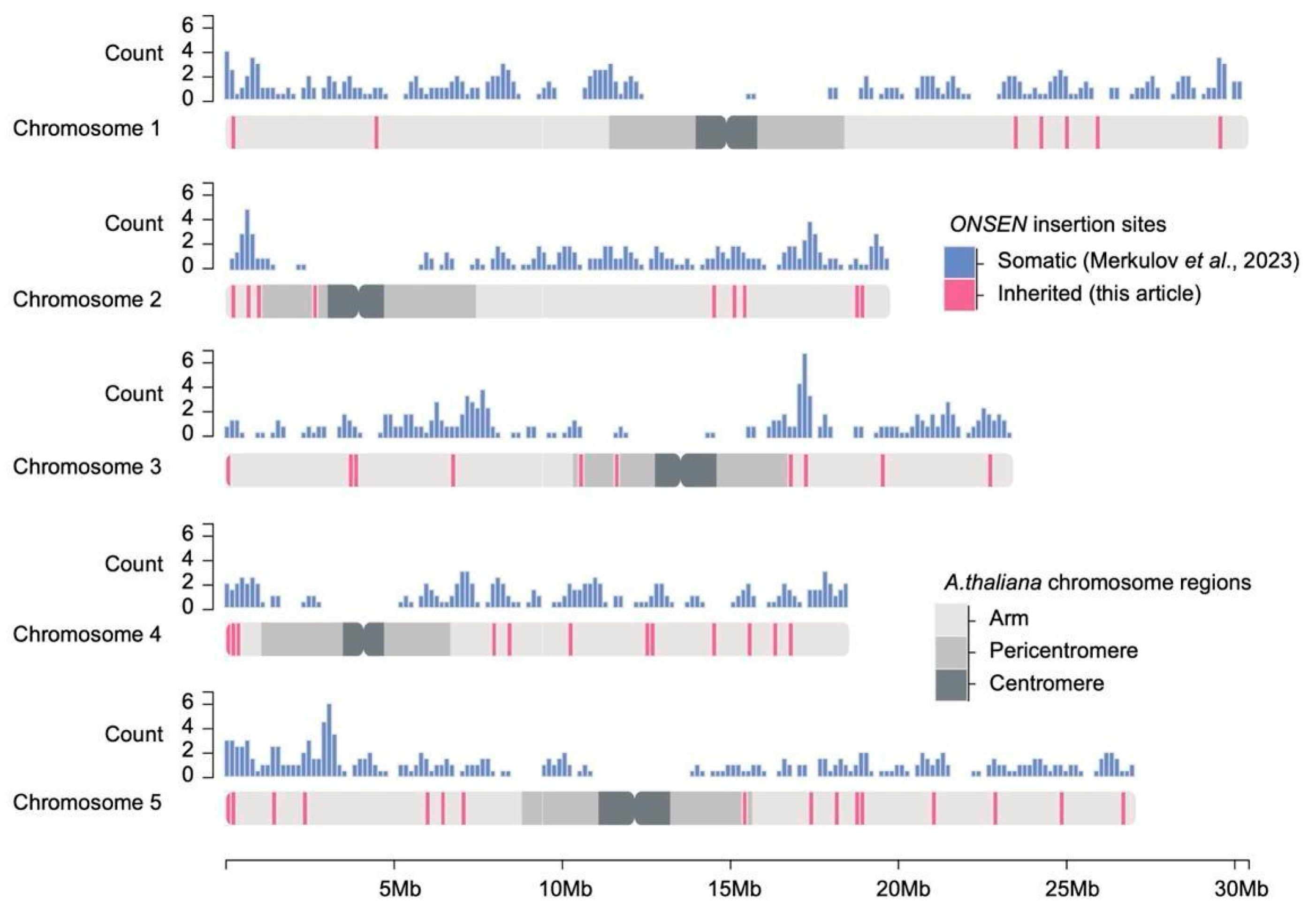
2.2. Whole-Genome Analysis of Novel ONSEN Insertions
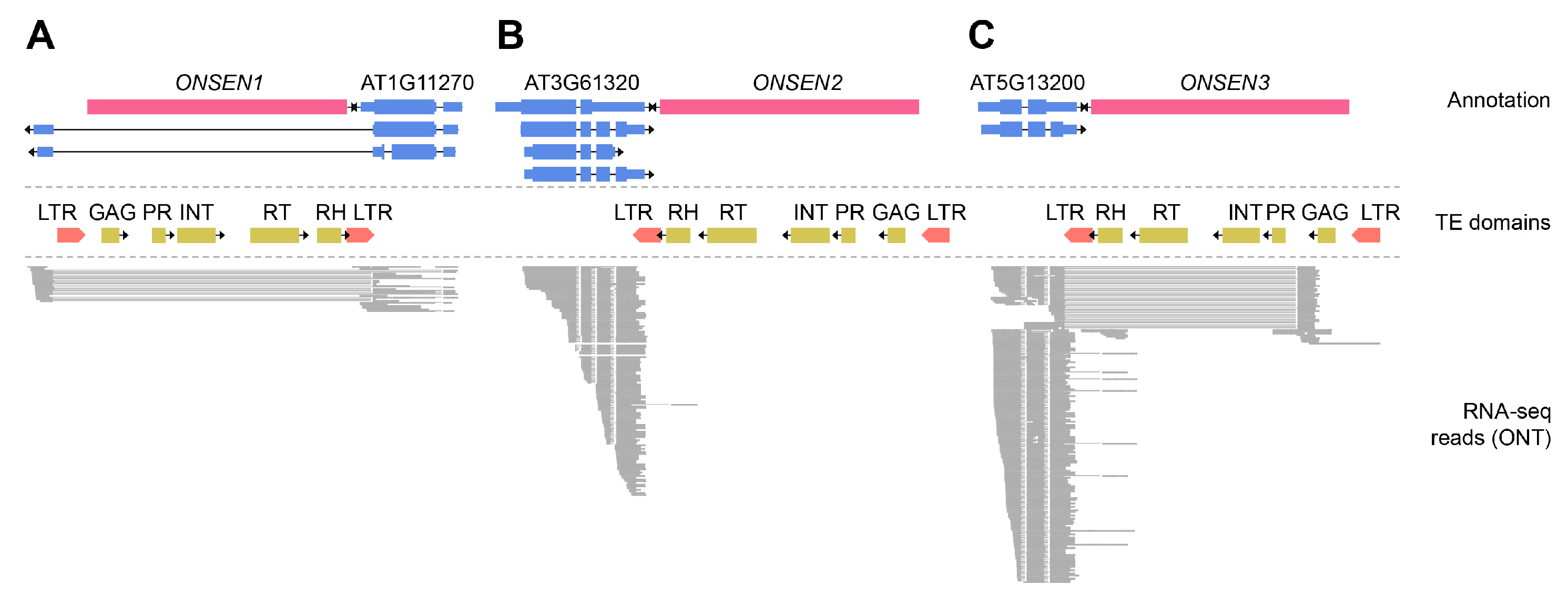
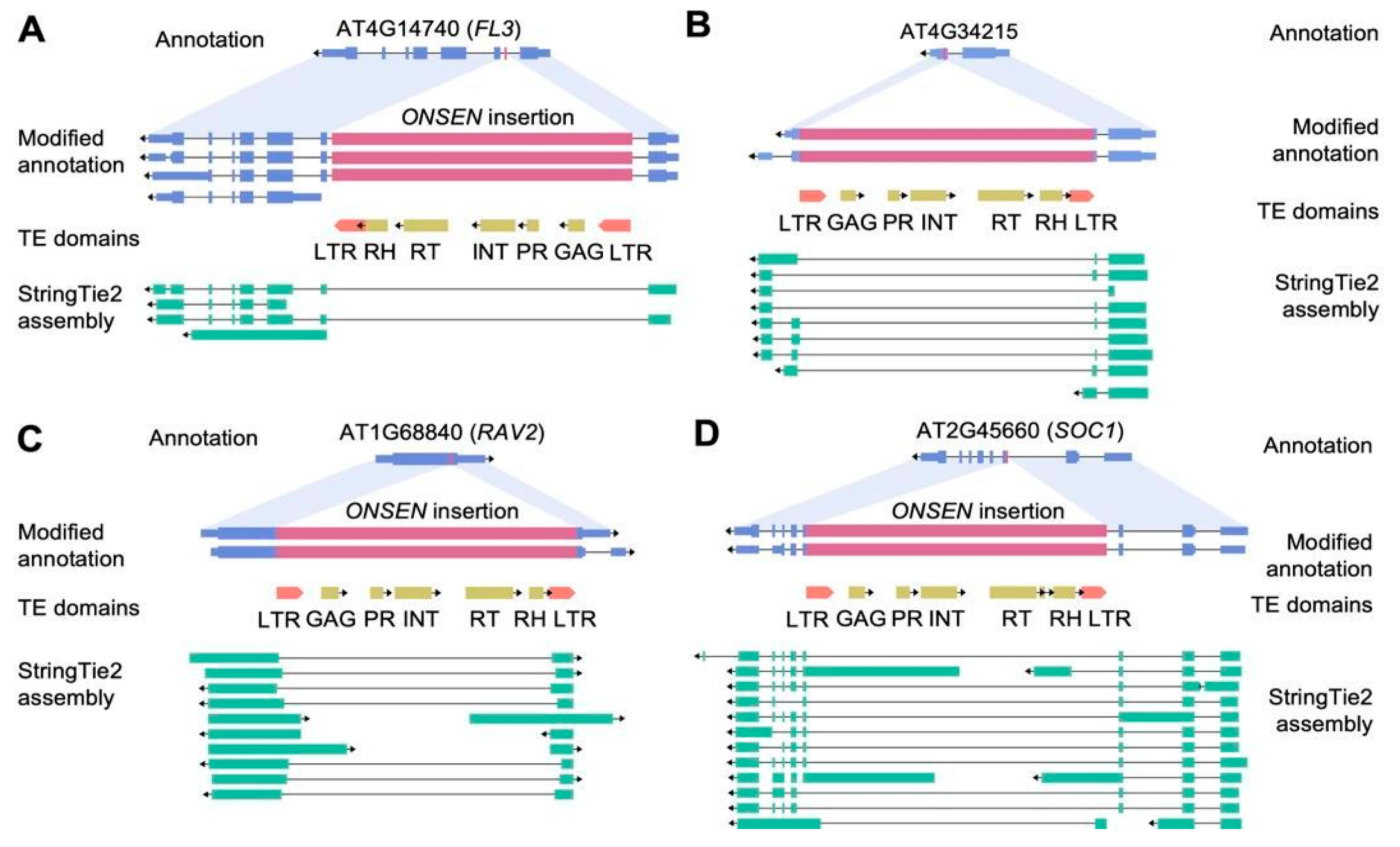
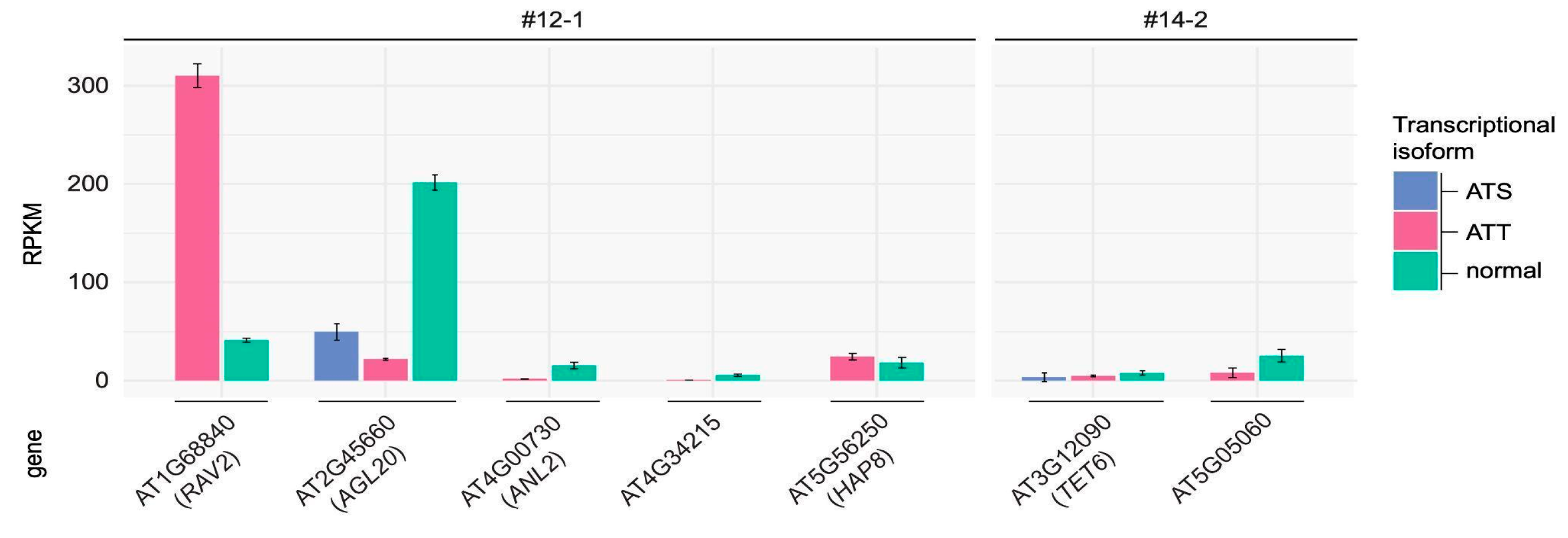
2.3. Transcription Changes Induced by ONSEN Insertions
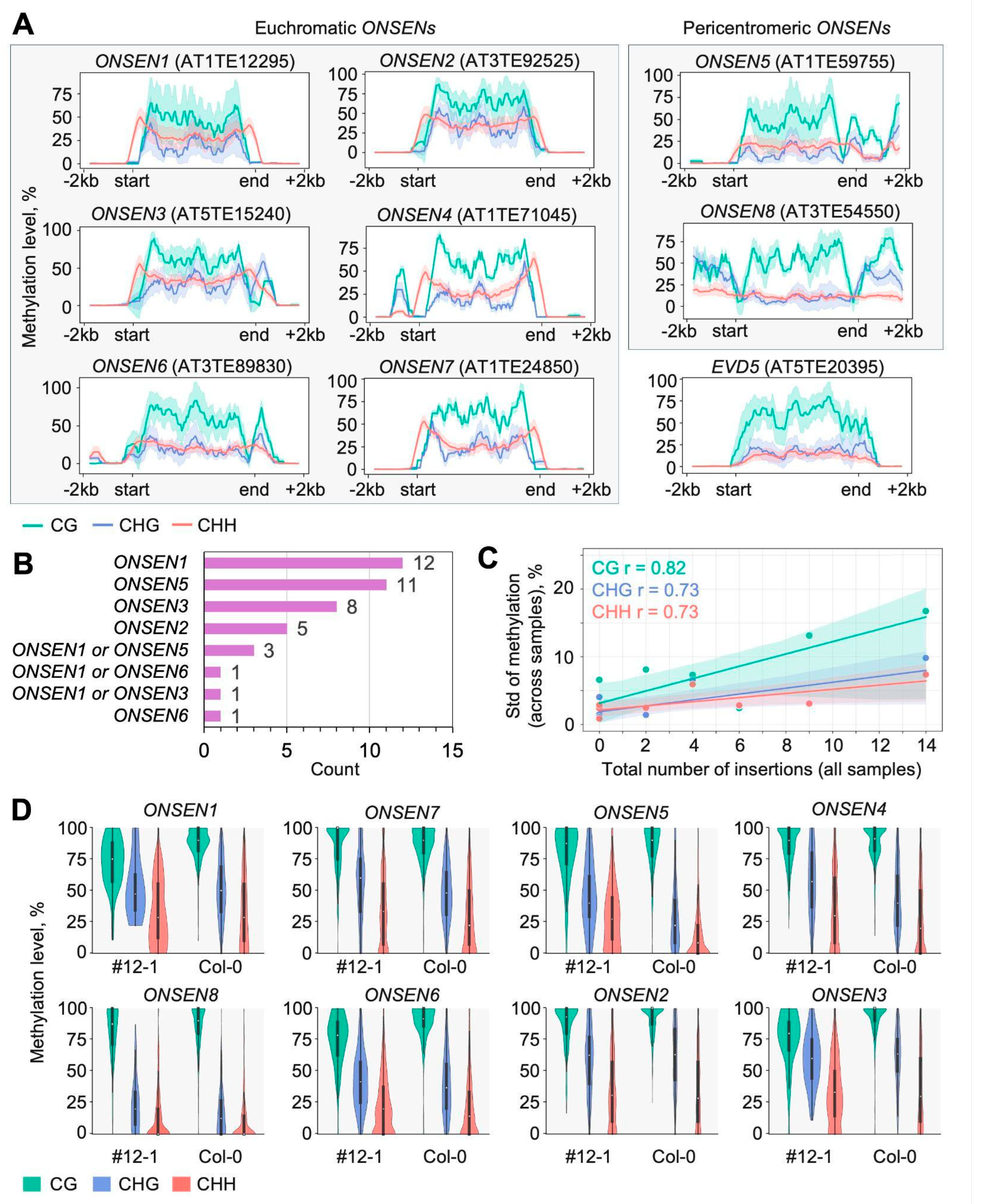
2.4. DNA Methylation of ONSEN Elements and Their Insertions
2.4.1. Methylation in Original Full-Length ONSEN Elements
2.4.2. Methylation in Novel ONSEN Insertions
3. Discussion
3.1. Convergent ONSEN Insertions in Different Arabidopsis Collections
3.2. Genomic Location Dictates DNA Methylation Landscapes of Arabidopsis ONSEN Elements
3.3. Distinct Phases of DNA Methylation of Novel ONSEN Insertions
3.4. Dual Defense: DNA Methylation and Alternative Splicing Rapidly Mitigate ONSEN Insertions in Arabidopsis
4. Materials and Methods
4.1. Plant Material and Seeds Sterilization
4.2. Transposon Activation and M1 Seed Production
4.3. M1 and M2 Plants Growth Conditions
4.4. DNA, RNA Isolation, and cDNA Synthesis
4.5. qPCR, Transposon Display and PCR Analysis
4.6. Nanopore Sequencing
4.7. Genome Assembly
4.8. Novel Insertions Discovery, Assembly, and Pseudoreference Generation
4.9. Novel Insertions Annotation
4.10. Distribution of Novel Insertion Sites in the Arabidopsis Genome and Chromatin States
4.11. Gene Ontology Analysis of Genes Harboring Novel ONSEN Insertions
4.12. Determination of TEI Parental ONSEN Copies
4.13. Methylation Calling
4.14. Methylation Analysis
4.15. Chimeric Gene-TE Transcripts Detection
4.16. Analysis of Differential Isoform and Gene Expression
4.17. Visualization
4.18. Data and Code Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wells, J.N.; Feschotte, C. A Field Guide to Eukaryotic Transposable Elements. Annu. Rev. Genet. 2020, 54, 539–561. [Google Scholar] [CrossRef]
- Mhiri, C.; Borges, F.; Grandbastien, M.A. Specificities and Dynamics of Transposable Elements in Land Plants. Biology 2022, 11, 488. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, D.J. Eukaryotic transposable elements and genome evolution. Trends Genet. 1989, 5, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Kapitonov, V.V.; Jurka, J. A universal classification of eukaryotic transposable elements implemented in Repbase. Nat. Rev. Genet. 2008, 9, 411–412. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Galindo-González, L.; Mhiri, C.; Deyholos, M.K.; Grandbastien, M.-A. LTR-retrotransposons in plants: Engines of evolution. Gene 2017, 626, 14–25. [Google Scholar] [CrossRef]
- Arvas, Y.E.; Marakli, S.; Kaya, Y.; Kalendar, R. The power of retrotransposons in high-throughput genotyping and sequencing. Front. Plant Sci. 2023, 14, 1174339. [Google Scholar] [CrossRef]
- Hassan, A.H.; Mokhtar, M.M.; El Allali, A. Transposable elements: Multifunctional players in the plant genome. Front. Plant Sci. 2024, 14, 1330127. [Google Scholar] [CrossRef]
- Kalendar, R.; Flavell, A.J.; Ellis, T.H.N.; Sjakste, T.; Moisy, C.; Schulman, A.H. Analysis of plant diversity with retrotransposon-based molecular markers. Heredity 2011, 106, 520–530. [Google Scholar] [CrossRef]
- Fultz, D.; Choudury, S.G.; Slotkin, R.K. Silencing of active transposable elements in plants. Curr. Opin. Plant Biol. 2015, 27, 67–76. [Google Scholar] [CrossRef]
- Sigman, M.J.; Slotkin, R.K. The First Rule of Plant Transposable Element Silencing: Location, Location, Location. Plant Cell 2016, 28, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Lippman, Z.; Gendrel, A.V.; Black, M.; Vaughn, M.W.; Dedhia, N.; McCombie, W.R.; Lavine, K.; Mittal, V.; May, B.; Kasschau, K.D.; et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004, 430, 471–476. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Hsieh, P.-H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis Nucleosome Remodeler DDM1 Allows DNA Methyltransferases to Access H1-Containing Heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef]
- Erdmann, R.M.; Picard, C.L. RNA-directed DNA Methylation. PLoS Genet. 2020, 16, e1009034. [Google Scholar] [CrossRef]
- Novikova, O. Chromodomains and LTR retrotransposons in plants. Commun. Integr. Biol. 2009, 2, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Quadrana, L.; Etcheverry, M.; Gilly, A.; Caillieux, E.; Madoui, M.-A.; Guy, J.; Bortolini Silveira, A.; Engelen, S.; Baillet, V.; Wincker, P.; et al. Transposition favors the generation of large effect mutations that may facilitate rapid adaption. Nat. Commun. 2019, 10, 3421. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, S.; Bousios, A.; Perez-Roman, E.; Yamaguchi, S.; Leduque, B.; Nakano, A.; Naish, M.; Osakabe, A.; Toyoda, A.; Ito, H.; et al. Centrophilic retrotransposon integration via CENH3 chromatin in Arabidopsis. Nature 2025, 637, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Gaubert, H.; Bucher, E.; Mirouze, M.; Vaillant, I.; Paszkowski, J. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 2011, 472, 115–119. [Google Scholar] [CrossRef]
- Mirouze, M.; Reinders, J.; Bucher, E.; Nishimura, T.; Schneeberger, K.; Ossowski, S.; Cao, J.; Weigel, D.; Paszkowski, J.; Mathieu, O. Selective epigenetic control of retrotransposition in Arabidopsis. Nature 2009, 461, 427–430. [Google Scholar] [CrossRef]
- Papolu, P.K.; Ramakrishnan, M.; Mullasseri, S.; Kalendar, R.; Wei, Q.; Zou, L.H.; Ahmad, Z.; Vinod, K.K.; Yang, P.; Zhou, M. Retrotransposons: How the continuous evolutionary front shapes plant genomes for response to heat stress. Front. Plant Sci. 2022, 13, 1064847. [Google Scholar] [CrossRef]
- Butelli, E.; Licciardello, C.; Zhang, Y.; Liu, J.; Mackay, S.; Bailey, P.; Reforgiato-Recupero, G.; Martin, C. Retrotransposons Control Fruit-Specific, Cold-Dependent Accumulation of Anthocyanins in Blood Oranges. Plant Cell 2012, 24, 1242–1255. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, J.; Han, X.; Li, J.; Gao, Y.; Richards, C.M.; Zhang, C.; Tian, Y.; Liu, G.; Gul, H.; et al. A high-quality apple genome assembly reveals the association of a retrotransposon and red fruit colour. Nat. Commun. 2019, 10, 1494. [Google Scholar] [CrossRef] [PubMed]
- Zervudacki, J.; Yu, A.; Amesefe, D.; Wang, J.; Drouaud, J.; Navarro, L.; Deleris, A. Transcriptional control and exploitation of an immune-responsive family of plant retrotransposons. EMBO J. 2018, 37, e98482. [Google Scholar] [CrossRef]
- Hollister, J.D.; Smith, L.M.; Guo, Y.-L.; Ott, F.; Weigel, D.; Gaut, B.S. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2011, 108, 2322–2327. [Google Scholar] [CrossRef] [PubMed]
- Kashkush, K.; Khasdan, V. Large-scale survey of cytosine methylation of retrotransposons and the impact of readout transcription from long terminal repeats on expression of adjacent rice genes. Genetics 2007, 177, 1975–1985. [Google Scholar] [CrossRef] [PubMed]
- Berthelier, J.; Furci, L.; Asai, S.; Sadykova, M.; Shimazaki, T.; Shirasu, K.; Saze, H. Long-read direct RNA sequencing reveals epigenetic regulation of chimeric gene-transposon transcripts in Arabidopsis thaliana. Nat. Commun. 2023, 14, 3248. [Google Scholar] [CrossRef]
- Gaubert, H.; Sanchez, D.H.; Drost, H.G.; Paszkowski, J. Developmental Restriction of Retrotransposition Activated in Arabidopsis by Environmental Stress. Genetics 2017, 207, 813–821. [Google Scholar] [CrossRef]
- Roquis, D.; Robertson, M.; Yu, L.; Thieme, M.; Julkowska, M.; Bucher, E. Genomic impact of stress-induced transposable element mobility in Arabidopsis. Nucleic Acids Res. 2021, 49, 10431–10447. [Google Scholar] [CrossRef]
- Parker, M.T.; Knop, K.; Zacharaki, V.; Sherwood, A.V.; Tomé, D.; Yu, X.; Martin, P.G.P.; Beynon, J.; Michaels, S.D.; Barton, G.J.; et al. Widespread premature transcription termination of Arabidopsis thaliana NLR genes by the spen protein FPA. eLife 2021, 10, e65537. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Eulgem, T. An alternative polyadenylation mechanism coopted to the Arabidopsis RPP7 gene through intronic retrotransposon domestication. Proc. Natl. Acad. Sci. USA 2013, 110, E3535–E3543. [Google Scholar] [CrossRef]
- Le, T.N.; Miyazaki, Y.; Takuno, S.; Saze, H. Epigenetic regulation of intragenic transposable elements impacts gene transcription in Arabidopsis thaliana. Nucleic Acids Res. 2015, 43, 3911–3921. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, L.; Li, Q.; Xu, W.; Li, K.; Wang, Z.-W.; Sun, Q. Intronic heterochromatin prevents cryptic transcription initiation in Arabidopsis. Plant J. 2020, 101, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.-G.; Wang, X.; Zhang, L.; Xiong, X.; Zhang, Z.; Tang, K.; Pan, L.; Hsu, C.-C.; Xu, H.; Tao, W.A.; et al. A protein complex regulates RNA processing of intronic heterochromatin-containing genes in Arabidopsis. Proc. Natl. Acad. Sci. USA 2017, 114, E7377–E7384. [Google Scholar] [CrossRef] [PubMed]
- Raingeval, M.; Leduque, B.; Baduel, P.; Edera, A.; Roux, F.; Colot, V.; Quadrana, L. Retrotransposon-driven environmental regulation of FLC leads to adaptive response to herbicide. Nat. Plants 2024, 10, 1672–1681. [Google Scholar] [CrossRef]
- Baduel, P.; Leduque, B.; Ignace, A.; Gy, I.; Gil, J.; Loudet, O.; Colot, V.; Quadrana, L. Genetic and environmental modulation of transposition shapes the evolutionary potential of Arabidopsis thaliana. Genome Biol. 2021, 22, 138. [Google Scholar] [CrossRef]
- Del Toro-De León, G.; van Boven, J.; Santos-González, J.; Jiao, W.B.; Peng, H.; Schneeberger, K.; Köhler, C. Epigenetic and transcriptional consequences in the endosperm of chemically induced transposon mobilization in Arabidopsis. Nucleic Acids Res. 2024, 52, 8833–8848. [Google Scholar] [CrossRef]
- Merkulov, P.; Gvaramiya, S.; Dudnikov, M.; Komakhin, R.; Omarov, M.; Kocheshkova, A.; Konstantinov, Z.; Soloviev, A.; Karlov, G.; Divashuk, M.; et al. Cas9-targeted Nanopore sequencing rapidly elucidates the transposition preferences and DNA methylation profiles of mobile elements in plants. J. Integr. Plant Biol. 2023, 65, 2242–2261. [Google Scholar] [CrossRef]
- Cavrak, V.V.; Lettner, N.; Jamge, S.; Kosarewicz, A.; Bayer, L.M.; Mittelsten Scheid, O. How a Retrotransposon Exploits the Plant’s Heat Stress Response for Its Activation. PLoS Genet. 2014, 10, e1004115. [Google Scholar] [CrossRef]
- Matsunaga, W.; Kobayashi, A.; Kato, A.; Ito, H. The effects of heat induction and the siRNA biogenesis pathway on the transgenerational transposition of ONSEN, a copia-like retrotransposon in Arabidopsis thaliana. Plant Cell Physiol. 2011, 53, 824–833. [Google Scholar] [CrossRef]
- Niu, X.; Chen, L.; Kato, A.; Ito, H. Regulatory mechanism of a heat-activated retrotransposon by DDR complex in Arabidopsis thaliana. Front. Plant Sci. 2022, 13, 1048957. [Google Scholar] [CrossRef]
- Niu, X.; Ge, Z.; Ito, H. Regulatory mechanism of heat-active retrotransposons by the SET domain protein SUVH2. Front. Plant Sci. 2024, 15, 1355626. [Google Scholar] [CrossRef] [PubMed]
- Thieme, M.; Lanciano, S.; Balzergue, S.; Daccord, N.; Mirouze, M.; Bucher, E. Inhibition of RNA polymerase II allows controlled mobilisation of retrotransposons for plant breeding. Genome Biol. 2017, 18, 134. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-X.; Liu, Z.-D.; Bai, X.; Wu, B.; Song, R.; Yao, H.-C.; Chen, Y.; Chi, W.; Hua, Q.; Cheng, L.; et al. Accurate cross-species 5mC detection for Oxford Nanopore sequencing in plants with DeepPlant. Nat. Commun. 2025, 16, 3227. [Google Scholar] [CrossRef]
- Panda, K.; Slotkin, R.K. Long-Read cDNA Sequencing Enables a “Gene-Like” Transcript Annotation of Transposable Elements. Plant Cell 2020, 32, 2687–2698. [Google Scholar] [CrossRef]
- Kirov, I.; Merkulov, P.; Dudnikov, M.; Polkhovskaya, E.; Komakhin, R.A.; Konstantinov, Z.; Gvaramiya, S.; Ermolaev, A.; Kudryavtseva, N.; Gilyok, M.; et al. Transposons Hidden in Arabidopsis thaliana Genome Assembly Gaps and Mobilization of Non-Autonomous LTR Retrotransposons Unravelled by Nanotei Pipeline. Plants 2021, 10, 2681. [Google Scholar] [CrossRef]
- Sanchez, D.H.; Gaubert, H.; Drost, H.-G.; Zabet, N.R.; Paszkowski, J. High-frequency recombination between members of an LTR retrotransposon family during transposition bursts. Nat. Commun. 2017, 8, 1283. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tian, T.; Zhang, K.; You, Q.; Yan, H.; Zhao, N.; Yi, X.; Xu, W.; Su, Z. PCSD: A plant chromatin state database. Nucleic Acids Res. 2017, 46, D1157–D1167. [Google Scholar] [CrossRef]
- Sequeira-Mendes, J.; Aragüez, I.; Peiró, R.; Mendez-Giraldez, R.; Zhang, X.; Jacobsen, S.E.; Bastolla, U.; Gutierrez, C. The Functional Topography of the Arabidopsis Genome Is Organized in a Reduced Number of Linear Motifs of Chromatin States. Plant Cell 2014, 26, 2351–2366. [Google Scholar] [CrossRef]
- Xu, W.; Thieme, M.; Roulin, A.C. Natural Diversity of Heat-Induced Transcription of Retrotransposons in Arabidopsis thaliana. Genome Biol. Evol. 2024, 16, evae242. [Google Scholar] [CrossRef]
- Sigman, M.J.; Panda, K.; Kirchner, R.; McLain, L.L.; Payne, H.; Peasari, J.R.; Husbands, A.Y.; Slotkin, R.K.; McCue, A.D. An siRNA-guided ARGONAUTE protein directs RNA polymerase V to initiate DNA methylation. Nat. Plants 2021, 7, 1461–1474. [Google Scholar] [CrossRef]
- Ahmed, I.; Sarazin, A.; Bowler, C.; Colot, V.; Quesneville, H. Genome-wide evidence for local DNA methylation spreading from small RNA-targeted sequences in Arabidopsis. Nucleic Acids Res. 2011, 39, 6919–6931. [Google Scholar] [CrossRef] [PubMed]
- Quadrana, L.; Bortolini Silveira, A.; Mayhew, G.F.; LeBlanc, C.; Martienssen, R.A.; Jeddeloh, J.A.; Colot, V. The Arabidopsis thaliana mobilome and its impact at the species level. eLife 2016, 5, e15716. [Google Scholar] [CrossRef] [PubMed]
- To, T.K.; Nishizawa, Y.; Inagaki, S.; Tarutani, Y.; Tominaga, S.; Toyoda, A.; Fujiyama, A.; Berger, F.; Kakutani, T. RNA interference-independent reprogramming of DNA methylation in Arabidopsis. Nat. Plants 2020, 6, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Panda, K.; Slotkin, R.K. Proposed mechanism for the initiation of transposable element silencing by the RDR6-directed DNA methylation pathway. Plant Signal. Behav. 2013, 8, e25206. [Google Scholar] [CrossRef]
- Panda, K.; Ji, L.; Neumann, D.A.; Daron, J.; Schmitz, R.J.; Slotkin, R.K. Full-length autonomous transposable elements are preferentially targeted by expression-dependent forms of RNA-directed DNA methylation. Genome Biol. 2016, 17, 170. [Google Scholar] [CrossRef]
- Fultz, D.; Slotkin, R.K. Exogenous Transposable Elements Circumvent Identity-Based Silencing, Permitting the Dissection of Expression-Dependent Silencing. Plant Cell 2017, 29, 360–376. [Google Scholar] [CrossRef]
- Nuthikattu, S.; McCue, A.D.; Panda, K.; Fultz, D.; DeFraia, C.; Thomas, E.N.; Slotkin, R.K. The Initiation of Epigenetic Silencing of Active Transposable Elements Is Triggered by RDR6 and 21–22 Nucleotide Small Interfering RNAs. Plant Physiol. 2013, 162, 116–131. [Google Scholar] [CrossRef]
- Griffin, P.T.; Niederhuth, C.E.; Schmitz, R.J. A Comparative Analysis of 5-Azacytidine- and Zebularine-Induced DNA Demethylation. G3 2016, 6, 2773–2780. [Google Scholar] [CrossRef]
- Huc, J.; Dziasek, K.; Pachamuthu, K.; Woh, T.; Köhler, C.; Borges, F. Bypassing reproductive barriers in hybrid seeds using chemically induced epimutagenesis. Plant Cell 2021, 34, 989–1001. [Google Scholar] [CrossRef]
- Cuerda-Gil, D.; Slotkin, R.K. Non-canonical RNA-directed DNA methylation. Nat. Plants 2016, 2, 16163. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Freeling, M.; Lisch, D. Heritable transposon silencing initiated by a naturally occurring transposon inverted duplication. Nat. Genet. 2005, 37, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lin, R.; Liang, J.; King, G.J.; Wu, J.; Wang, X. Transposable element insertion: A hidden major source of domesticated phenotypic variation in Brassica rapa. Plant Biotechnol. J. 2022, 20, 1298–1310. [Google Scholar] [CrossRef]
- Liu, Z.; Fan, M.; Yue, E.-K.; Li, Y.; Tao, R.-F.; Xu, H.-M.; Duan, M.-H.; Xu, J.-H. Natural variation and evolutionary dynamics of transposable elements in Brassica oleracea based on next-generation sequencing data. Hortic. Res. 2020, 7, 145. [Google Scholar] [CrossRef] [PubMed]
- Gozashti, L.; Roy, S.W.; Thornlow, B.; Kramer, A.; Ares, M.; Corbett-Detig, R. Transposable elements drive intron gain in diverse eukaryotes. Proc. Natl. Acad. Sci. USA 2022, 119, e2209766119. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Kim, J.-M.; Matsunaga, W.; Saze, H.; Matsui, A.; Endo, T.A.; Harukawa, Y.; Takagi, H.; Yaegashi, H.; Masuta, Y.; et al. A Stress-Activated Transposon in Arabidopsis Induces Transgenerational Abscisic Acid Insensitivity. Sci. Rep. 2016, 6, 23181. [Google Scholar] [CrossRef]
- Rigal, M.; Kevei, Z.; Pélissier, T.; Mathieu, O. DNA methylation in an intron of the IBM1 histone demethylase gene stabilizes chromatin modification patterns. EMBO J. 2012, 31, 2981–2993. [Google Scholar] [CrossRef]
- He, C.; Chen, Z.; Zhao, Y.; Yu, Y.; Wang, H.; Wang, C.; Copenhaver, G.P.; Qi, J.; Wang, Y. Histone demethylase IBM1-mediated meiocyte gene expression ensures meiotic chromosome synapsis and recombination. PLoS Genet. 2022, 18, e1010041. [Google Scholar] [CrossRef]
- Brody, Y.; Neufeld, N.; Bieberstein, N.; Causse, S.Z.; Böhnlein, E.M.; Neugebauer, K.M.; Darzacq, X.; Shav-Tal, Y. The in vivo kinetics of RNA polymerase II elongation during co-transcriptional splicing. PLoS Biol. 2011, 9, e1000573. [Google Scholar] [CrossRef]
- Muniz, L.; Nicolas, E.; Trouche, D. RNA polymerase II speed: A key player in controlling and adapting transcriptome composition. EMBO J. 2021, 40, e105740. [Google Scholar] [CrossRef]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef]
- Schor, I.E.; Rascovan, N.; Pelisch, F.; Alló, M.; Kornblihtt, A.R. Neuronal cell depolarization induces intragenic chromatin modifications affecting NCAM alternative splicing. Proc. Natl. Acad. Sci. USA 2009, 106, 4325–4330. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wu, Z.; Duan, H.C.; Fang, X.; Jia, G.; Dean, C. R-loop resolution promotes co-transcriptional chromatin silencing. Nat. Commun. 2021, 12, 1790. [Google Scholar] [CrossRef]
- Xu, W.; Xu, H.; Li, K.; Fan, Y.; Liu, Y.; Yang, X.; Sun, Q. The R-loop is a common chromatin feature of the Arabidopsis genome. Nat. Plants 2017, 3, 704–714. [Google Scholar] [CrossRef]
- Lahmy, S.; Pontier, D.; Bies-Etheve, N.; Laudié, M.; Feng, S.; Jobet, E.; Hale, C.J.; Cooke, R.; Hakimi, M.A.; Angelov, D.; et al. Evidence for ARGONAUTE4-DNA interactions in RNA-directed DNA methylation in plants. Genes Dev. 2016, 30, 2565–2570. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, T.; Pérez-Pérez, J.M.; Ruperti, B.; Aleksandrova, T.; Palme, K. A New In Vitro Growth System for Phenotypic Characterization and Seed Propagation of Arabidopsis thaliana. J. Plant Growth Regul. 2024, 43, 652–658. [Google Scholar] [CrossRef]
- Pucker, B.; Kleinbölting, N.; Weisshaar, B. Large scale genomic rearrangements in selected Arabidopsis thaliana T-DNA lines are caused by T-DNA insertion mutagenesis. BMC Genom. 2021, 22, 599. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.Y.; Fan, W.; Cho, J. Determination of TE Insertion Positions Using Transposon Display. In Plant Transposable Elements: Methods and Protocols; Cho, J., Ed.; Springer: New York, NY, USA, 2021; pp. 115–121. [Google Scholar]
- Cheng, C.-Y.; Krishnakumar, V.; Chan, A.P.; Thibaud-Nissen, F.; Schobel, S.; Town, C.D. Araport11: A complete reannotation of the Arabidopsis thaliana reference genome. Plant J. 2017, 89, 789–804. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Diesh, C.; Stevens, G.J.; Xie, P.; De Jesus Martinez, T.; Hershberg, E.A.; Leung, A.; Guo, E.; Dider, S.; Zhang, J.; Bridge, C.; et al. JBrowse 2: A modular genome browser with views of synteny and structural variation. Genome Biol. 2023, 24, 74. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Ellinghaus, D.; Kurtz, S.; Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinform. 2008, 9, 18. [Google Scholar] [CrossRef]
- Novák, P.; Hoštáková, N.; Neumann, P.; Macas, J. DANTE and DANTE_LTR: Lineage-centric annotation pipelines for long terminal repeat retrotransposons in plant genomes. NAR Genom. Bioinform. 2024, 6, lqae113. [Google Scholar] [CrossRef]
- Underwood, C.J.; Choi, K.; Lambing, C.; Zhao, X.; Serra, H.; Borges, F.; Simorowski, J.; Ernst, E.; Jacob, Y.; Henderson, I.R.; et al. Epigenetic activation of meiotic recombination near Arabidopsis thaliana centromeres via loss of H3K9me2 and non-CG DNA methylation. Genome Res. 2018, 28, 519–531. [Google Scholar] [CrossRef]
- Anand, L.; Rodriguez Lopez, C.M. ChromoMap: An R package for interactive visualization of multi-omics data and annotation of chromosomes. BMC Bioinform. 2022, 23, 33. [Google Scholar] [CrossRef] [PubMed]
- Ge, S.X.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2019, 36, 2628–2629. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef]
- Reddy, A.S.N.; Marquez, Y.; Kalyna, M.; Barta, A. Complexity of the Alternative Splicing Landscape in Plants. Plant Cell 2013, 25, 3657–3683. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2013, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2. WIREs Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Cheetham, S.W.; Kindlova, M.; Ewing, A.D. Methylartist: Tools for visualizing modified bases from nanopore sequence data. Bioinformatics 2022, 38, 3109–3112. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M2 Plant | New ONSEN Insertions | Intragenic Insertions, % | Homozygous Insertions | Heterozygous Insertions |
|---|---|---|---|---|
| #1-1 | 5 | 80.0 | 5 | 0 |
| #3-6 | 2 | 100 | 0 | 2 |
| #7-5 | 3 | 66.6 | 1 | 2 |
| #12-1 | 32 | 90.6 | 18 | 14 |
| #13-1 | 8 | 100.0 | 3 | 5 |
| #14-2 | 6 | 100.0 | 2 | 4 |
| Total | 57 | 89.6 | 30 | 27 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Merkulov, P.; Latypova, A.; Tiurin, K.; Serganova, M.; Kirov, I. DNA Methylation and Alternative Splicing Safeguard Genome and Transcriptome After a Retrotransposition Burst in Arabidopsis thaliana. Int. J. Mol. Sci. 2025, 26, 4816. https://doi.org/10.3390/ijms26104816
Merkulov P, Latypova A, Tiurin K, Serganova M, Kirov I. DNA Methylation and Alternative Splicing Safeguard Genome and Transcriptome After a Retrotransposition Burst in Arabidopsis thaliana. International Journal of Molecular Sciences. 2025; 26(10):4816. https://doi.org/10.3390/ijms26104816
Chicago/Turabian StyleMerkulov, Pavel, Anastasiia Latypova, Kirill Tiurin, Melania Serganova, and Ilya Kirov. 2025. "DNA Methylation and Alternative Splicing Safeguard Genome and Transcriptome After a Retrotransposition Burst in Arabidopsis thaliana" International Journal of Molecular Sciences 26, no. 10: 4816. https://doi.org/10.3390/ijms26104816
APA StyleMerkulov, P., Latypova, A., Tiurin, K., Serganova, M., & Kirov, I. (2025). DNA Methylation and Alternative Splicing Safeguard Genome and Transcriptome After a Retrotransposition Burst in Arabidopsis thaliana. International Journal of Molecular Sciences, 26(10), 4816. https://doi.org/10.3390/ijms26104816