
MixOmics Integration of Biological Datasets Identifies Highly Correlated Variables of COVID-19 Severity

Abstract
1. Introduction
2. Results
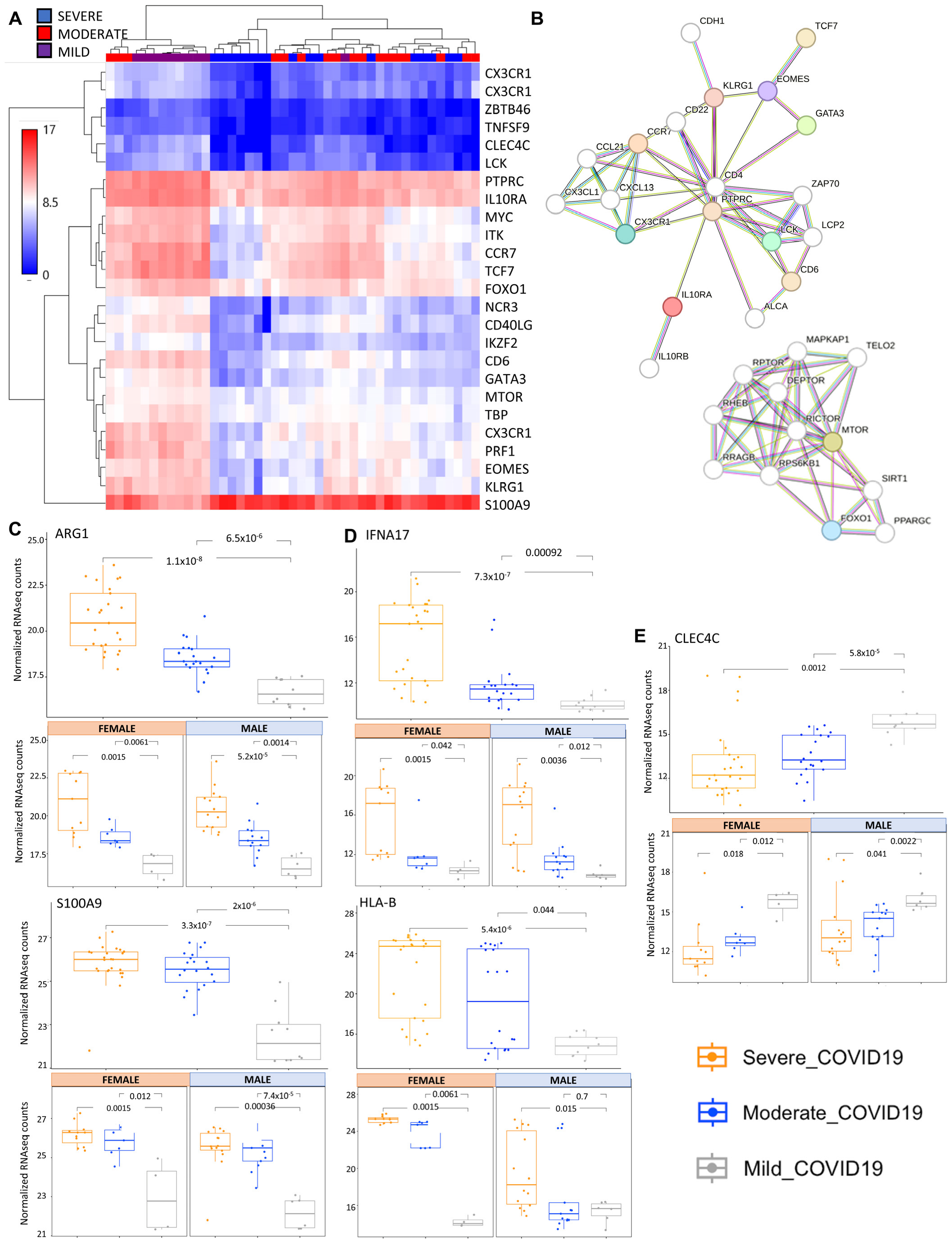
2.1. Transcriptomic Analysis of COVID-19 Severity
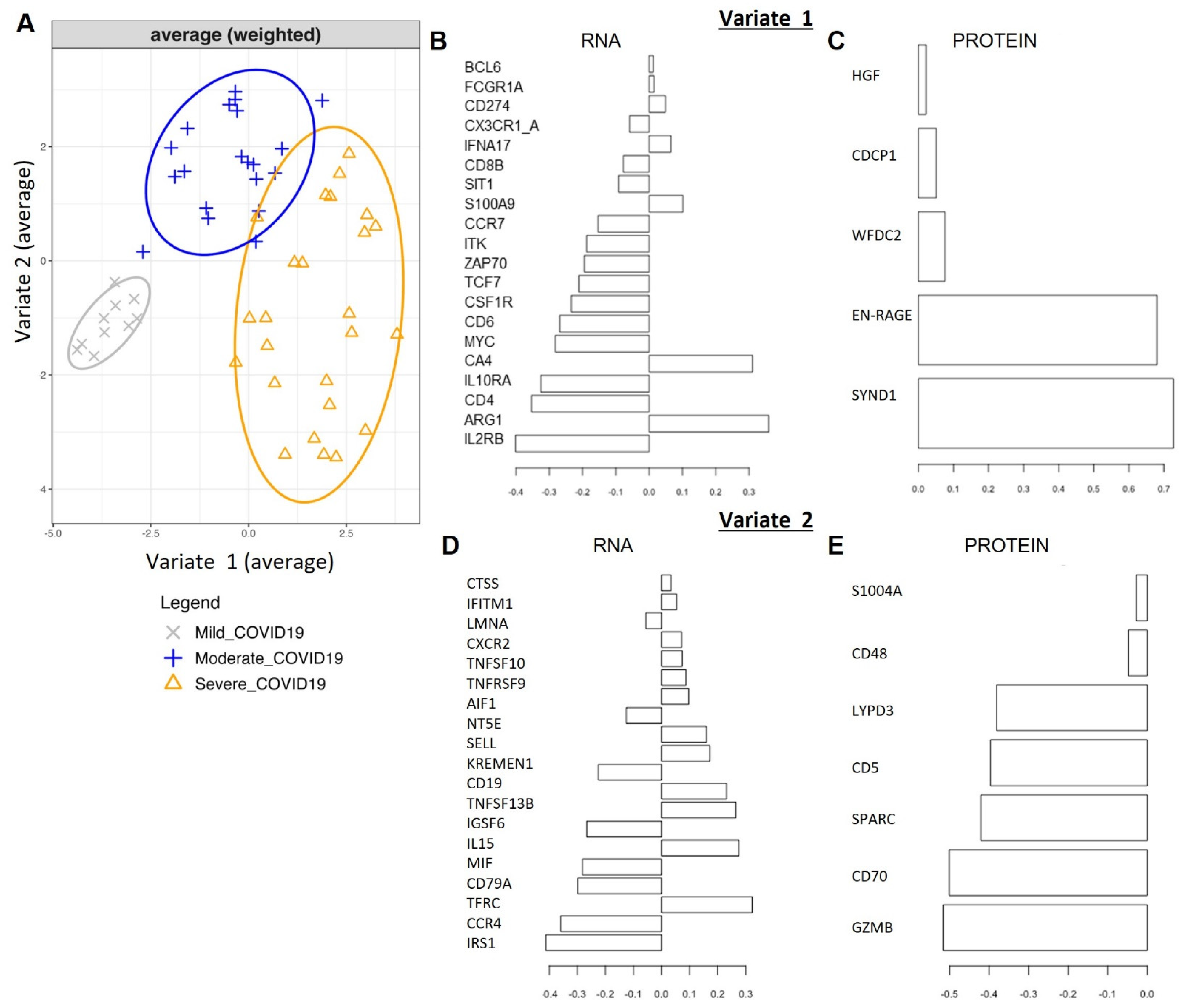
2.2. Single ‘Omics’ Modeling with Sparse PLS-Discriminant Analysis
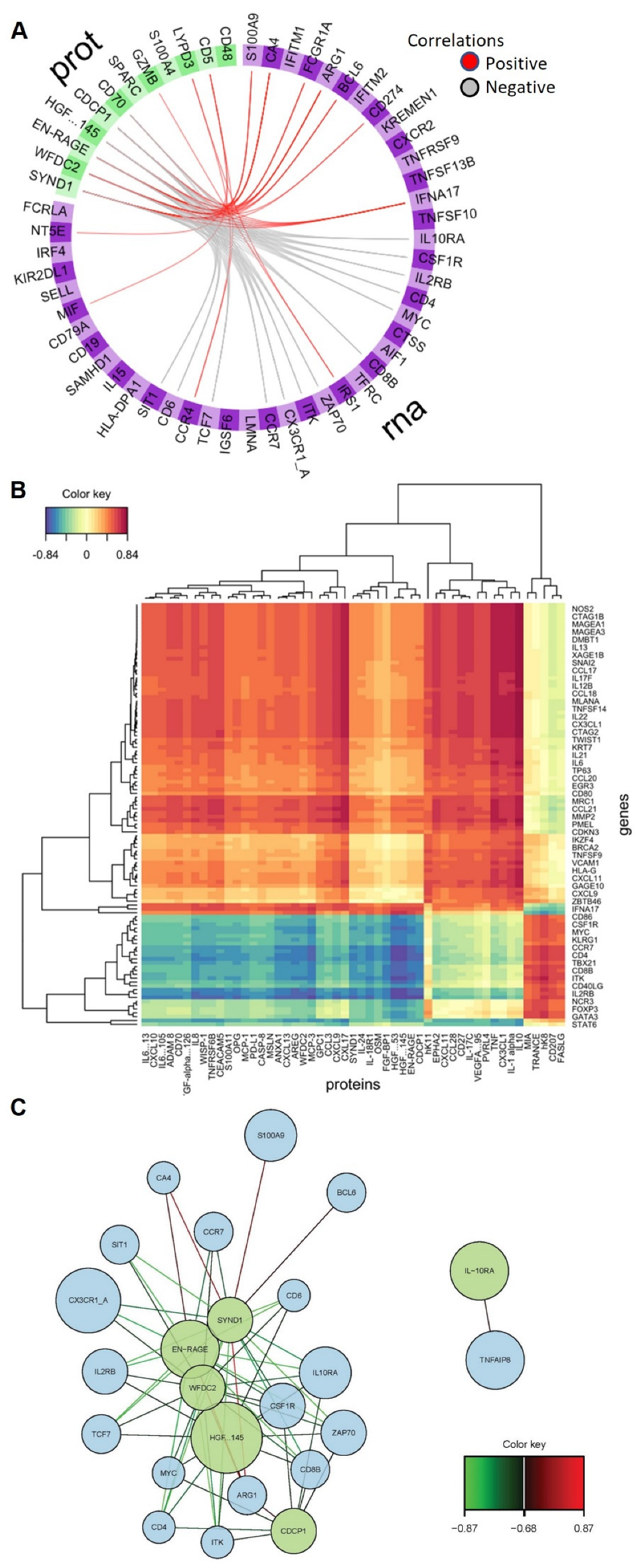
2.3. Omics’ Integrative Modeling with DIABLO
2.4. GO Term and Pathway Analysis of Selected Biomarkers of COVID-19 Severity
3. Discussion
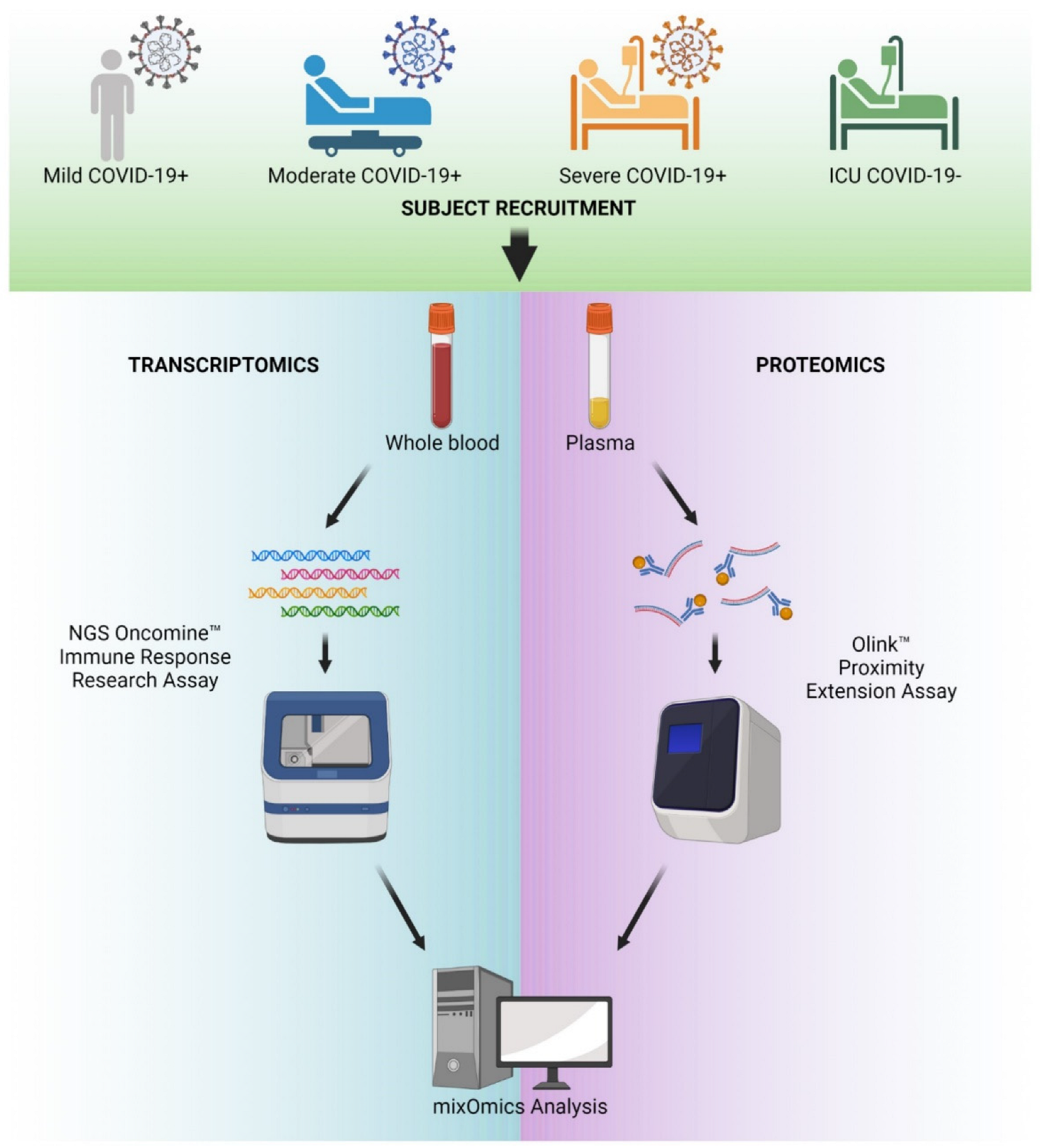
4. Materials and Methods
4.1. Patient Recruitment and Sample Collection
4.2. Proteomics
4.3. RNA Extraction and Quantitation
4.4. Library Preparation and Next-Generation Sequencing
4.5. NGS Analysis Pipeline and QC
4.6. Single-Omics Data Analysis
4.7. Dataset Preparation for MixOmics Analysis
4.8. Sparse PLS Modeling of RNA-Seq Data Alone
4.9. MixOmics Multi-Omics Data Integration
4.10. Gene Ontology and Reactome Pathway Analysis
4.11. Study Approval
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BER | Balanced Error Rate |
| COVID-19 | Coronavirus disease 2019 |
| DIABLO | Data Integration Analysis for Biomarker Discovery using Latent components |
| DNA | Deoxyribonucleic acid |
| FDR | False Discovery Rate |
| ICU | Intensive Care Unit |
| ISP | Ion sphere particle |
| LOD | Limit of detection |
| NPX | Normalized protein expression |
| PEA | Proximity extension assay |
| RNA | Ribonucleic acid |
| ROC | Receiver operating characteristic |
| SARS-CoV-2 | Severe acute respiratory distress syndrome—coronavirus 2 |
References
- Houston, H.; Hakki, S.; Pillay, T.D.; Madon, K.; Derqui-Fernandez, N.; Koycheva, A.; Singanayagam, A.; Fenn, J.; Kundu, R.; Conibear, E.; et al. Broadening symptom criteria improves early case identification in SARS-CoV-2 contacts. Eur. Respir. J. 2022, 60, 2102308. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Moeller, A.L.; Mills, E.H.A.; Collatz Christensen, H.; Gnesin, F.; Blomberg, S.; Zylyftari, N.; Jensen, B.; Ringgren, K.B.; Broccia, M.D.; Boggild, H.; et al. Symptom presentation of SARS-CoV-2-positive and negative patients: A nested case-control study among patients calling the emergency medical service and medical helpline. BMJ Open 2021, 11, e044208. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Elliott, J.; Whitaker, M.; Bodinier, B.; Eales, O.; Riley, S.; Ward, H.; Cooke, G.; Darzi, A.; Chadeau-Hyam, M.; Elliott, P. Predictive symptoms for COVID-19 in the community: REACT-1 study of over 1 million people. PLoS Med. 2021, 18, e1003777. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mizrahi, B.; Shilo, S.; Rossman, H.; Kalkstein, N.; Marcus, K.; Barer, Y.; Keshet, A.; Shamir-Stein, N.; Shalev, V.; Zohar, A.E.; et al. Longitudinal symptom dynamics of COVID-19 infection. Nat. Commun. 2020, 11, 6208. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Song, W.J.; Hui, C.K.M.; Hull, J.H.; Birring, S.S.; McGarvey, L.; Mazzone, S.B.; Chung, K.F. Confronting COVID-19-associated cough and the post-COVID syndrome: Role of viral neurotropism, neuroinflammation, and neuroimmune responses. Lancet Respir. Med. 2021, 9, 533–544. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mayne, E.S.; George, J.A.; Louw, S. Assessing Biomarkers in Viral Infection. Adv. Exp. Med. Biol. 2023, 1412, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Wanhella, K.J.; Fernandez-Patron, C. Biomarkers of ageing and frailty may predict COVID-19 severity. Ageing Res. Rev. 2022, 73, 101513. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, B.B.; Huang, S.J.; Fu, Y.L.; Li, Z.L.; Wang, J.; Wang, J.L. Laboratory Biomarkers for the Diagnosis and Management of Patients with COVID-19: An Updated Review. Discov. Med. 2021, 31, 61–68. [Google Scholar] [PubMed]
- Al-Mterin, M.A.; Alsalman, A.; Elkord, E. Inhibitory Immune Checkpoint Receptors and Ligands as Prognostic Biomarkers in COVID-19 Patients. Front. Immunol. 2022, 13, 870283. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Battaglini, D.; Lopes-Pacheco, M.; Castro-Faria-Neto, H.C.; Pelosi, P.; Rocco, P.R.M. Laboratory Biomarkers for Diagnosis and Prognosis in COVID-19. Front. Immunol. 2022, 13, 857573. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Karimi, A.; Shobeiri, P.; Kulasinghe, A.; Rezaei, N. Novel Systemic Inflammation Markers to Predict COVID-19 Prognosis. Front. Immunol. 2021, 12, 741061. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Varikasuvu, S.R.; Varshney, S.; Dutt, N.; Munikumar, M.; Asfahan, S.; Kulkarni, P.P.; Gupta, P. D-dimer, disease severity, and deaths (3D-study) in patients with COVID-19: A systematic review and meta-analysis of 100 studies. Sci. Rep. 2021, 11, 21888. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tang, Z.; Fan, W.; Li, Q.; Wang, D.; Wen, M.; Wang, J.; Li, X.; Zhou, Y. MVIP: Multi-omics portal of viral infection. Nucleic Acids Res. 2022, 50, D817–D827. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Neogi, U.; Elaldi, N.; Appelberg, S.; Ambikan, A.; Kennedy, E.; Dowall, S.; Bagci, B.K.; Gupta, S.; Rodriguez, J.E.; Svensson-Akusjärvi, S.; et al. Multi-omics insights into host-viral response and pathogenesis in Crimean-Congo hemorrhagic fever viruses for novel therapeutic target. Elife 2022, 11, e76071. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Amemiya, T.; Horimoto, K.; Fukui, K. Application of multiple omics and network projection analyses to drug repositioning for pathogenic mosquito-borne viruses. Sci. Rep. 2021, 11, 10136. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, D.; Kumar, V.; Burnham, K.L.; Mentzer, A.J.; Marsden, B.D.; Knight, J.C. COMBATdb: A database for the COVID-19 Multi-Omics Blood ATlas. Nucleic Acids Res. 2023, 51, D896–D905. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lu, T.; Wang, Y.; Guo, T. Multi-omics in COVID-19: Seeing the unseen but overlooked in the clinic. Cell Rep. Med. 2022, 3, 100580. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lipman, D.; Safo, S.E.; Chekouo, T. Multi-omic analysis reveals enriched pathways associated with COVID-19 and COVID-19 severity. PLoS ONE 2022, 17, e0267047. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Z.N.; Yang, X.S.; Sun, J.; Zhao, J.C.; Zhong, N.S.; Tang, X.X. Multi-omics evaluation of SARS-CoV-2 infected mouse lungs reveals dynamics of host responses. iScience 2022, 25, 103967. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Unterman, A.; Sumida, T.S.; Nouri, N.; Yan, X.; Zhao, A.Y.; Gasque, V.; Schupp, J.C.; Asashima, H.; Liu, Y.; Cosme, C.; et al. Single-cell multi-omics reveals dyssynchrony of the innate and adaptive immune system in progressive COVID-19. Nat. Commun. 2022, 13, 440. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, S.; Yao, X.; Ma, S.; Ping, Y.; Fan, Y.; Sun, S.; He, Z.; Shi, Y.; Sun, L.; Xiao, S.; et al. A single-cell transcriptomic landscape of the lungs of patients with COVID-19. Nat. Cell Biol. 2021, 23, 1314–1328. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hernandez Cordero, A.I.; Li, X.; Milne, S.; Yang, C.X.; Bosse, Y.; Joubert, P.; Timens, W.; van den Berge, M.; Nickle, D.; Hao, K.; et al. Multi-omics highlights ABO plasma protein as a causal risk factor for COVID-19. Hum. Genet. 2021, 140, 969–979. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bernardes, J.P.; Mishra, N.; Tran, F.; Bahmer, T.; Best, L.; Blase, J.I.; Bordoni, D.; Franzenburg, J.; Geisen, U.; Josephs-Spaulding, J.; et al. Longitudinal Multi-omics Analyses Identify Responses of Megakaryocytes, Erythroid Cells, and Plasmablasts as Hallmarks of Severe COVID-19. Immunity 2020, 53, 1296–1314. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Su, Y.; Chen, D.; Yuan, D.; Lausted, C.; Choi, J.; Dai, C.L.; Voillet, V.; Duvvuri, V.R.; Scherler, K.; Troisch, P.; et al. Multi-Omics Resolves a Sharp Disease-State Shift between Mild and Moderate COVID-19. Cell 2020, 183, 1479–1495. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Barh, D.; Tiwari, S.; Weener, M.E.; Azevedo, V.; Goes-Neto, A.; Gromiha, M.M.; Ghosh, P. Multi-omics-based identification of SARS-CoV-2 infection biology and candidate drugs against COVID-19. Comput. Biol. Med. 2020, 126, 104051. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Overmyer, K.A.; Shishkova, E.; Miller, I.J.; Balnis, J.; Bernstein, M.N.; Peters-Clarke, T.M.; Meyer, J.G.; Quan, Q.; Muehlbauer, L.K.; Trujillo, E.A.; et al. Large-Scale Multi-omic Analysis of COVID-19 Severity. Cell Syst. 2021, 12, 23–40. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Le Cao, K.A. DIABLO: An integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sameh, M.; Khalaf, H.M.; Anwar, A.M.; Osama, A.; Ahmed, E.A.; Mahgoub, S.; Ezzeldin, S.; Tanios, A.; Alfishawy, M.; Said, A.F.; et al. Integrated multiomics analysis to infer COVID-19 biological insights. Sci. Rep. 2023, 13, 1802. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Harriott, N.C.; Ryan, A.L. Proteomic profiling identifies biomarkers of COVID-19 severity. Heliyon. 2023, 10, e23320. [Google Scholar] [CrossRef]
- Seki, K.; Sueyoshi, K.; Miyoshi, Y.; Nakamura, Y.; Ishihara, T.; Kondo, Y.; Kuroda, Y.; Yonekura, A.; Iwabuchi, K.; Okamoto, K.; et al. Complement activation and lung injury in Japanese patients with COVID-19: A prospective observational study. Sci. Rep. 2024, 14, 24895. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, Q.; Ye, Z.; Bignotti, A.; Zheng, X.L. Longitudinal Assessment of Plasma Syndecan-1 Predicts 60-Day Mortality in Patients with COVID-19. J. Clin. Med. 2023, 12, 552. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Goonewardena, S.N.; Grushko, O.G.; Wells, J.; Herty, L.; Rosenson, R.S.; Haus, J.M.; Hummel, S.L. Immune-Mediated Glycocalyx Remodeling in Hospitalized COVID-19 Patients. Cardiovasc. Drugs Ther. 2023, 37, 307–313. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rajan, R.; Hanifah, M.; Mariappan, V.; Anand, M.; Balakrishna Pillai, A. Soluble Endoglin and Syndecan-1 levels predicts the clinical outcome in COVID-19 patients. Microb. Pathog. 2024, 188, 106558. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Koyama, D.; Hamazaki, Y.; Kamiyama, T.; Yamada, S.; Furukawa, M.; Tanino, Y.; Shibata, Y.; Ikezoe, T. Syndecan-1 as a prognostic biomarker in COVID-19 patients: A retrospective study of a Japanese cohort. Thromb. J. 2024, 22, 52. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mohebbi, A.; Haybar, H.; Nakhaei Moghaddam, F.; Rasti, Z.; Vahid, M.A.; Saki, N. Biomarkers of endothelial dysfunction are associated with poor outcome in COVID-19 patients: A systematic review and meta-analysis. Rev. Med. Virol. 2023, 33, e2442. [Google Scholar] [CrossRef] [PubMed]
- Ghondaghsaz, E.; Khalaji, A.; Norouzi, M.; Fraser, D.D.; Alilou, S.; Behnoush, A.H. The utility of syndecan-1 circulating levels as a biomarker in patients with previous or active COVID-19: A systematic review and meta-analysis. BMC Infect. Dis. 2023, 23, 510. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Munguia, L.; Najera, N.; Martinez, F.J.; Diaz-Chiguer, D.; Jimenez-Ponce, F.; Ortiz-Flores, M.; Villarreal, F.; Ceballos, G. Correlation of Biomarkers of Endothelial Injury and Inflammation to Outcome in Hospitalized COVID-19 Patients. J. Clin. Med. 2022, 11, 7436. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Karampoor, S.; Zahednasab, H.; Farahmand, M.; Mirzaei, R.; Zamani, F.; Tabibzadeh, A.; Bouzari, B.; Ajdarkosh, H.; Nikkhah, M.; Hashemi, M.R.; et al. A possible pathogenic role of Syndecan-1 in the pathogenesis of coronavirus disease 2019 (COVID-19). Int. Immunopharmacol. 2021, 97, 107684. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ebihara, T.; Matsubara, T.; Togami, Y.; Matsumoto, H.; Tachino, J.; Matsuura, H.; Kojima, T.; Sugihara, F.; Seno, S.; Okuzaki, D.; et al. Combination of WFDC2, CHI3L1, and KRT19 in Plasma Defines a Clinically Useful Molecular Phenotype Associated with Prognosis in Critically Ill COVID-19 Patients. J. Clin. Immunol. 2023, 43, 286–298. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, Z.; Zeng, X.; Liao, J.; Dong, X.; Deng, Y.; Wang, Y.; Zhou, M. Immune Characteristic Genes and Neutrophil Immune Transformation Studies in Severe COVID-19. Microorganisms 2024, 12, 737. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sganzerla Martinez, G.; Garduno, A.; Toloue Ostadgavahi, A.; Hewins, B.; Dutt, M.; Kumar, A.; Martin-Loeches, I.; Kelvin, D.J. Identification of Marker Genes in Infectious Diseases from ScRNA-seq Data Using Interpretable Machine Learning. Int. J. Mol. Sci. 2024, 25, 5920. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lei, H. A single transcript for the prognosis of disease severity in COVID-19 patients. Sci. Rep. 2021, 11, 12174. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mester, P.; Keller, D.; Kunst, C.; Rath, U.; Rusch, S.; Schmid, S.; Krautbauer, S.; Muller, M.; Buechler, C.; Pavel, V. High Serum S100A12 as a Diagnostic and Prognostic Biomarker for Severity, Multidrug-Resistant Bacteria Superinfection and Herpes Simplex Virus Reactivation in COVID-19. Viruses 2024, 16, 1084. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ma, Y.; Qiu, F.; Deng, C.; Li, J.; Huang, Y.; Wu, Z.; Zhou, Y.; Zhang, Y.; Xiong, Y.; Yao, Y.; et al. Integrating single-cell sequencing data with GWAS summary statistics reveals CD16+monocytes and memory CD8+T cells involved in severe COVID-19. Genome Med. 2022, 14, 16. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Boucher, J.; Gilbert, C.; Bose, S.; Tessier, P.A. S100A9: The Unusual Suspect Connecting Viral Infection and Inflammation. J. Immunol. 2024, 212, 1523–1529. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gupta, A.; Al-Tamimi, A.O.; Halwani, R.; Alsaidi, H.; Kannan, M.; Ahmad, F. Lipocalin-2, S100A8/A9, and cystatin C: Potential predictive biomarkers of cardiovascular complications in COVID-19. Exp. Biol. Med. 2022, 247, 1205–1213. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, Z.; Wang, Y.; Yan, Q.; Cai, C.; Feng, Y.; Huang, Q.; Li, T.; Yuan, S.; Huang, J.; Luo, Z.H.; et al. FPR1 signaling aberrantly regulates S100A8/A9 production by CD14(+)FCN1(hi) macrophages and aggravates pulmonary pathology in severe COVID-19. Commun. Biol. 2024, 7, 1321. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hudak, A.; Letoha, A.; Szilak, L.; Letoha, T. Contribution of Syndecans to the Cellular Entry of SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 5336. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Singh, P.; Ali, S.A. Multifunctional Role of S100 Protein Family in the Immune System: An Update. Cells 2022, 11, 2274. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tserel, L.; Jogi, P.; Naaber, P.; Maslovskaja, J.; Haling, A.; Salumets, A.; Zusinaite, E.; Soeorg, H.; Lattekivi, F.; Ingerainen, D.; et al. Long-Term Elevated Inflammatory Protein Levels in Asymptomatic SARS-CoV-2 Infected Individuals. Front. Immunol. 2021, 12, 709759. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Perreau, M.; Suffiotti, M.; Marques-Vidal, P.; Wiedemann, A.; Levy, Y.; Laouenan, C.; Ghosn, J.; Fenwick, C.; Comte, D.; Roger, T.; et al. The cytokines HGF and CXCL13 predict the severity and the mortality in COVID-19 patients. Nat. Commun. 2021, 12, 4888. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guo, J.; Wang, S.; Xia, H.; Shi, D.; Chen, Y.; Zheng, S.; Chen, Y.; Gao, H.; Guo, F.; Ji, Z.; et al. Cytokine Signature Associated with Disease Severity in COVID-19. Front. Immunol. 2021, 12, 681516. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gorgojo-Galindo, O.; Martin-Fernandez, M.; Penarrubia-Ponce, M.J.; Alvarez, F.J.; Ortega-Loubon, C.; Gonzalo-Benito, H.; Martinez-Paz, P.; Miramontes-Gonzalez, J.P.; Gomez-Sanchez, E.; Poves-Alvarez, R.; et al. Predictive Modeling of Poor Outcome in Severe COVID-19: A Single-Center Observational Study Based on Clinical, Cytokine and Laboratory Profiles. J. Clin. Med. 2021, 10, 5431. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blanco, J.R.; Cobos-Ceballos, M.J.; Navarro, F.; Sanjoaquin, I.; Arminanzas, C.; Bernal, E.; Buzon-Martin, L.; Viribay, M.; Perez-Martinez, L.; Espejo-Perez, S.; et al. Elevated levels of serum CDCP1 in individuals recovering from severe COVID-19 disease. Aging 2022, 14, 1597–1610. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nienhold, R.; Ciani, Y.; Koelzer, V.H.; Tzankov, A.; Haslbauer, J.D.; Menter, T.; Schwab, N.; Henkel, M.; Frank, A.; Zsikla, V.; et al. Two distinct immunopathological profiles in autopsy lungs of COVID-19. Nat. Commun. 2020, 11, 5086. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lopez-Martinez, C.; Martin-Vicente, P.; Gomez de Ona, J.; Lopez-Alonso, I.; Gil-Pena, H.; Cuesta-Llavona, E.; Fernandez-Rodriguez, M.; Crespo, I.; Salgado Del Riego, E.; Rodriguez-Garcia, R.; et al. Transcriptomic clustering of critically ill COVID-19 patients. Eur. Respir. J. 2023, 61, 2200592. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Derakhshani, A.; Hemmat, N.; Asadzadeh, Z.; Ghaseminia, M.; Shadbad, M.A.; Jadideslam, G.; Silvestris, N.; Racanelli, V.; Baradaran, B. Arginase 1 (Arg1) as an Up-Regulated Gene in COVID-19 Patients: A Promising Marker in COVID-19 Immunopathy. J. Clin. Med. 2021, 10, 1051. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dean, M.J.; Ochoa, J.B.; Sanchez-Pino, M.D.; Zabaleta, J.; Garai, J.; Del Valle, L.; Wyczechowska, D.; Baiamonte, L.B.; Philbrook, P.; Majumder, R.; et al. Severe COVID-19 Is Characterized by an Impaired Type I Interferon Response and Elevated Levels of Arginase Producing Granulocytic Myeloid Derived Suppressor Cells. Front. Immunol. 2021, 12, 695972. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fakhkhari, M.; Caidi, H.; Sadki, K. HLA alleles associated with COVID-19 susceptibility and severity in different populations: A systematic review. Egypt. J. Med. Hum. Genet. 2023, 24, 10. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Migliorini, F.; Torsiello, E.; Spiezia, F.; Oliva, F.; Tingart, M.; Maffulli, N. Association between HLA genotypes and COVID-19 susceptibility, severity and progression: A comprehensive review of the literature. Eur. J. Med. Res. 2021, 26, 84. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liu, T.; Balzano-Nogueira, L.; Lleo, A.; Conesa, A. Transcriptional Differences for COVID-19 Disease Map Genes between Males and Females Indicate a Different Basal Immunophenotype Relevant to the Disease. Genes 2020, 11, 1447. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- COVID-19 Multi-omics Blood ATlas (COMBAT) Consortium. A blood atlas of COVID-19 defines hallmarks of disease severity and specificity. Cell 2022, 185, 916–938. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Donlan, A.N.; Sutherland, T.E.; Marie, C.; Preissner, S.; Bradley, B.T.; Carpenter, R.M.; Sturek, J.M.; Ma, J.Z.; Moreau, G.B.; Donowitz, J.R.; et al. IL-13 is a driver of COVID-19 severity. JCI Insight 2021, 6, e150107. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Liang, J.; Jiang, D.; Jung, Y.; Xie, T.; Ingram, J.; Church, T.; Degan, S.; Leonard, M.; Kraft, M.; Noble, P.W. Role of hyaluronan and hyaluronan-binding proteins in human asthma. J. Allergy Clin. Immunol. 2011, 128, 403–411. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- R Bonam, S.; Hu, H.; Bayry, J. Role of the PD-1 and PD-L1 axis in COVID-19. Future Microbiol. 2022, 17, 985–988. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aghbash, P.S.; Eslami, N.; Shamekh, A.; Entezari-Maleki, T.; Baghi, H.B. SARS-CoV-2 infection: The role of PD-1/PD-L1 and CTLA-4 axis. Life Sci. 2021, 270, 119124. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Duhalde Vega, M.; Olivera, D.; Gastao Davanzo, G.; Bertullo, M.; Noya, V.; Fabiano de Souza, G.; Primon Muraro, S.; Castro, I.; Arevalo, A.P.; Crispo, M.; et al. PD-1/PD-L1 blockade abrogates a dysfunctional innate-adaptive immune axis in critical beta-coronavirus disease. Sci. Adv. 2022, 8, eabn6545. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schonrich, G.; Raftery, M.J. The PD-1/PD-L1 Axis and Virus Infections: A Delicate Balance. Front. Cell Infect. Microbiol. 2019, 9, 207. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jubel, J.M.; Barbati, Z.R.; Burger, C.; Wirtz, D.C.; Schildberg, F.A. The Role of PD-1 in Acute and Chronic Infection. Front. Immunol. 2020, 11, 487. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Von Mering, C.; Jensen, L.J.; Snel, B.; Hooper, S.D.; Krupp, M.; Foglierini, M.; Jouffre, N.; Huynen, M.A.; Bork, P. STRING: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005, 33, D433–D437. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Navratil, P.; Sahi, S.; Hruba, P.; Ticha, A.; Timkova, K.; Viklicky, O.; Cerny, V.; Astapenko, D. Syndecan-1 in the Serum of Deceased Kidney Donors as a Potential Biomarker of Kidney Function. Transplant. Proc. 2025, 57, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Okada, H.; Sumi, K.; Tomita, H.; Kobayashi, R.; Ishihara, T.; Kakino, Y.; Suzuki, K.; Yoshiyama, N.; Yasuda, R.; et al. Serum syndecan-1 reflects organ dysfunction in critically ill patients. Sci. Rep. 2021, 11, 8864. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Beltran, J.V.B.; Lin, F.P.; Chang, C.L.; Ko, T.M. Single-Cell Meta-Analysis of Neutrophil Activation in Kawasaki Disease and Multisystem Inflammatory Syndrome in Children Reveals Potential Shared Immunological Drivers. Circulation 2023, 148, 1778–1796. [Google Scholar] [CrossRef] [PubMed]
- Eddins, D.J.; Yang, J.; Kosters, A.; Giacalone, V.D.; Pechuan-Jorge, X.; Chandler, J.D.; Eum, J.; Babcock, B.R.; Dobosh, B.S.; Hernandez, M.R.; et al. Transcriptional reprogramming of infiltrating neutrophils drives lung pathology in severe COVID-19 despite low viral load. Blood Adv. 2023, 7, 778–799. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- De Pasquale, V.; Quiccione, M.S.; Tafuri, S.; Avallone, L.; Pavone, L.M. Heparan Sulfate Proteoglycans in Viral Infection and Treatment: A Special Focus on SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 6574. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Svobodova, B.; Lofdahl, A.; Nybom, A.; Wigen, J.; Hirdman, G.; Olm, F.; Brunnstrom, H.; Lindstedt, S.; Westergren-Thorsson, G.; Elowsson, L. Overlapping Systemic Proteins in COVID-19 and Lung Fibrosis Associated with Tissue Remodeling and Inflammation. Biomedicines 2024, 12, 2893. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gruber, C.N.; Patel, R.S.; Trachtman, R.; Lepow, L.; Amanat, F.; Krammer, F.; Wilson, K.M.; Onel, K.; Geanon, D.; Tuballes, K.; et al. Mapping Systemic Inflammation and Antibody Responses in Multisystem Inflammatory Syndrome in Children (MIS-C). Cell 2020, 183, 982–995. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lun, Y.; Borjini, N.; Miura, N.N.; Ohno, N.; Singer, N.G.; Lin, F. CDCP1 on Dendritic Cells Contributes to the Development of a Model of Kawasaki Disease. J. Immunol. 2021, 206, 2819–2827. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Fan, C.; Wu, Y.; Rui, X.; Yang, Y.; Ling, C.; Liu, S.; Liu, S.; Wang, Y. Animal models for COVID-19: Advances, gaps and perspectives. Signal Transduct. Target. Ther. 2022, 7, 220. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO Biological Process Complete | Homo Sapiens—REFLIST (20,589) | Upload # (110) | Upload (Expected) | Upload (Over/ Under) | Upload (Fold Enriched) | Upload (Raw p-Value) | Upload (FDR) |
|---|---|---|---|---|---|---|---|
| immune system process (GO:0002376) | 2429 | 73 | 12.98 | + | 5.63 | 1.08 × 10−40 | 1.69 × 10−36 |
| regulation of immune system process (GO:0002682) | 1520 | 56 | 8.12 | + | 6.9 | 1.57 × 10−33 | 1.23 × 10−29 |
| immune response (GO:0006955) | 1621 | 55 | 8.66 | + | 6.35 | 4.74 × 10−31 | 2.48 × 10−27 |
| positive regulation of immune system process (GO:0002684) | 967 | 43 | 5.17 | + | 8.32 | 5.24 × 10−28 | 2.05 × 10−24 |
| regulation of lymphocyte activation (GO:0051249) | 591 | 34 | 3.16 | + | 10.77 | 2.86 × 10−25 | 8.98 × 10−22 |
| regulation of T-cell activation (GO:0050863) | 377 | 29 | 2.01 | + | 14.4 | 8.19 × 10−25 | 2.14 × 10−21 |
| regulation of leukocyte activation (GO:0002694) | 684 | 35 | 3.65 | + | 9.58 | 2.00 × 10−24 | 4.47 × 10−21 |
| regulation of cell activation (GO:0050865) | 741 | 35 | 3.96 | + | 8.84 | 2.49 × 10−23 | 4.89 × 10−20 |
| regulation of immune response (GO:0050776) | 935 | 37 | 5 | + | 7.41 | 3.54 × 10−22 | 6.16 × 10−19 |
| leukocyte activation (GO:0045321) | 581 | 31 | 3.1 | + | 9.99 | 4.52 × 10−22 | 7.09 × 10−19 |
| response to stimulus (GO:0050896) | 8209 | 93 | 43.86 | + | 2.12 | 7.35 × 10−22 | 1.05 × 10−18 |
| response to organic substance (GO:0010033) | 2704 | 55 | 14.45 | + | 3.81 | 2.32 × 10−20 | 3.03 × 10−17 |
| cell activation (GO:0001775) | 700 | 31 | 3.74 | + | 8.29 | 8.05 × 10−20 | 8.41 × 10−17 |
| regulation of cell population proliferation (GO:0042127) | 1674 | 44 | 8.94 | + | 4.92 | 7.79 × 10−20 | 8.73 × 10−17 |
| cellular response to stimulus (GO:0051716) | 6569 | 82 | 35.1 | + | 2.34 | 7.34 × 10−20 | 8.86 × 10−17 |
| cell surface receptor signaling pathway (GO:0007166) | 2174 | 49 | 11.61 | + | 4.22 | 1.38 × 10−19 | 1.35 × 10−16 |
| regulation of leukocyte proliferation (GO:0070663) | 271 | 22 | 1.45 | + | 15.19 | 2.36 × 10−19 | 2.18 × 10−16 |
| defense response (GO:0006952) | 1478 | 41 | 7.9 | + | 5.19 | 3.50 × 10−19 | 3.05 × 10−16 |
| regulation of leukocyte cell-cell adhesion (GO:1903037) | 369 | 24 | 1.97 | + | 12.17 | 5.69 × 10−19 | 4.46 × 10−16 |
| lymphocyte activation (GO:0046649) | 465 | 26 | 2.48 | + | 10.47 | 5.54 × 10−19 | 4.57 × 10−16 |
| signal transduction (GO:0007165) | 4887 | 70 | 26.11 | + | 2.68 | 9.53 × 10−19 | 7.12 × 10−16 |
| positive regulation of T-cell activation (GO:0050870) | 253 | 21 | 1.35 | + | 15.54 | 1.10 × 10−18 | 7.84 × 10−16 |
| regulation of response to stimulus (GO:0048583) | 4034 | 63 | 21.55 | + | 2.92 | 4.41 × 10−18 | 3.00 × 10−15 |
| positive regulation of leukocyte cell-cell adhesion (GO:1903039) | 276 | 21 | 1.47 | + | 14.24 | 5.78 × 10−18 | 3.78 × 10−15 |
| positive regulation of leukocyte proliferation (GO:007066) | 168 | 18 | 0.9 | + | 20.05 | 6.15 × 10−18 | 3.86 × 10−15 |
| Pathway Identifier | Pathway Name | #Entities Found | #Entities Total | #Interactors Found | #Interactors Total | Entities Ratio | Entities p-Value | Entities FDR | #Reaction Found | #Reaction Total |
|---|---|---|---|---|---|---|---|---|---|---|
| R-HSA-6785807 | Interleukin-4 and Interleukin-13 signaling | 20 | 211 | 3 | 162 | 0.013845 | 1.37 × 10−10 | 1.76 × 10−7 | 9 | 47 |
| R-HSA-1280215 | Cytokine Signaling in immune system | 65 | 1115 | 50 | 2999 | 0.073162 | 4.59 × 10−8 | 2.95 × 10−5 | 290 | 740 |
| R-HSA-168256 | Immune System | 88 | 2703 | 62 | 4209 | 0.177362 | 3.38 × 10−7 | 1.44 × 10−4 | 530 | 1659 |
| R-HSA-6783783 | Interleukin-10 signaling | 11 | 86 | 2 | 93 | 0.005643 | 6.72 × 10−7 | 2.16 × 10−4 | 13 | 15 |
| R-HSA-380108 | Chemokine receptors bind chemokines | 8 | 57 | 2 | 70 | 0.003740 | 4.74 × 10−6 | 0.001212 | 12 | 19 |
| R-HSA-449147 | Signaling by Interleukins | 43 | 658 | 35 | 2161 | 0.043176 | 5.08 × 10−5 | 0.010863 | 187 | 505 |
| R-HSA-389948 | PD-1 signaling | 5 | 45 | 1 | 4 | 0.002953 | 6.68 × 10−5 | 0.012223 | 4 | 5 |
| R-HSA-202430 | Translocation of ZAP-70 to Immunological synapse | 5 | 42 | 3 | 14 | 0.002756 | 1.18 × 10−4 | 0.018894 | 4 | 4 |
| R-HSA-9012546 | Interleukin-18 signaling | 3 | 11 | 1 | 5 | 7.22 × 10−4 | 2.93 × 10−4 | 0.041670 | 4 | 4 |
| Protein | COVID Status | Sample | Data Summary | Reference |
|---|---|---|---|---|
| Syndecan-1 | Healthy vs. COVID-19 | Serum/Plasma Proteomics | Elevated | [30,31,32] |
| Syndecan-1 | Healthy vs. COVID-19 (moderate and severe) | Plasma Proteomics | Elevated and increasing with severity | [33,34,35,36,37,38] |
| WFDC2 | Healthy vs. COVID-19 (moderate and severe) | Plasma Proteomics | Elevated and increasing with severity | [39] |
| S100A12 | Healthy vs. COVID-19 (moderate and severe) | Peripheral blood mRNA-seq/BAL fluid scRNA-seq | Elevated and increasing with severity | [40,41,42] |
| S100A12 | Healthy vs. COVID-19 (moderate and severe) | Serum/Plasma Proteomics | Elevated and increasing with severity | [43] |
| S100A12 | Healthy vs. COVID-19 (moderate and severe) | GWAS and scRNA-seq | Elevated and increasing with severity | [44] |
| S100A9 | Healthy vs. COVID-19 | PBMC protein | Elevated and increasing with severity | [45,46,47] |
| RNA | Proteomics | |
|---|---|---|
| RNA | 0 | 0.1 |
| Proteomics | 0.1 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harriott, N.C.; Chimenti, M.S.; Bonde, G.; Ryan, A.L. MixOmics Integration of Biological Datasets Identifies Highly Correlated Variables of COVID-19 Severity. Int. J. Mol. Sci. 2025, 26, 4743. https://doi.org/10.3390/ijms26104743
Harriott NC, Chimenti MS, Bonde G, Ryan AL. MixOmics Integration of Biological Datasets Identifies Highly Correlated Variables of COVID-19 Severity. International Journal of Molecular Sciences. 2025; 26(10):4743. https://doi.org/10.3390/ijms26104743
Chicago/Turabian StyleHarriott, Noa C., Michael S. Chimenti, Gregory Bonde, and Amy L. Ryan. 2025. "MixOmics Integration of Biological Datasets Identifies Highly Correlated Variables of COVID-19 Severity" International Journal of Molecular Sciences 26, no. 10: 4743. https://doi.org/10.3390/ijms26104743
APA StyleHarriott, N. C., Chimenti, M. S., Bonde, G., & Ryan, A. L. (2025). MixOmics Integration of Biological Datasets Identifies Highly Correlated Variables of COVID-19 Severity. International Journal of Molecular Sciences, 26(10), 4743. https://doi.org/10.3390/ijms26104743