
Excessive Existence of Positively Charged Amino Acids Caused Off-Target Recognition in the Seed Region of Clostridium butyricum Argonaute



Abstract
1. Introduction
2. Results and Discussion
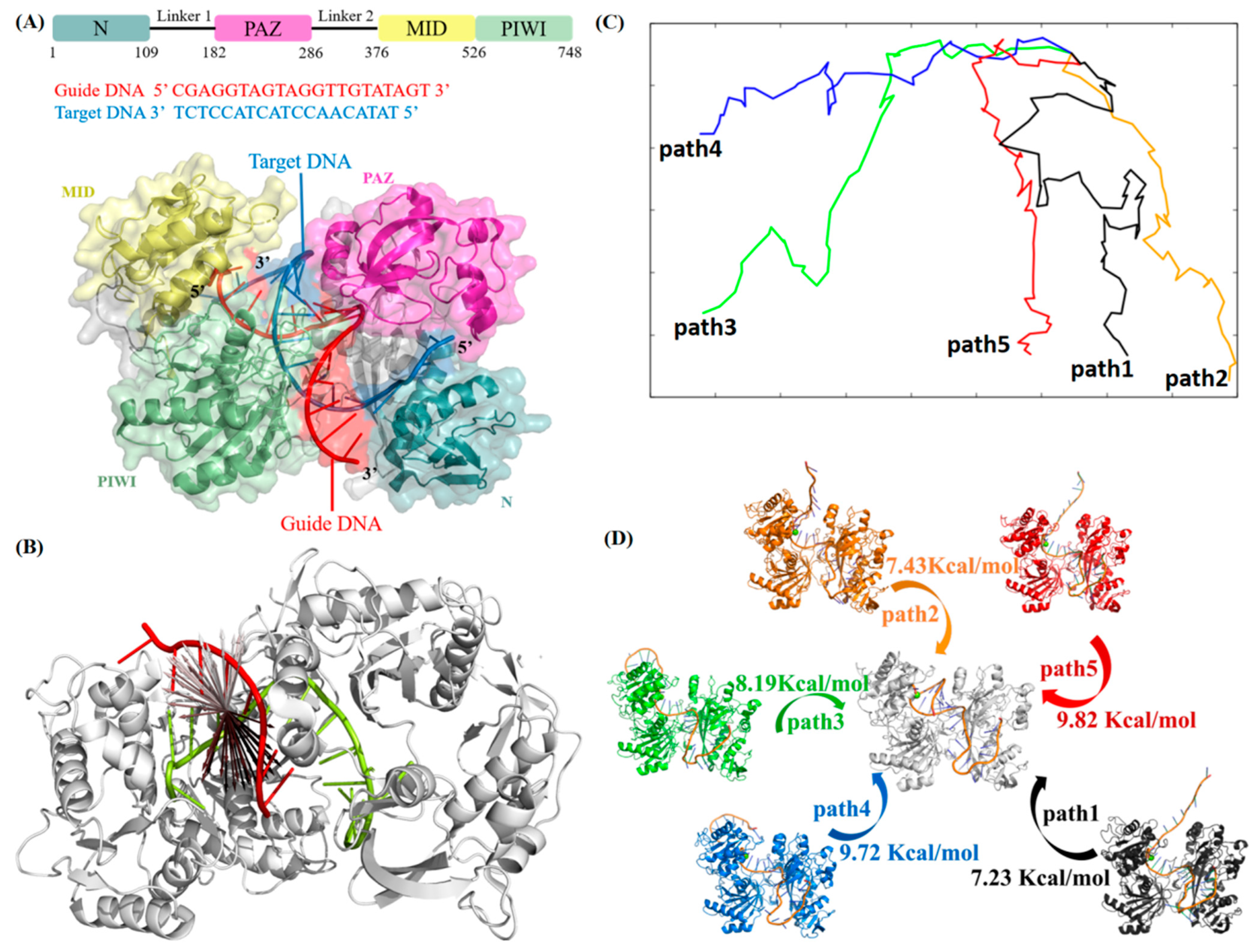
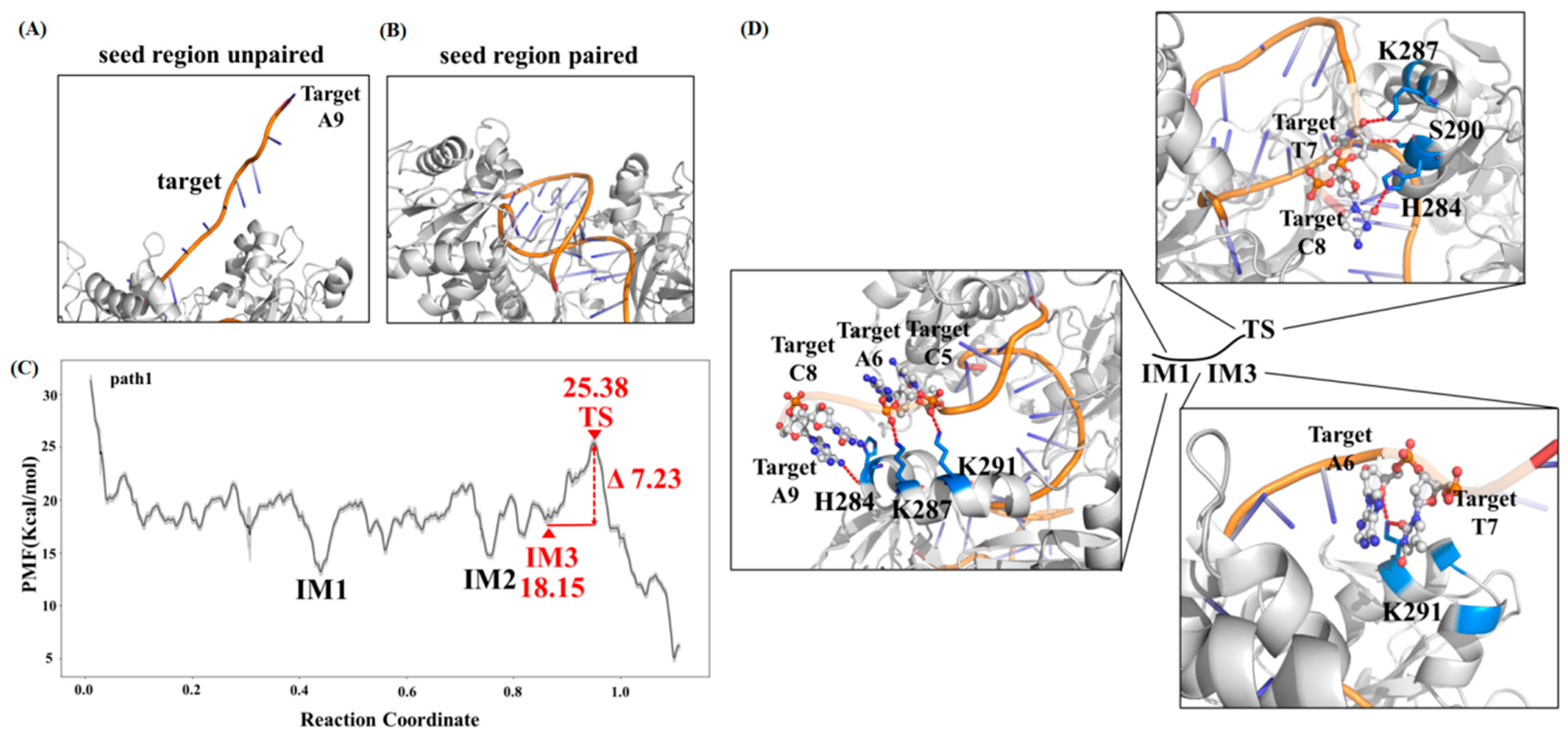
2.1. Target DNA Recognition in the Seed Region of CbAgo
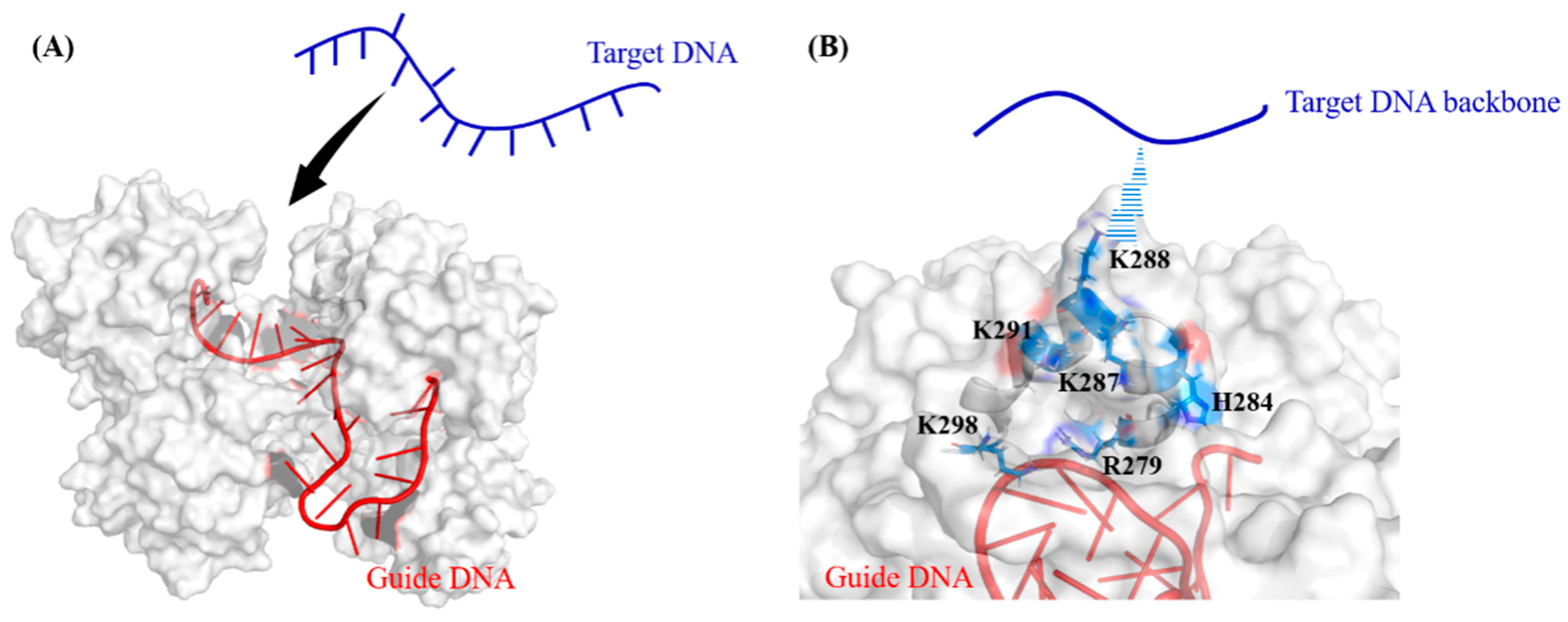
2.2. Simulation Study of a Charge-Attenuated CbAgo Mutant
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hegge, J.W.; Swarts, D.C.; Chandradoss, S.D.; Cui, T.J.; Kneppers, J.; Jinek, M.; Joo, C.; van der Oost, J. DNA-guided DNA cleavage at moderate temperatures by Clostridium butyricum Argonaute. Nucleic Acids Res. 2019, 47, 5809–5821. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Sun, W.; Wang, J.; Sheng, G.; Xiang, G.; Zhang, T.; Shi, W.; Li, C.; Wang, Y.; Zhao, F.; et al. Argonaute proteins from human gastrointestinal bacteria catalyze DNA-guided cleavage of single- and double-stranded DNA at 37 °C. Cell Discov. 2019, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Kuzmenko, A.; Yudin, D.; Ryazansky, S.; Kulbachinskiy, A.; Aravin, A.A. Programmable DNA cleavage by Ago nucleases from mesophilic bacteria Clostridium butyricum and Limnothrix rosea. Nucleic Acids Res. 2019, 47, 5822–5836. [Google Scholar] [CrossRef] [PubMed]
- Vaiskunaite, R.; Vainauskas, J.; Morris, J.J.L.; Potapov, V.; Bitinaite, J. Programmable cleavage of linear double-stranded DNA by combined action of Argonaute CbAgo from Clostridium butyricum and nuclease deficient RecBC helicase from E. coli. Nucleic Acids Res. 2022, 50, 4616–4629. [Google Scholar] [CrossRef]
- Mishra, G.; Levy, Y. Molecular determinants of the interactions between proteins and ssDNA. Proc. Natl. Acad. Sci. USA 2015, 112, 5033–5038. [Google Scholar] [CrossRef]
- Plumridge, A.; Meisburger, S.P.; Andresen, K.; Pollack, L. The impact of base stacking on the conformations and electrostatics of single-stranded DNA. Nucleic Acids Res. 2017, 45, 3932–3943. [Google Scholar] [CrossRef]
- Yin, Y.; Zhao, X.S. Kinetics and Dynamics of DNA Hybridization. Acc. Chem. Res. 2011, 44, 1172–1181. [Google Scholar] [CrossRef]
- Wolff, A.M.; Nango, E.; Young, I.D.; Brewster, A.S.; Kubo, M.; Nomura, T.; Sugahara, M.; Owada, S.; Barad, B.A.; Ito, K.; et al. Mapping protein dynamics at high spatial resolution with temperature-jump X-ray crystallography. Nat. Chem. 2023, 15, 1549–1558. [Google Scholar] [CrossRef]
- Klyshko, E.; Kim, J.S.-H.; McGough, L.; Valeeva, V.; Lee, E.; Ranganathan, R.; Rauscher, S. Functional protein dynamics in a crystal. Nat. Commun. 2024, 15, 3244. [Google Scholar] [CrossRef]
- Kleckner, I.R.; Foster, M.P. An introduction to NMR-based approaches for measuring protein dynamics. Biochim. Biophys. Acta 2011, 1814, 942–968. [Google Scholar] [CrossRef]
- Wayment-Steele, H.K.; Nesr, G.E.; Hettiarachchi, R.; Kariyawasam, H.; Ovchinnikov, S.; Kern, D. Learning millisecond protein dynamics from what is missing in NMR spectra. bioRxiv 2025. [Google Scholar] [CrossRef]
- Markwick, P.R.L.; Andrew McCammon, J. Studying functional dynamics in bio-molecules using accelerated molecular dynamics. Phys. Chem. Chem. Phys. 2011, 13, 20053–20065. [Google Scholar] [CrossRef] [PubMed]
- Adcock, S.A.; McCammon, J.A. Molecular Dynamics: Survey of Methods for Simulating the Activity of Proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, W.; Pang, L.; Zhu, W. Unveiling conformational dynamics changes of H-Ras induced by mutations based on accelerated molecular dynamics. Phys. Chem. Chem. Phys. 2020, 22, 21238–21250. [Google Scholar] [CrossRef]
- Saladino, G.; Gervasio, F.L. Modeling the effect of pathogenic mutations on the conformational landscape of protein kinases. Curr. Opin. Struct. Biol. 2016, 37, 108–114. [Google Scholar] [CrossRef]
- Mahita, J.; Sowdhamini, R. Investigating the effect of key mutations on the conformational dynamics of toll-like receptor dimers through molecular dynamics simulations and protein structure networks. Proteins Struct. Funct. Bioinform. 2018, 86, 475–490. [Google Scholar] [CrossRef]
- Zhu, L.; Sheong, F.K.; Cao, S.; Liu, S.; Unarta, I.C.; Huang, X. TAPS: A traveling-salesman based automated path searching method for functional conformational changes of biological macromolecules. J. Chem. Phys. 2019, 150, 124105. [Google Scholar] [CrossRef]
- Kästner, J. Umbrella sampling. WIREs Comput. Mol. Sci. 2011, 1, 932–942. [Google Scholar] [CrossRef]
- Isralewitz, B.; Gao, M.; Schulten, K. Steered molecular dynamics and mechanical functions of proteins. Curr. Opin. Struct. Biol. 2001, 11, 224–230. [Google Scholar] [CrossRef]
- Douglas Carroll, J.; Arabie, P. Chapter 3—Multidimensional Scaling. In Measurement, Judgment and Decision Making; Birnbaum, M.H., Ed.; Academic Press: San Diego, CA, USA, 1998; pp. 179–250. ISBN 978-0-12-099975-0. [Google Scholar]
- Jolly, S.M.; Gainetdinov, I.; Jouravleva, K.; Zhang, H.; Strittmatter, L.; Bailey, S.M.; Hendricks, G.M.; Dhabaria, A.; Ueberheide, B.; Zamore, P.D. Thermus thermophilus Argonaute Functions in the Completion of DNA Replication. Cell 2020, 182, 1545–1559.e18. [Google Scholar] [CrossRef]
- Swarts, D.C.; Jore, M.M.; Westra, E.R.; Zhu, Y.; Janssen, J.H.; Snijders, A.P.; Wang, Y.; Patel, D.J.; Berenguer, J.; Brouns, S.J.J.; et al. DNA-guided DNA interference by a prokaryotic Argonaute. Nature 2014, 507, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Sheng, G.; Zhao, H.; Wang, J.; Rao, Y.; Tian, W.; Swarts, D.C.; van der Oost, J.; Patel, D.J.; Wang, Y. Structure-based cleavage mechanism of Thermus thermophilus Argonaute DNA guide strand-mediated DNA target cleavage. Proc. Natl. Acad. Sci. USA 2014, 111, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Galindo-Murillo, R.; Robertson, J.C.; Zgarbová, M.; Šponer, J.; Otyepka, M.; Jurečka, P.; Cheatham, T.E. Assessing the Current State of Amber Force Field Modifications for DNA. J. Chem. Theory Comput. 2016, 12, 4114–4127. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal Structure and Pair Potentials: A Molecular-Dynamics Study. Phys. Rev. Lett. 1980, 45, 1196–1199. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- Bonomi, M.; Branduardi, D.; Bussi, G.; Camilloni, C.; Provasi, D.; Raiteri, P.; Donadio, D.; Marinelli, F.; Pietrucci, F.; Broglia, R.A.; et al. PLUMED: A portable plugin for free-energy calculations with molecular dynamics. Comput. Phys. Commun. 2009, 180, 1961–1972. [Google Scholar] [CrossRef]
- Branduardi, D.; Gervasio, F.L.; Parrinello, M. From A to B in free energy space. J. Chem. Phys. 2007, 126, 054103. [Google Scholar] [CrossRef]
- Souaille, M.; Roux, B. Extension to the weighted histogram analysis method: Combining umbrella sampling with free energy calculations. Comput. Phys. Commun. 2001, 135, 40–57. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dG | dG | ddG | dG | ddG | dG | ddG | |
|---|---|---|---|---|---|---|---|
| Path | Mpath | Mpath-Path | M1mpath | M1mpath-Mpath | M2mpath | M2mpath-Mpath | |
| #1 | 7.23 | 6.8 | −0.43 | 8.34 | 1.54 | 9.06 | 2.26 |
| #2 | 7.43 | 5.36 | −2.07 | 7.44 | 2.08 | 8.17 | 2.81 |
| #3 | 8.19 | 8.37 | 0.18 | 10.31 | 1.94 | 9.48 | 1.11 |
| #4 | 9.72 | 6.15 | −3.57 | 7.65 | 1.5 | 9.49 | 3.34 |
| #5 | 9.82 | 6.82 | −3.0 | 10.26 | 3.34 | 8.4 | 1.58 |
| mean | 8.48 | 6.7 | −1.78 | 8.8 | 2.08 | 8.92 | 2.22 |
| Std. | 1.23 | 1.11 | 1.62 | 1.40 | 0.75 | 0.61 | 0.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, W.; Lyu, W.; Zhu, L. Excessive Existence of Positively Charged Amino Acids Caused Off-Target Recognition in the Seed Region of Clostridium butyricum Argonaute. Int. J. Mol. Sci. 2025, 26, 4738. https://doi.org/10.3390/ijms26104738
Ma W, Lyu W, Zhu L. Excessive Existence of Positively Charged Amino Acids Caused Off-Target Recognition in the Seed Region of Clostridium butyricum Argonaute. International Journal of Molecular Sciences. 2025; 26(10):4738. https://doi.org/10.3390/ijms26104738
Chicago/Turabian StyleMa, Wenzhuo, Wenping Lyu, and Lizhe Zhu. 2025. "Excessive Existence of Positively Charged Amino Acids Caused Off-Target Recognition in the Seed Region of Clostridium butyricum Argonaute" International Journal of Molecular Sciences 26, no. 10: 4738. https://doi.org/10.3390/ijms26104738
APA StyleMa, W., Lyu, W., & Zhu, L. (2025). Excessive Existence of Positively Charged Amino Acids Caused Off-Target Recognition in the Seed Region of Clostridium butyricum Argonaute. International Journal of Molecular Sciences, 26(10), 4738. https://doi.org/10.3390/ijms26104738