
Alleviation of Neurological Disorders by Targeting Neurodegenerative-Associated Enzymes: Natural and Synthetic Molecules




Abstract
1. Introduction
2. Role of Multiple Enzymes Linked in the Progression of Neurological Disorders
2.1. Sphingomyelinases
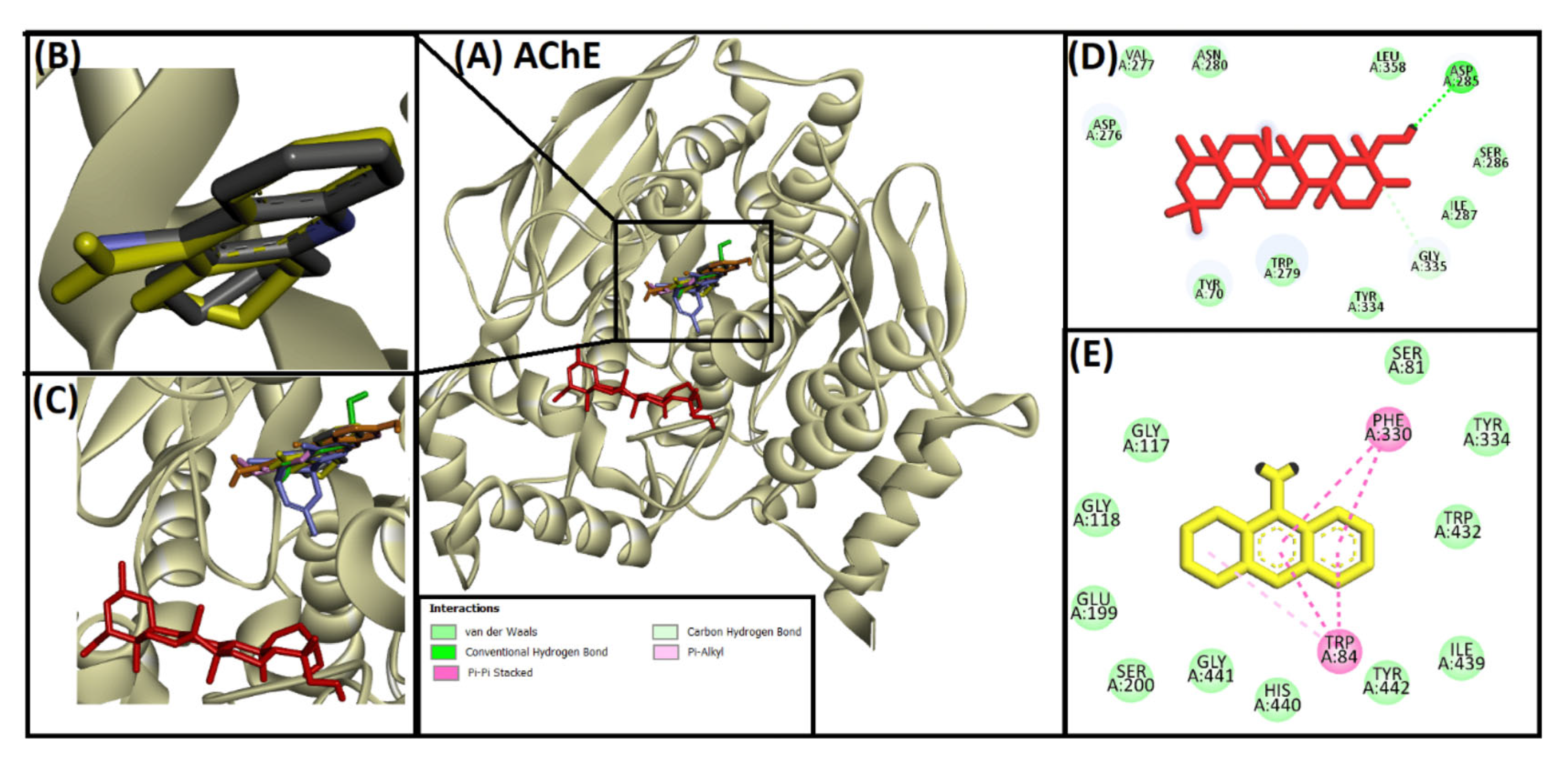
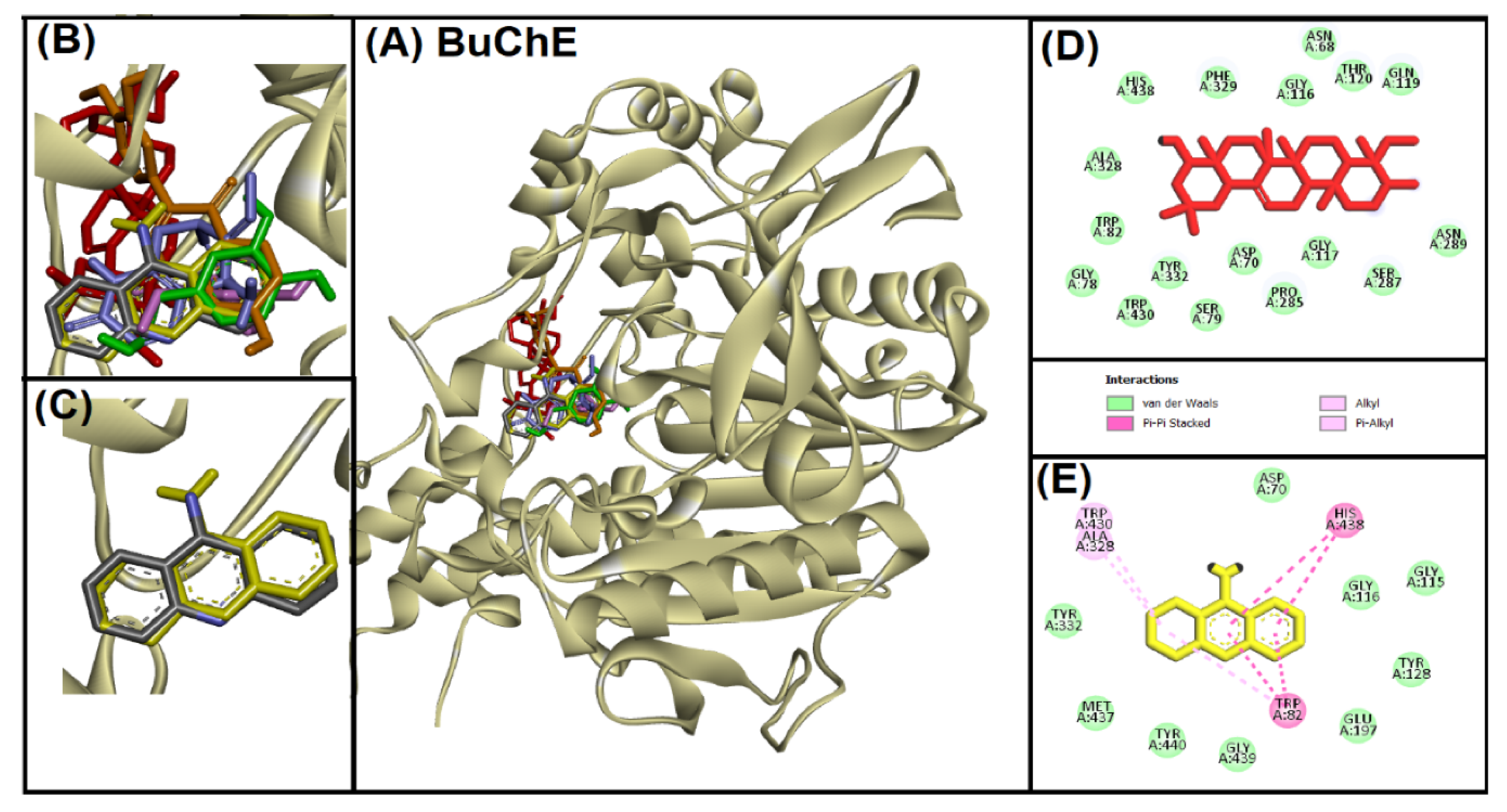
2.2. Acetylcholinesterase (AChE)
2.3. Monoamine Oxidase
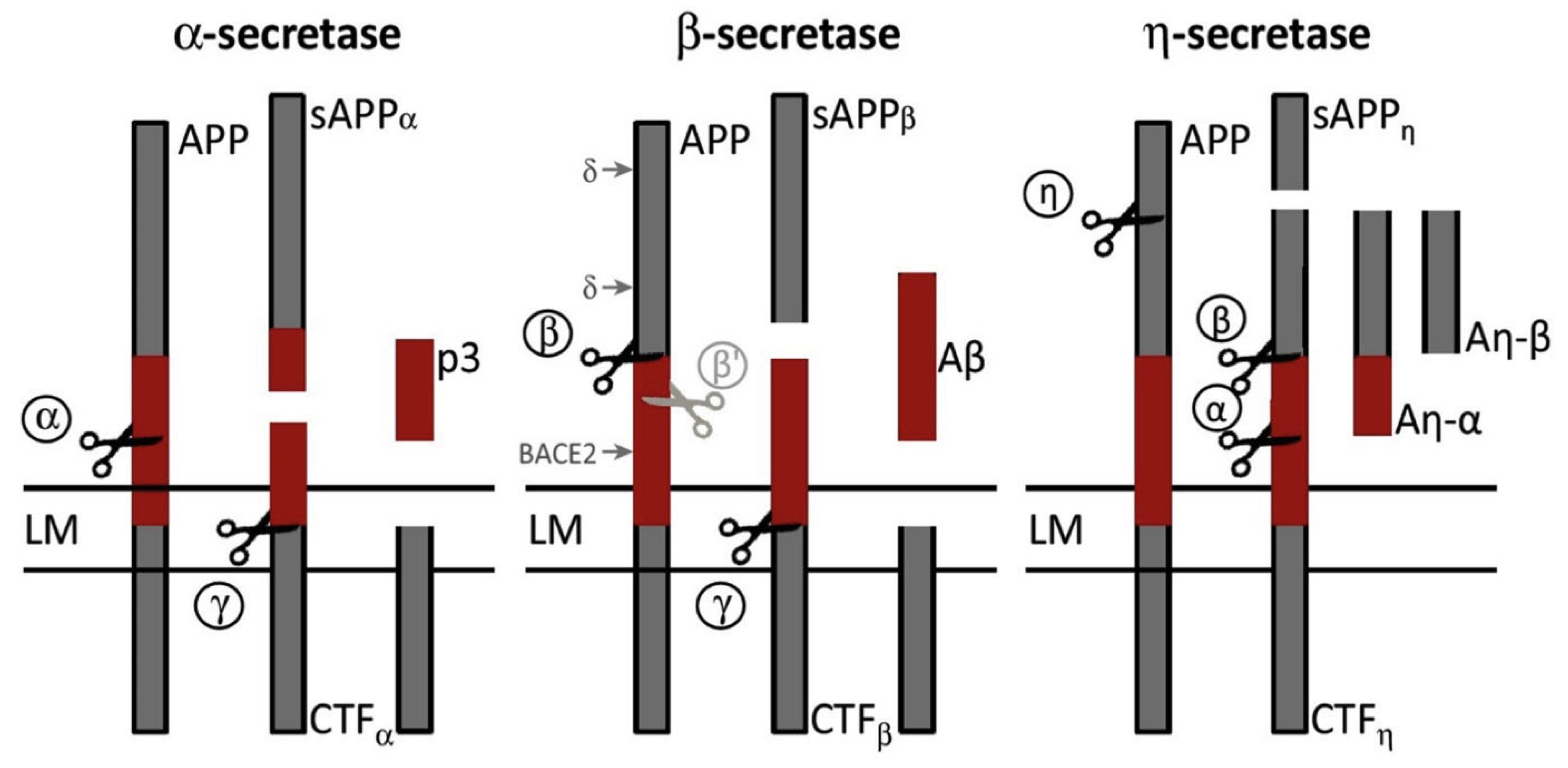
2.4. Beta-Secretase (BACE1)
2.5. Tau Kinases
2.6. Caspases
2.7. Cyclooxygenase-2 (COX-2)
3. Multiple Strategies for Controlling Neurological Disorders by Targeting Associated Enzymes
3.1. Naturally Derived Molecules for Controlling Neurological Disorders
3.1.1. Plant-Derived Compounds
Polyphenols
- Examples and Mechanisms (e.g., Resveratrol, Curcumin)
- Efficacy and Safety Profile
Flavonoids
Prominent Compounds (e.g., Quercetin, EGCG)
- Impact on Enzyme Activity and Disease Progression
Alkaloids
- Notable Alkaloids (e.g., Berberine)
- Therapeutic Potential and Challenges
3.1.2. Microbial-Derived Natural Molecules
3.1.3. Bacterial- and Fungal-Derived Molecules
3.1.4. Marine Organism-Derived Molecules
3.1.5. Animal-Derived Natural Compound
3.2. Chemically Synthesized Molecules for Controlling Neurological Disorders by Targeting Associated Enzymes
3.2.1. Small-Molecule Inhibitors
3.2.2. Peptide Inhibitors
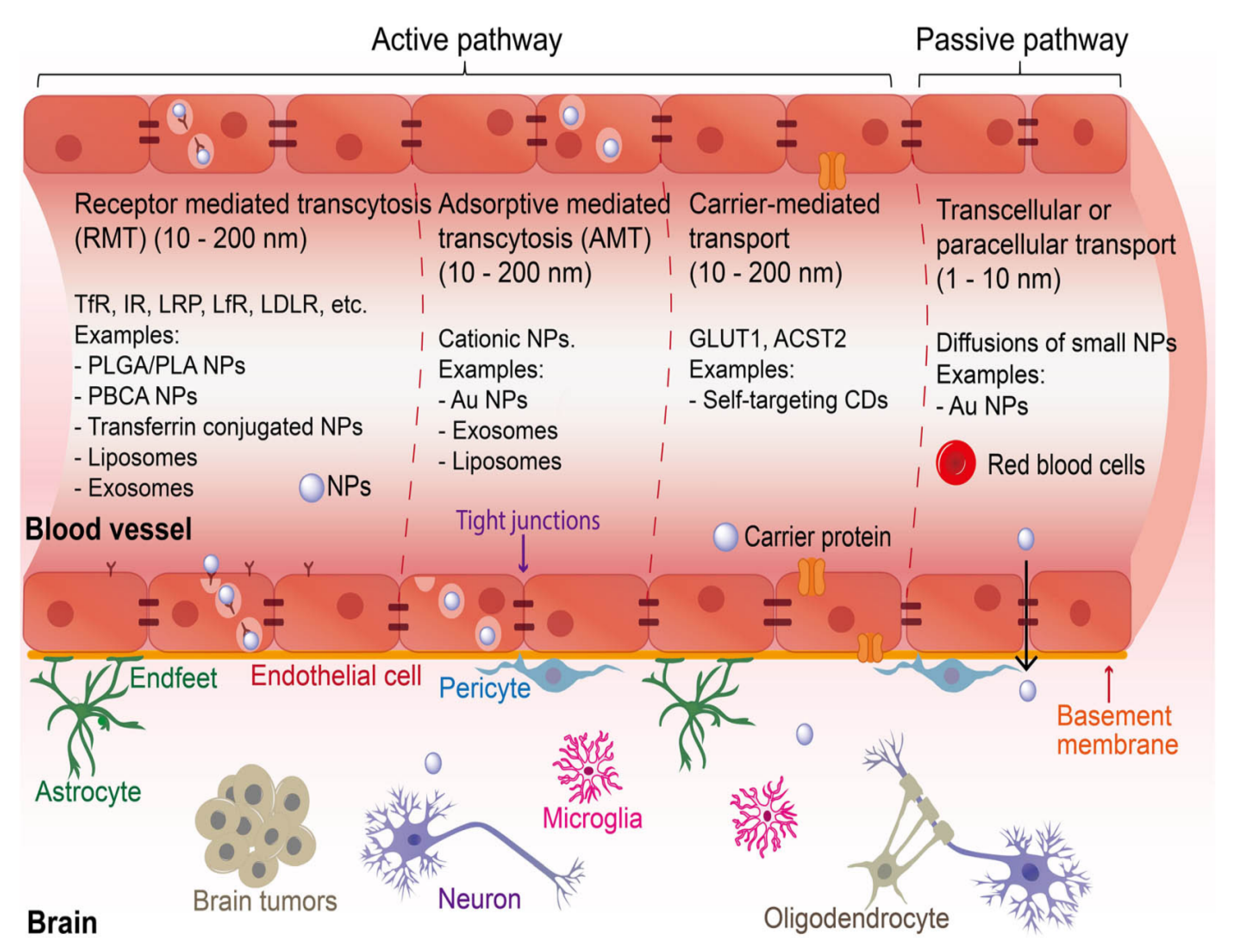
3.3. Advanced Drug Delivery Systems
3.3.1. Nanomaterials-Mediated Control of Neurological Disorders by Associated Targeting Enzymes
3.3.2. Nanomaterials and Action Mechanisms Targeting Neurodegenerative Enzymes
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Ningrum, D.N.A.; Kung, W.-M. Challenges and perspectives of neurological disorders. Brain Sci. 2023, 13, 676. [Google Scholar] [CrossRef] [PubMed]
- Giri, P.M.; Banerjee, A.; Ghosal, A.; Layek, B. Neuroinflammation in neurodegenerative disorders: Current knowledge and therapeutic implications. Int. J. Mol. Sci. 2024, 25, 3995. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.E.; Lee, J.S. Mechanisms and Emerging Regulators of Neuroinflammation: Exploring New Therapeutic Strategies for Neurological Disorders. Curr. Issues Mol. Biol. 2024, 47, 8. [Google Scholar] [CrossRef] [PubMed]
- Pizza, V.; Agresta, A.; D’Acunto, C.W.; Festa, M.; Capasso, A. Neuroinflamm-aging and neurodegenerative diseases: An overview. CNS Neurol. Disord.-Drug Targets (Former. Curr. Drug Targets-CNS Neurol. Disord.) 2011, 10, 621–634. [Google Scholar] [CrossRef]
- Adamu, A.; Li, S.; Gao, F.; Xue, G. The role of neuroinflammation in neurodegenerative diseases: Current understanding and future therapeutic targets. Front. Aging Neurosci. 2024, 16, 1347987. [Google Scholar] [CrossRef]
- Rauf, A.; Badoni, H.; Abu-Izneid, T.; Olatunde, A.; Rahman, M.M.; Painuli, S.; Semwal, P.; Wilairatana, P.; Mubarak, M.S. Neuroinflammatory markers: Key indicators in the pathology of neurodegenerative diseases. Molecules 2022, 27, 3194. [Google Scholar] [CrossRef]
- Amor, S.; Peferoen, L.A.; Vogel, D.Y.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases–an update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef]
- Vysakh, V.; Roshni, P.R. Role of Enzymes in Causing Neurological Disorders. Int. J. Res. Pharm. Sci. 2021, 12, 466–476. [Google Scholar] [CrossRef]
- Kumar, S.; Saha, S.; Singh, T.; Tripathi, R.B.; Singh, K.; Dwivedi, V.; Singh, S.; Chandra, D. Focusing on Enzyme Suppression in Neurodegenerative Disorders: Promising Approaches for Therapeutic Measures. New Emir. Med. J. 2024, 5, e02506882311325. [Google Scholar] [CrossRef]
- Alam, J.; Sharma, L. Potential enzymatic targets in Alzheimer’s: A comprehensive review. Curr. Drug Targets 2019, 20, 316–339. [Google Scholar] [CrossRef] [PubMed]
- Fronza, M.G.; Alves, D.; Praticò, D.; Savegnago, L. The neurobiology and therapeutic potential of multi-targeting β-secretase, glycogen synthase kinase 3β and acetylcholinesterase in Alzheimer’s disease. Ageing Res. Rev. 2023, 90, 102033. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.S.; Younis, K.; Philippe, J.; Aschner, M.; Khan, H. Strategic approaches to target the enzymes using natural compounds for the management of Alzheimer’s disease: A review. CNS Neurol. Disord.-Drug Targets-CNS Neurol. Disord. 2022, 21, 610–620. [Google Scholar] [CrossRef]
- Walczak-Nowicka, Ł.J.; Herbet, M. Acetylcholinesterase inhibitors in the treatment of neurodegenerative diseases and the role of acetylcholinesterase in their pathogenesis. Int. J. Mol. Sci. 2021, 22, 9290. [Google Scholar] [CrossRef]
- Rees, T.M.; Brimijoin, S. The role of acetylcholinesterase in the pathogenesis of Alzheimer’s disease. Drugs Today 2003, 39, 75–83. [Google Scholar] [CrossRef]
- García-Ayllón, M.-S.; Small, D.H.; Avila, J.; Sáez-Valero, J. Revisiting the role of acetylcholinesterase in Alzheimer’s disease: Cross-talk with P-tau and β-amyloid. Front. Mol. Neurosci. 2011, 4, 22. [Google Scholar] [CrossRef]
- Youdim, M.B.; Riederer, P.F. A review of the mechanisms and role of monoamine oxidase inhibitors in Parkinson’s disease. Neurology 2004, 63, S32–S35. [Google Scholar] [CrossRef]
- Kumar, B.; Prakash Gupta, V.; Kumar, V. A perspective on monoamine oxidase enzyme as drug target: Challenges and opportunities. Curr. Drug Targets 2017, 18, 87–97. [Google Scholar] [CrossRef]
- Shi, R.; Wu, Q.; Xin, C.; Yu, H.; Lim, K.L.; Li, X.; Shi, Z.; Zhang, C.W.; Qian, L.; Li, L. Structure-Based Specific Detection and Inhibition of Monoamine Oxidases and Their Applications in Central Nervous System Diseases. ChemBioChem 2019, 20, 1487–1497. [Google Scholar] [CrossRef]
- Nam, M.-H.; Sa, M.; Ju, Y.H.; Park, M.G.; Lee, C.J. Revisiting the role of astrocytic MAOB in Parkinson’s disease. Int. J. Mol. Sci. 2022, 23, 4453. [Google Scholar] [CrossRef]
- Cohen, G.; Farooqui, R.; Kesler, N. Parkinson disease: A new link between monoamine oxidase and mitochondrial electron flow. Proc. Natl. Acad. Sci. USA 1997, 94, 4890–4894. [Google Scholar] [CrossRef] [PubMed]
- Khezri, M.R.; Ghasemnejad-Berenji, M. The role of caspases in Alzheimer’s disease: Pathophysiology implications and pharmacologic modulation. J. Alzheimer’s Dis. 2023, 91, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Cotman, C.W.; Poon, W.W.; Rissman, R.A.; Blurton-Jones, M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J. Neuropathol. Exp. Neurol. 2005, 64, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Kumasaka, D.K.; Galvan, V.; Head, E.; Rohn, T.T. Caspase cleavage of the amyloid precursor protein is prevented after overexpression of bcl-2 in a triple transgenic mouse model of Alzheimer’s disease. Int. J. Physiol. Pathophysiol. Pharmacol. 2009, 1, 48. [Google Scholar]
- Rohn, T.T.; Rissman, R.A.; Head, E.; Cotman, C.W. Caspase activation in the Alzheimer’s disease brain: Tortuous and torturous. Drug News Perspect. 2002, 15, 549–557. [Google Scholar] [CrossRef]
- Venero, J.L.; Burguillos, M.; Brundin, P.; Joseph, B. The executioners sing a new song: Killer caspases activate microglia. Cell Death Differ. 2011, 18, 1679–1691. [Google Scholar] [CrossRef]
- Phillis, J.W.; Horrocks, L.A.; Farooqui, A.A. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: Their role and involvement in neurological disorders. Brain Res. Rev. 2006, 52, 201–243. [Google Scholar] [CrossRef]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef]
- Aïd, S.; Bosetti, F. Targeting cyclooxygenases-1 and -2 in neuroinflammation: Therapeutic implications. Biochimie 2011, 93, 46–51. [Google Scholar] [CrossRef]
- Yang, H.; Chen, C. Cyclooxygenase-2 in synaptic signaling. Curr. Pharm. Des. 2008, 14, 1443–1451. [Google Scholar] [CrossRef]
- Rose, J.W.; Hill, K.E.; Watt, H.E.; Carlson, N.G. Inflammatory cell expression of cyclooxygenase-2 in the multiple sclerosis lesion. J. Neuroimmunol. 2004, 149, 40–49. [Google Scholar] [CrossRef]
- Palumbo, S. Pathogenesis and progression of multiple sclerosis: The role of arachidonic acid-mediated neuroinflammation. Exon Publ. 2017, 111–123. [Google Scholar] [CrossRef]
- Palumbo, S.; Bosetti, F. Alterations of brain eicosanoid synthetic pathway in multiple sclerosis and in animal models of demyelination: Role of cyclooxygenase-2. Prostaglandins Leukot. Essent. Fat. Acids 2013, 89, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-H.; Aid, S.; Bosetti, F. The distinct roles of cyclooxygenase-1 and-2 in neuroinflammation: Implications for translational research. Trends Pharmacol. Sci. 2009, 30, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Adamek, R.N.; Dick, B.L.; Credille, C.V.; Morrison, C.N.; Cohen, S.M. Targeting metalloenzymes for therapeutic intervention. Chem. Rev. 2018, 119, 1323–1455. [Google Scholar] [CrossRef]
- Ferreira, J.P.; Albuquerque, H.M.; Cardoso, S.M.; Silva, A.M.; Silva, V.L. Dual-target compounds for Alzheimer’s disease: Natural and synthetic AChE and BACE-1 dual-inhibitors and their structure-activity relationship (SAR). Eur. J. Med. Chem. 2021, 221, 113492. [Google Scholar] [CrossRef]
- Ibrahim, M.M.; Gabr, M.T. Multitarget therapeutic strategies for Alzheimer’s disease. Neural Regen. Res. 2019, 14, 437–440. [Google Scholar]
- Wang, S.; Kong, X.; Chen, Z.; Wang, G.; Zhang, J.; Wang, J. Role of natural compounds and target enzymes in the treatment of Alzheimer’s disease. Molecules 2022, 27, 4175. [Google Scholar] [CrossRef]
- Saxena, M.; Dubey, R. Target enzyme in Alzheimer’s disease: Acetylcholinesterase inhibitors. Curr. Top. Med. Chem. 2019, 19, 264–275. [Google Scholar] [CrossRef]
- Silva, T.; Reis, J.; Teixeira, J.; Borges, F. Alzheimer’s disease, enzyme targets and drug discovery struggles: From natural products to drug prototypes. Ageing Res. Rev. 2014, 15, 116–145. [Google Scholar] [CrossRef]
- Islam, B.U.; Tabrez, S. Management of Alzheimer’s disease-An insight of the enzymatic and other novel potential targets. Int. J. Biol. Macromol. 2017, 97, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.H.; Bajgai, J.; Fadriquela, A.; Sharma, S.; Trinh, T.T.; Akter, R.; Jeong, Y.J.; Goh, S.H.; Kim, C.-S.; Lee, K.-J. Therapeutic potential of natural products in treating neurodegenerative disorders and their future prospects and challenges. Molecules 2021, 26, 5327. [Google Scholar] [CrossRef] [PubMed]
- Elufioye, T.O.; Berida, T.I.; Habtemariam, S. Plants-derived neuroprotective agents: Cutting the cycle of cell death through multiple mechanisms. Evid.-Based Complement. Altern. Med. 2017, 2017, 3574012. [Google Scholar] [CrossRef]
- Hussain, G.; Rasul, A.; Anwar, H.; Aziz, N.; Razzaq, A.; Wei, W.; Ali, M.; Li, J.; Li, X. Role of plant derived alkaloids and their mechanism in neurodegenerative disorders. Int. J. Biol. Sci. 2018, 14, 341. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.R.; Tay, K.C.; Su, Y.X.; Wong, C.K.; Tan, W.N.; Khaw, K.Y. Potential of naturally derived alkaloids as multi-targeted therapeutic agents for neurodegenerative diseases. Molecules 2021, 26, 728. [Google Scholar] [CrossRef]
- Parashar, A.; Mehta, V.; Chauhan, B.; Ghosh, P.; Deb, P.K.; Jaiswal, M.; Prajapati, S.K. Sonic hedgehog signalling pathway contributes in age-related disorders and Alzheimer’s disease. Ageing Res. Rev. 2024, 96, 102271. [Google Scholar] [CrossRef]
- Gaudreault, R.; Mousseau, N. Mitigating Alzheimer’s disease with natural polyphenols: A review. Curr. Alzheimer Res. 2019, 16, 529–543. [Google Scholar] [CrossRef]
- Andrade, S.; Ramalho, M.J.; Loureiro, J.A.; Pereira, M.d.C. Natural compounds for Alzheimer’s disease therapy: A systematic review of preclinical and clinical studies. Int. J. Mol. Sci. 2019, 20, 2313. [Google Scholar] [CrossRef]
- Stacchiotti, A.; Corsetti, G. Natural compounds and autophagy: Allies against neurodegeneration. Front. Cell Dev. Biol. 2020, 8, 555409. [Google Scholar] [CrossRef]
- Sadhukhan, P.; Saha, S.; Dutta, S.; Mahalanobish, S.; Sil, P.C. Nutraceuticals: An emerging therapeutic approach against the pathogenesis of Alzheimer’s disease. Pharmacol. Res. 2018, 129, 100–114. [Google Scholar] [CrossRef]
- Sanghai, N.; Vuong, B.; Burak Berk, A.; Afridi, M.S.K.; Tranmer, G.K. Current small molecule–based medicinal chemistry approaches for neurodegeneration therapeutics. ChemMedChem 2024, 19, e202300705. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, W.J.; Van der Schyf, C.J. Rationally designed multi-targeted agents against neurodegenerative diseases. Curr. Med. Chem. 2013, 20, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Agustín-Pavón, C.; Isalan, M. Synthetic biology and therapeutic strategies for the degenerating brain: Synthetic biology approaches can transform classical cell and gene therapies, to provide new cures for neurodegenerative diseases. Bioessays 2014, 36, 979–990. [Google Scholar] [CrossRef]
- Ryan, P. An Investigation Into Novel Molecular Strategies Targeting Neurodegenerative Diseases. Ph.D. Thesis, Griffith University, Brisbane South, Australia, 2020. [Google Scholar]
- Coimbra, J.R.; Marques, D.F.; Baptista, S.J.; Pereira, C.M.; Moreira, P.I.; Dinis, T.C.; Santos, A.E.; Salvador, J.A. Highlights in BACE1 inhibitors for Alzheimer’s disease treatment. Front. Chem. 2018, 6, 178. [Google Scholar] [CrossRef]
- Gupta, S.P.; Patil, V.M. Recent studies on design and development of drugs against Alzheimer’s disease (AD) based on inhibition of BACE-1 and other AD-causative agents. Curr. Top. Med. Chem. 2020, 20, 1195–1213. [Google Scholar] [CrossRef]
- Guo, T.; Hobbs, D.W. Development of BACE1 inhibitors for Alzheimer’s disease. Curr. Med. Chem. 2006, 13, 1811–1829. [Google Scholar] [CrossRef]
- Huang, W.-H.; Sheng, R.; Hu, Y.-Z. Progress in the development of nonpeptidomimetic BACE 1 inhibitors for Alzheimer’s disease. Curr. Med. Chem. 2009, 16, 1806–1820. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Osswald, H.L. BACE1 (β-secretase) inhibitors for the treatment of Alzheimer’s disease. Chem. Soc. Rev. 2014, 43, 6765–6813. [Google Scholar] [CrossRef]
- Mazanetz, M.P.; Fischer, P.M. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat. Rev. Drug Discov. 2007, 6, 464–479. [Google Scholar] [CrossRef]
- Moussa-Pacha, N.M.; Abdin, S.M.; Omar, H.A.; Alniss, H.; Al-Tel, T.H. BACE1 inhibitors: Current status and future directions in treating Alzheimer’s disease. Med. Res. Rev. 2020, 40, 339–384. [Google Scholar] [CrossRef]
- Prati, F.; Bottegoni, G.; Bolognesi, M.L.; Cavalli, A. BACE-1 inhibitors: From recent single-target molecules to multitarget compounds for Alzheimer’s disease: Miniperspective. J. Med. Chem. 2018, 61, 619–637. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.-Y.; Herr, D.R.; Farooqui, T.; Ling, E.-A.; Farooqui, A.A. Role of sphingomyelinases in neurological disorders. Expert Opin. Ther. Targets 2015, 19, 1725–1742. [Google Scholar] [CrossRef] [PubMed]
- Bilousova, T.; Simmons, B.J.; Knapp, R.R.; Elias, C.J.; Campagna, J.; Melnik, M.; Chandra, S.; Focht, S.; Zhu, C.; Vadivel, K.; et al. Dual Neutral Sphingomyelinase-2/Acetylcholinesterase Inhibitors for the Treatment of Alzheimer’s Disease. ACS Chem. Biol. 2020, 15, 1671–1684. [Google Scholar] [CrossRef]
- Marchesini, N.; Hannun, Y.A. Acid and neutral sphingomyelinases: Roles and mechanisms of regulation. Biochem. Cell Biol. 2004, 82, 27–44. [Google Scholar] [CrossRef]
- Hofmann, K.; Tomiuk, S.; Wolff, G.; Stoffel, W. Cloning and characterization of the mammalian brain-specific, Mg2+-dependent neutral sphingomyelinase. Proc. Natl. Acad. Sci. USA 2000, 97, 5895–5900. [Google Scholar] [CrossRef]
- Gu, L.; Huang, B.; Shen, W.; Gao, L.; Ding, Z.; Wu, H.; Guo, J. Early activation of nSMase2/ceramide pathway in astrocytes is involved in ischemia-associated neuronal damage via inflammation in rat hippocampi. J. NeuroInflamm. 2013, 10, 1–16. [Google Scholar] [CrossRef]
- He, X.; Huang, Y.; Li, B.; Gong, C.-X.; Schuchman, E.H. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 398–408. [Google Scholar] [CrossRef]
- Dias, I.H.; Mistry, J.; Fell, S.; Reis, A.; Spickett, C.M.; Polidori, M.C.; Lip, G.Y.; Griffiths, H.R. Oxidized LDL lipids increase β-amyloid production by SH-SY5Y cells through glutathione depletion and lipid raft formation. Free Radic. Biol. Med. 2014, 75, 48–59. [Google Scholar] [CrossRef]
- Lee, J.K.; Jin, H.K.; Park, M.H.; Kim, B.-R.; Lee, P.H.; Nakauchi, H.; Carter, J.E.; He, X.; Schuchman, E.H.; Bae, J.-S. Acid sphingomyelinase modulates the autophagic process by controlling lysosomal biogenesis in Alzheimer’s disease. J. Exp. Med. 2014, 211, 1551–1570. [Google Scholar] [CrossRef]
- Fonteh, A.N.; Ormseth, C.; Chiang, J.; Cipolla, M.; Arakaki, X.; Harrington, M.G. Sphingolipid metabolism correlates with cerebrospinal fluid Beta amyloid levels in Alzheimer’s disease. PLoS ONE 2015, 10, e0125597. [Google Scholar] [CrossRef]
- Krejci, E.; Duval, N.; Chatonnet, A.; Vincens, P.; Massoulie, J. Cholinesterase-like domains in enzymes and structural proteins: Functional and evolutionary relationships and identification of a catalytically essential aspartic acid. Proc. Natl. Acad. Sci. USA 1991, 88, 6647–6651. [Google Scholar] [CrossRef] [PubMed]
- Talesa, V.N. Acetylcholinesterase in Alzheimer’s disease. Mech. Ageing Dev. 2001, 122, 1961–1969. [Google Scholar] [CrossRef] [PubMed]
- Sáez-Valero, J.; Sberna, G.; McLean, C.A.; Small, D.H. Molecular isoform distribution and glycosylation of acetylcholinesterase are altered in brain and cerebrospinal fluid of patients with Alzheimer’s disease. J. Neurochem. 1999, 72, 1600–1608. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer’s disease. Prim. Care Companion CNS Disord. 2013, 15, 26731. [Google Scholar] [CrossRef]
- Perry, E.; McKeith, I.; Ballard, C. Butyrylcholinesterase and progression of cognitive deficits in dementia with Lewy bodies. Neurology 2003, 60, 1852–1853. [Google Scholar] [CrossRef]
- Zhao, X.; Hu, Q.; Wang, X.; Li, C.; Chen, X.; Zhao, D.; Qiu, Y.; Xu, H.; Wang, J.; Ren, L. Dual-Target Inhibitors Based on Acetylcholinesterase: Novel Agents for Alzheimer’s Disease. Eur. J. Med. Chem. 2024, 279, 116810. [Google Scholar] [CrossRef]
- Nagori, K.; Pradhan, M.; Sharma, M.; Ajazuddin; Badwaik, H.R.; Nakhate, K.T. Current progress on central cholinergic receptors as therapeutic targets for Alzheimer’s disease. Curr. Alzheimer Res. 2024, 21, 50–68. [Google Scholar] [CrossRef]
- Luque, F.J.; Muñoz-Torrero, D. Acetylcholinesterase: A versatile template to coin potent modulators of multiple therapeutic targets. Acc. Chem. Res. 2024, 57, 450–467. [Google Scholar] [CrossRef]
- Pollak, Y.; Gilboa, A.; Ben-Menachem, O.; Ben-Hur, T.; Soreq, H.; Yirmiya, R. Acetylcholinesterase inhibitors reduce brain and blood interleukin-1β production. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2005, 57, 741–745. [Google Scholar] [CrossRef]
- Reale, M.; Iarlori, C.; Gambi, F.; Lucci, I.; Salvatore, M.; Gambi, D. Acetylcholinesterase inhibitors effects on oncostatin-M, interleukin-1β and interleukin-6 release from lymphocytes of Alzheimer’s disease patients. Exp. Gerontol. 2005, 40, 165–171. [Google Scholar] [CrossRef]
- Reale, M.; Iarlori, C.; Gambi, F.; Feliciani, C.; Salone, A.; Toma, L.; DeLuca, G.; Salvatore, M.; Conti, P.; Gambi, D. Treatment with an acetylcholinesterase inhibitor in Alzheimer patients modulates the expression and production of the pro-inflammatory and anti-inflammatory cytokines. J. Neuroimmunol. 2004, 148, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Kokras, N.; Stamouli, E.; Sotiropoulos, I.; Katirtzoglou, E.A.; Siarkos, K.T.; Dalagiorgou, G.; Alexandraki, K.I.; Coulocheri, S.; Piperi, C.; Politis, A.M. Acetyl cholinesterase inhibitors and cell-derived peripheral inflammatory cytokines in early stages of Alzheimer’s disease. J. Clin. Psychopharmacol. 2018, 38, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, H.-j.; Park, S.K.; Park, J.-H.; Jeong, H.-R.; Lee, S.; Lee, H.; Seol, E.; Hoe, H.-S. Donepezil regulates LPS and Aβ-stimulated neuroinflammation through MAPK/NLRP3 inflammasome/STAT3 signaling. Int. J. Mol. Sci. 2021, 22, 10637. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, D.; Mattevi, A.; Binda, C.; Li, M.; Hubalek, F. Structure and mechanism of monoamine oxidase. Curr. Med. Chem. 2004, 11, 1983–1993. [Google Scholar] [CrossRef]
- Parambi, D.G. Treatment of parkinson’s disease by MAO-B inhibitors, new therapies and future challenges-A mini-review. Comb. Chem. High Throughput Screen. 2020, 23, 847–861. [Google Scholar] [CrossRef]
- Riederer, P.; Laux, G. MAO-inhibitors in Parkinson’s Disease. Exp. Neurobiol. 2011, 20, 1. [Google Scholar] [CrossRef]
- Tan, Y.-Y.; Jenner, P.; Chen, S.-D. Monoamine oxidase-B inhibitors for the treatment of Parkinson’s disease: Past, present, and future. J. Park. Dis. 2022, 12, 477–493. [Google Scholar] [CrossRef]
- Özdemir, Z.; Alagöz, M.A.; Bahçecioğlu, Ö.F.; Gök, S. Monoamine oxidase-B (MAO-B) inhibitors in the treatment of Alzheimer’s and Parkinson’s disease. Curr. Med. Chem. 2021, 28, 6045–6065. [Google Scholar] [CrossRef]
- Jost, W.H. A critical appraisal of MAO-B inhibitors in the treatment of Parkinson’s disease. J. Neural Transm. 2022, 129, 723–736. [Google Scholar] [CrossRef]
- Hampel, H.; Vassar, R.; De Strooper, B.; Hardy, J.; Willem, M.; Singh, N.; Zhou, J.; Yan, R.; Vanmechelen, E.; De Vos, A. The β-secretase BACE1 in Alzheimer’s disease. Biol. Psychiatry 2021, 89, 745–756. [Google Scholar] [CrossRef]
- Zhu, K.; Xiang, X.; Filser, S.; Marinković, P.; Dorostkar, M.M.; Crux, S.; Neumann, U.; Shimshek, D.R.; Rammes, G.; Haass, C. Beta-site amyloid precursor protein cleaving enzyme 1 inhibition impairs synaptic plasticity via seizure protein 6. Biol. Psychiatry 2018, 83, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Fu, Y.; Yasvoina, M.; Shao, P.; Hitt, B.; O’Connor, T.; Logan, S.; Maus, E.; Citron, M.; Berry, R. β-Site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: Implications for Alzheimer’s disease pathogenesis. J. Neurosci. 2007, 27, 3639–3649. [Google Scholar] [CrossRef] [PubMed]
- Kandalepas, P.C.; Sadleir, K.R.; Eimer, W.A.; Zhao, J.; Nicholson, D.A.; Vassar, R. The Alzheimer’s β-secretase BACE1 localizes to normal presynaptic terminals and to dystrophic presynaptic terminals surrounding amyloid plaques. Acta Neuropathol. 2013, 126, 329–352. [Google Scholar] [CrossRef]
- Sadleir, K.R.; Kandalepas, P.C.; Buggia-Prévot, V.; Nicholson, D.A.; Thinakaran, G.; Vassar, R. Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Aβ generation in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 235–256. [Google Scholar] [CrossRef]
- Barão, S.; Moechars, D.; Lichtenthaler, S.F.; De Strooper, B. BACE1 physiological functions may limit its use as therapeutic target for Alzheimer’s disease. Trends Neurosci. 2016, 39, 158–169. [Google Scholar] [CrossRef]
- Kandalepas, P.C.; Vassar, R. The normal and pathologic roles of the Alzheimer’s β-secretase, BACE1. Curr. Alzheimer Res. 2014, 11, 441–449. [Google Scholar] [CrossRef]
- Willem, M.; Lammich, S.; Haass, C. Function, regulation and therapeutic properties of β-secretase (BACE1). In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2009; pp. 175–182. [Google Scholar]
- Citron, M.; Teplow, D.B.; Selkoe, D.J. Generation of amyloid β protein from its precursor is sequence specific. Neuron 1995, 14, 661–670. [Google Scholar] [CrossRef]
- Sinha, S.; Anderson, J.P.; Barbour, R.; Basi, G.S.; Caccavello, R.; Davis, D.; Doan, M.; Dovey, H.F.; Frigon, N.; Hong, J. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 1999, 402, 537–540. [Google Scholar] [CrossRef]
- Haass, C.; Koo, E.H.; Mellon, A.; Hung, A.Y.; Selkoe, D.J. Targeting of cell-surface β-amyloid precursor protein to lysosomes: Alternative processing into amyloid-bearing fragments. Nature 1992, 357, 500–503. [Google Scholar] [CrossRef]
- Liebsch, F.; Kulic, L.; Teunissen, C.; Shobo, A.; Ulku, I.; Engelschalt, V.; Hancock, M.A.; van der Flier, W.M.; Kunach, P.; Rosa-Neto, P. Aβ34 is a BACE1-derived degradation intermediate associated with amyloid clearance and Alzheimer’s disease progression. Nat. Commun. 2019, 10, 2240. [Google Scholar] [CrossRef]
- Ulku, I.; Liebsch, F.; Akerman, S.C.; Schulz, J.F.; Kulic, L.; Hock, C.; Pietrzik, C.; Di Spiezio, A.; Thinakaran, G.; Saftig, P. Mechanisms of amyloid-β34 generation indicate a pivotal role for BACE1 in amyloid homeostasis. Sci. Rep. 2023, 13, 2216. [Google Scholar] [CrossRef] [PubMed]
- Nicsanu, R.; Cervellati, C.; Benussi, L.; Squitti, R.; Zanardini, R.; Rosta, V.; Trentini, A.; Ferrari, C.; Saraceno, C.; Longobardi, A. Increased serum beta-secretase 1 activity is an early marker of Alzheimer’s disease. J. Alzheimer’s Dis. 2022, 87, 433–441. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.-Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef]
- Mareck, A.; Fellous, A.; Francon, J.; Nunez, J. Changes in composition and activity of microtubule-associated proteins during brain development. Nature 1980, 284, 353–355. [Google Scholar] [CrossRef]
- Wood, J.G.; Mirra, S.S.; Pollock, N.J.; Binder, L.I. Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc. Natl. Acad. Sci. USA 1986, 83, 4040–4043. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.-C.; Quinlan, M.; Wisniewski, H.M.; Binder, L.I. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef]
- Stoothoff, W.H.; Johnson, G.V. Tau phosphorylation: Physiological and pathological consequences. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2005, 1739, 280–297. [Google Scholar] [CrossRef]
- Yu, Y.; Run, X.; Liang, Z.; Li, Y.; Liu, F.; Liu, Y.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J. Neurochem. 2009, 108, 1480–1494. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef]
- Brion, J.P.; Smith, C.; Couck, A.M.; Gallo, J.M.; Anderton, B.H. Developmental Changes in τ Phosphorylation: Fetal τ Is Transiently Phosphorylated in a Manner Similar to Paired Helical Filament-τ Characteristic of Alzheimer’s Disease. J. Neurochem. 1993, 61, 2071–2080. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Gomez-Isla, T.; Puig, B.; Freixes, M.; Ribe, E.; Dalfo, E.; Avila, J. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer’s disease and tauopathies. Curr. Alzheimer Res. 2005, 2, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, B.; Tung, E.J.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur. J. Neurosci. 2007, 26, 3429–3436. [Google Scholar] [CrossRef] [PubMed]
- Abraha, A.; Ghoshal, N.; Gamblin, T.C.; Cryns, V.; Berry, R.W.; Kuret, J.; Binder, L.I. C-terminal inhibition of tau assembly in vitro and in Alzheimer’s disease. J. Cell Sci. 2000, 113, 3737–3745. [Google Scholar] [CrossRef]
- Haase, C.; Stieler, J.; Arendt, T.; Holzer, M. Pseudophosphorylation of tau protein alters its ability for self-aggregation. J. Neurochem. 2004, 88, 1509–1520. [Google Scholar] [CrossRef]
- Ferrari, A.; Hoerndli, F.; Baechi, T.; Nitsch, R.M.; Götz, J.r. β-Amyloid induces paired helical filament-like tau filaments in tissue culture. J. Biol. Chem. 2003, 278, 40162–40168. [Google Scholar] [CrossRef]
- Kasana, S.; Kumar, S.; Patel, P.; Kurmi, B.D.; Jain, S.; Sahu, S.; Vaidya, A. Caspase inhibitors: A review on recently patented compounds (2016–2023). Expert Opin. Ther. Pat. 2024, 34, 1047–1072. [Google Scholar] [CrossRef]
- Zhang, W.; Zhu, C.; Liao, Y.; Zhou, M.; Xu, W.; Zou, Z. Caspase-8 in inflammatory diseases: A potential therapeutic target. Cell. Mol. Biol. Lett. 2024, 29, 130. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in cell death, inflammation, and disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Flores, J.; Noël, A.; Foveau, B.; Lynham, J.; Lecrux, C.; LeBlanc, A.C. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 2018, 9, 3916. [Google Scholar] [CrossRef]
- Kirby, L.; Jin, J.; Cardona, J.G.; Smith, M.D.; Martin, K.A.; Wang, J.; Strasburger, H.; Herbst, L.; Alexis, M.; Karnell, J. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 2019, 10, 3887. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, S.K.; Pathak, A.; Samaiya, P.K. Alzheimer’s disease: From early pathogenesis to novel therapeutic approaches. Metab. Brain Dis. 2024, 39, 1231–1254. [Google Scholar] [CrossRef] [PubMed]
- Berta, T.; Park, C.-K.; Xu, Z.-Z.; Xie, R.-G.; Liu, T.; Lü, N.; Liu, Y.-C.; Ji, R.-R. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-α secretion. J. Clin. Investig. 2014, 124, 1173–1186. [Google Scholar] [CrossRef]
- Berta, T.; Lee, J.E.; Park, C.-K. Unconventional Role of Caspase-6 in Spinal Microglia Activation and Chronic Pain. Mediat. Inflamm. 2017, 2017, 9383184. [Google Scholar] [CrossRef]
- Gao, F.; Xiang, H.-C.; Li, H.-p.; Jia, M.; Pan, X.-l.; Pan, H.-L.; Li, M. Electroacupuncture inhibits NLRP3 inflammasome activation through CB2 receptors in inflammatory pain. Brain Behav. Immun. 2018, 67, 91–100. [Google Scholar] [CrossRef]
- Chen, G.; Luo, X.; Qadri, M.Y.; Berta, T.; Ji, R.-R. Sex-dependent glial signaling in pathological pain: Distinct roles of spinal microglia and astrocytes. Neurosci. Bull. 2018, 34, 98–108. [Google Scholar] [CrossRef]
- Friedlander, R.M.; Gagliardini, V.; Hara, H.; Fink, K.B.; Li, W.; MacDonald, G.; Fishman, M.C.; Greenberg, A.H.; Moskowitz, M.A.; Yuan, J. Expression of a dominant negative mutant of interleukin-1β converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J. Exp. Med. 1997, 185, 933–940. [Google Scholar] [CrossRef]
- Loddick, S.A.; MacKenzie, A.; Rothwell, N.J. An ICE inhibitor, z-VAD-DCB attenuates ischaemic brain damage in the rat. Neuroreport 1996, 7, 1465–1468. [Google Scholar] [CrossRef]
- Rabuffetti, M.; Sciorati, C.; Tarozzo, G.; Clementi, E.; Manfredi, A.A.; Beltramo, M. Inhibition of caspase-1-like activity by Ac-Tyr-Val-Ala-Asp-chloromethyl ketone induces long-lasting neuroprotection in cerebral ischemia through apoptosis reduction and decrease of proinflammatory cytokines. J. Neurosci. 2000, 20, 4398–4404. [Google Scholar] [CrossRef]
- Zhu, S.; Stavrovskaya, I.G.; Drozda, M.; Kim, B.Y.; Ona, V.; Li, M.; Sarang, S.; Liu, A.S.; Hartley, D.M.; Wu, D.C. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature 2002, 417, 74–78. [Google Scholar] [CrossRef]
- Plesnila, N.; Zinkel, S.; Le, D.A.; Amin-Hanjani, S.; Wu, Y.; Qiu, J.; Chiarugi, A.; Thomas, S.S.; Kohane, D.S.; Korsmeyer, S.J. BID mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2001, 98, 15318–15323. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, R.M. Apoptosis and caspases in neurodegenerative diseases. N. Engl. J. Med. 2003, 348, 1365–1375. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ona, V.O.; Guegan, C.; Chen, M.; Jackson-Lewis, V.; Andrews, L.J.; Olszewski, A.J.; Stieg, P.E.; Lee, J.-P.; Przedborski, S. Functional role of caspase-1 and caspase-3 in an ALS transgenic mouse model. Science 2000, 288, 335–339. [Google Scholar] [CrossRef]
- Malhotra, S.; Deshmukh, S.S.; Dastidar, S.G. COX inhibitors for airway inflammation. Expert Opin. Ther. Targets 2012, 16, 195–207. [Google Scholar] [CrossRef]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, 106. [Google Scholar] [CrossRef]
- Yamagata, K.; Andreasson, K.I.; Kaufmann, W.E.; Barnes, C.A.; Worley, P.F. Expression of a mitogen-inducible cyclooxygenase in brain neurons: Regulation by synaptic activity and glucocorticoids. Neuron 1993, 11, 371–386. [Google Scholar] [CrossRef]
- Breder, C.D.; Dewitt, D.; Kraig, R.P. Characterization of inducible cyclooxygenase in rat brain. J. Comp. Neurol. 1995, 355, 296–315. [Google Scholar] [CrossRef]
- Smith, W.L.; Garavito, R.M.; DeWitt, D.L. Prostaglandin endoperoxide H synthases (cyclooxygenases)-1 and− 2. J. Biol. Chem. 1996, 271, 33157–33160. [Google Scholar] [CrossRef]
- O’Neill, G.P.; Ford-Hutchinson, A.W. Expression of mRNA for cyclooxygenase-1 and cyclooxygenase-2 in human tissues. FEBS Lett. 1993, 330, 157–160. [Google Scholar] [CrossRef]
- Yasojima, K.; Schwab, C.; McGeer, E.G.; McGeer, P.L. Distribution of cyclooxygenase-1 and cyclooxygenase-2 mRNAs and proteins in human brain and peripheral organs. Brain Res. 1999, 830, 226–236. [Google Scholar] [CrossRef]
- Hoozemans, J.; Rozemuller, A.; Janssen, I.; De Groot, C.; Veerhuis, R.; Eikelenboom, P. Cyclooxygenase expression in microglia and neurons in Alzheimer’s disease and control brain. Acta Neuropathol. 2001, 101, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Pasinetti, G.; Aisen, P. Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer’s disease brain. Neuroscience 1998, 87, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Fujimi, K.; Noda, K.; Sasaki, K.; Wakisaka, Y.; Tanizaki, Y.; Iida, M.; Kiyohara, Y.; Kanba, S.; Iwaki, T. Altered expression of COX-2 in subdivisions of the hippocampus during aging and in Alzheimer’s disease: The Hisayama Study. Dement. Geriatr. Cogn. Disord. 2007, 23, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Yermakova, A.V.; O’Banion, M.K. Downregulation of neuronal cyclooxygenase-2 expression in end stage Alzheimer’s disease. Neurobiol. Aging 2001, 22, 823–836. [Google Scholar] [CrossRef]
- Combrinck, M.; Williams, J.; De Berardinis, M.A.; Warden, D.; Puopolo, M.; Smith, A.D.; Minghetti, L. Levels of CSF prostaglandin E2, cognitive decline, and survival in Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2006, 77, 85–88. [Google Scholar] [CrossRef]
- Montine, T.; Sidell, K.; Crews, B.; Markesbery, W.; Marnett, L.; Roberts, L.; Morrow, J. Elevated CSF prostaglandin E2 levels in patients with probable AD. Neurology 1999, 53, 1495. [Google Scholar] [CrossRef]
- Hoozemans, J.; Rozemuller, J.; Van Haastert, E.; Veerhuis, R.; Eikelenboom, P. Cyclooxygenase-1 and-2 in the different stages of Alzheimer’s disease pathology. Curr. Pharm. Des. 2008, 14, 1419–1427. [Google Scholar] [CrossRef]
- Serrano, G.; Lelutiu, N.; Rojas, A.; Cochi, S.; Shaw, R.; Makinson, C.; Wang, D.; FitzGerald, G.; Dingledine, R. Ablation of cyclooxygenase-2 in forebrain neurons is neuroprotective and dampens brain inflammation after status epilepticus. J. Neurosci. 2011, 31, 14850–14860. [Google Scholar] [CrossRef]
- Teismann, P.; Tieu, K.; Choi, D.-K.; Wu, D.-C.; Naini, A.; Hunot, S.; Vila, M.; Jackson-Lewis, V.; Przedborski, S. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc. Natl. Acad. Sci. USA 2003, 100, 5473–5478. [Google Scholar] [CrossRef]
- Rushendran, R.; Begum, R.F.; Singh, A.; Narayanan, P.L.; Vellapandian, C.; Prajapati, B.G.; Paul, P.K. Navigating neurological disorders: Harnessing the power of natural compounds for innovative therapeutic breakthroughs. EXCLI J. 2024, 23, 534. [Google Scholar]
- Li, L.; Wang, L.; Zhang, L. Therapeutic potential of natural compounds from herbs and nutraceuticals in alleviating neurological disorders: Targeting the Wnt signaling pathway. J. Agric. Food Chem. 2024, 72, 2411–2433. [Google Scholar] [CrossRef]
- Yousaf, M.; Chang, D.; Liu, Y.; Liu, T.; Zhou, X. Neuroprotection of cannabidiol, its synthetic derivatives and combination preparations against microglia-mediated neuroinflammation in neurological disorders. Molecules 2022, 27, 4961. [Google Scholar] [CrossRef] [PubMed]
- Puri, V.; Kanojia, N.; Sharma, A.; Huanbutta, K.; Dheer, D.; Sangnim, T. Natural product-based pharmacological studies for neurological disorders. Front. Pharmacol. 2022, 13, 1011740. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Rahman, M.M. Potential therapeutics against neurological disorders: Natural products-based drugs. Front. Pharmacol. 2022, 13, 950457. [Google Scholar] [CrossRef]
- Spagnuolo, C.; Napolitano, M.; Tedesco, I.; Moccia, S.; Milito, A.; Luigi Russo, G. Neuroprotective role of natural polyphenols. Curr. Top. Med. Chem. 2016, 16, 1943–1950. [Google Scholar] [CrossRef]
- Ribaudo, G.; Memo, M.; Gianoncelli, A. Multi-target natural and nature-inspired compounds against neurodegeneration: A focus on dual cholinesterase and phosphodiesterase inhibitors. Appl. Sci. 2021, 11, 5044. [Google Scholar] [CrossRef]
- Majumdar, S.; Gupta, S.; Prajapati, S.K.; Krishnamurthy, S. Neuro-nutraceutical potential of Asparagus racemosus: A review. Neurochem. Int. 2021, 145, 105013. [Google Scholar] [CrossRef]
- Bertelli, A.; Biagi, M.; Corsini, M.; Baini, G.; Cappellucci, G.; Miraldi, E. Polyphenols: From theory to practice. Foods 2021, 10, 2595. [Google Scholar] [CrossRef]
- Froldi, G.; Ragazzi, E. Selected plant-derived polyphenols as potential therapeutic agents for peripheral artery disease: Molecular mechanisms, efficacy and safety. Molecules 2022, 27, 7110. [Google Scholar] [CrossRef]
- Breuss, J.M.; Atanasov, A.G.; Uhrin, P. Resveratrol and its effects on the vascular system. Int. J. Mol. Sci. 2019, 20, 1523. [Google Scholar] [CrossRef]
- Singh, A.P.; Singh, R.; Verma, S.S.; Rai, V.; Kaschula, C.H.; Maiti, P.; Gupta, S.C. Health benefits of resveratrol: Evidence from clinical studies. Med. Res. Rev. 2019, 39, 1851–1891. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, B.S.; Ijaz, M.; Buabeid, M.; Kharaba, Z.J.; Yaseen, H.S.; Murtaza, G. Therapeutic effects and safe uses of plant-derived polyphenolic compounds in cardiovascular diseases: A review. Drug Des. Dev. Ther. 2021, 15, 4713–4732. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, I.; Wilairatana, P.; Saqib, F.; Nasir, B.; Wahid, M.; Latif, M.F.; Iqbal, A.; Naz, R.; Mubarak, M.S. Plant polyphenols and their potential benefits on cardiovascular health: A review. Molecules 2023, 28, 6403. [Google Scholar] [CrossRef]
- Teng, H.; Chen, L. Polyphenols and bioavailability: An update. Crit. Rev. Food Sci. Nutr. 2019, 59, 2040–2051. [Google Scholar] [CrossRef]
- Yang, B.; Dong, Y.; Wang, F.; Zhang, Y. Nanoformulations to enhance the bioavailability and physiological functions of polyphenols. Molecules 2020, 25, 4613. [Google Scholar] [CrossRef]
- Rangel-Huerta, O.D.; Pastor-Villaescusa, B.; Aguilera, C.M.; Gil, A. A systematic review of the efficacy of bioactive compounds in cardiovascular disease: Phenolic compounds. Nutrients 2015, 7, 5177–5216. [Google Scholar] [CrossRef]
- Muhaisen, H.M. Introduction and interpretation of flavonoids. Adv. Sci. Eng. Med. 2014, 6, 1235–1250. [Google Scholar] [CrossRef]
- Khajuria, R.; Singh, S.; Bahl, A. General introduction and sources of flavonoids. In Current Aspects of Flavonoids: Their Role in Cancer Treatment; Springer: Berlin/Heidelberg, Germany, 2019; pp. 1–7. [Google Scholar]
- Ribaudo, G.; Coghi, P.; Zanforlin, E.; Law, B.Y.K.; Wu, Y.Y.J.; Han, Y.; Qiu, A.C.; Qu, Y.Q.; Zagotto, G.; Wong, V.K.W. Semi-synthetic isoflavones as BACE-1 inhibitors against Alzheimer’s disease. Bioorg. Chem. 2019, 87, 474–483. [Google Scholar] [CrossRef]
- Singh, M.; Kaur, M.; Silakari, O. Flavones: An important scaffold for medicinal chemistry. Eur. J. Med. Chem. 2014, 84, 206–239. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, A.Y.; Simonyi, A.; Jensen, M.D.; Shelat, P.B.; Rottinghaus, G.E.; MacDonald, R.S.; Miller, D.K.; Lubahn, D.E.; Weisman, G.A. Neuroprotective mechanisms of curcumin against cerebral ischemia-induced neuronal apoptosis and behavioral deficits. J. Neurosci. Res. 2005, 82, 138–148. [Google Scholar] [CrossRef]
- Wang, Q.; Yu, S.; Simonyi, A.; Rottinghaus, G.; Sun, G.Y.; Sun, A.Y. Resveratrol protects against neurotoxicity induced by kainic acid. Neurochem. Res. 2004, 29, 2105–2112. [Google Scholar] [CrossRef] [PubMed]
- Simeonova, R.; Zheleva, D.; Valkova, I.; Stavrakov, G.; Philipova, I.; Atanasova, M.; Doytchinova, I. A novel galantamine-curcumin hybrid as a potential multi-target agent against neurodegenerative disorders. Molecules 2021, 26, 1865. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.K.; Lee, P.; Park, J.A.; Oh, H.R.; Lee, S.Y.; Park, J.-H.; Lee, E.H.; Ryu, J.H.; Lee, K.R.; Kim, S.Y. Apigenin inhibits the production of NO and PGE2 in microglia and inhibits neuronal cell death in a middle cerebral artery occlusion-induced focal ischemia mice model. Neurochem. Int. 2008, 52, 878–886. [Google Scholar] [CrossRef] [PubMed]
- Madeswaran, A.; Mohan, S. Neuroprotective effects of terpenoids against streptozotocin-nicotinamide-induced diabetic rats: An in silico, in vitro and in vivo study. Int. J. Biol. Macromol. 2023, 247, 125817. [Google Scholar] [CrossRef]
- Ozawa, Y.; Sasaki, M.; Takahashi, N.; Kamoshita, M.; Miyake, S.; Tsubota, K. Neuroprotective effects of lutein in the retina. Curr. Pharm. Des. 2012, 18, 51–56. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, M.; Chen, W.; Wang, K.; Wang, Y. Assessing neuroprotective efficacy of phytochemical saponin ruscogenin in both in vitro and in vivo model. Arab. J. Chem. 2023, 16, 104693. [Google Scholar] [CrossRef]
- Rizvi, S.I.; Mishra, N. Anti-oxidant effect of quercetin on type 2 diabetic erythrocytes. J. Food Biochem. 2009, 33, 404–415. [Google Scholar] [CrossRef]
- Pandey, K.B.; Rizvi, S.I. Protection of protein carbonyl formation by quercetin in erythrocytes subjected to oxidative stress. Med. Chem. Res. 2010, 19, 186–192. [Google Scholar] [CrossRef]
- Aguirre, L.; Arias, N.; Teresa Macarulla, M.; Gracia, A.; P Portillo, M. Beneficial effects of quercetin on obesity and diabetes. Open Nutraceuticals J. 2011, 4, 189–198. [Google Scholar]
- Dajas, F.; Abin-Carriquiry, J.A.; Arredondo, F.; Blasina, F.; Echeverry, C.; Martínez, M.; Rivera, F.; Vaamonde, L. Quercetin in brain diseases: Potential and limits. Neurochem. Int. 2015, 89, 140–148. [Google Scholar] [CrossRef]
- Chung, J.E.; Tan, S.; Gao, S.J.; Yongvongsoontorn, N.; Kim, S.H.; Lee, J.H.; Choi, H.S.; Yano, H.; Zhuo, L.; Kurisawa, M. Self-assembled micellar nanocomplexes comprising green tea catechin derivatives and protein drugs for cancer therapy. Nat. Nanotechnol. 2014, 9, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Hou, L.; Gu, S.; Zuo, X.; Meng, D.; Luo, M.; Zhang, X.; Huang, S.; Zhao, X. Molecular mechanism of epigallocatechin-3-gallate in human esophageal squamous cell carcinoma in vitro and in vivo. Oncol. Rep. 2015, 33, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.S.; Kang, S.U.; Park, J.K.; Kim, Y.E.; Kim, Y.S.; Baek, S.J.; Lee, S.-H.; Kim, C.-H. Anti-cancer effect of (-)-epigallocatechin-3-gallate (EGCG) in head and neck cancer through repression of transactivation and enhanced degradation of β-catenin. Phytomedicine 2016, 23, 1344–1355. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Chang, X.; Yan, R.; Rong, C.; Yang, C.; Cheng, S.; Gu, X.; Yao, H.; Hou, X.; Mo, Y. (−)-Epigallocatechin-3-gallate induces cancer cell apoptosis via acetylation of amyloid precursor protein. Med. Oncol. 2015, 32, 1–11. [Google Scholar] [CrossRef]
- Lin, R.-W.; Chen, C.-H.; Wang, Y.-H.; Ho, M.-L.; Hung, S.-H.; Chen, I.-S.; Wang, G.-J. (−)-Epigallocatechin gallate inhibition of osteoclastic differentiation via NF-κB. Biochem. Biophys. Res. Commun. 2009, 379, 1033–1037. [Google Scholar] [CrossRef]
- Oka, Y.; Iwai, S.; Amano, H.; Irie, Y.; Yatomi, K.; Ryu, K.; Yamada, S.; Inagaki, K.; Oguchi, K. Tea polyphenols inhibit rat osteoclast formation and differentiation. J. Pharmacol. Sci. 2012, 118, 55–64. [Google Scholar] [CrossRef]
- Shen, C.-L.; Yeh, J.K.; Stoecker, B.J.; Chyu, M.-C.; Wang, J.-S. Green tea polyphenols mitigate deterioration of bone microarchitecture in middle-aged female rats. Bone 2009, 44, 684–690. [Google Scholar] [CrossRef]
- Sigler, K.; Ruch, R.J. Enhancement of gap junctional intercellular communication in tumor promoter-treated cells by components of green tea. Cancer Lett. 1993, 69, 15–19. [Google Scholar] [CrossRef]
- Mukhtar, H.; Ahmad, N. Tea polyphenols: Prevention of cancer and optimizing health. Am. J. Clin. Nutr. 2000, 71, 1698S–1702S. [Google Scholar] [CrossRef]
- Davis, J.M.; Murphy, E.A.; Carmichael, M.D. Effects of the dietary flavonoid quercetin upon performance and health. Curr. Sports Med. Rep. 2009, 8, 206–213. [Google Scholar] [CrossRef]
- Hirpara, K.V.; Aggarwal, P.; Mukherjee, A.J.; Joshi, N.; Burman, A.C. Quercetin and its derivatives: Synthesis, pharmacological uses with special emphasis on anti-tumor properties and prodrug with enhanced bio-availability. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem.-Anti-Cancer Agents) 2009, 9, 138–161. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, S.; Meganathan, K.; Wagh, V.; Winkler, J.; Hescheler, J.; Sachinidis, A. Chemoprotective mechanism of the natural compounds, epigallocatechin-3-O-gallate, quercetin and curcumin against cancer and cardiovascular diseases. Curr. Med. Chem. 2009, 16, 1451–1462. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.-Y.; Wu, Y.-C.; Chung, J.-G.; Yang, J.-S.; Lu, H.-F.; Tsou, M.-F.; Wood, W.; Kuo, S.-J.; Chen, D.-R. Quercetin-induced apoptosis acts through mitochondrial-and caspase-3-dependent pathways in human breast cancer MDA-MB-231 cells. Hum. Exp. Toxicol. 2009, 28, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Granado-Serrano, A.B.; Martiín, M.A.; Bravo, L.; Goya, L.; Ramos, S. Quercetin induces apoptosis via caspase activation, regulation of Bcl-2, and inhibition of PI-3-kinase/Akt and ERK pathways in a human hepatoma cell line (HepG2). J. Nutr. 2006, 136, 2715–2721. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhao, X.-H.; Wang, Z.-J. Cytotoxicity of flavones and flavonols to a human esophageal squamous cell carcinoma cell line (KYSE-510) by induction of G2/M arrest and apoptosis. Toxicol. Vitr. 2009, 23, 797–807. [Google Scholar] [CrossRef]
- Kim, M.K.; Jung, H.S.; Yoon, C.S.; Ko, J.H.; Chun, H.J.; Kim, T.K.; Kwon, M.J.; Lee, S.H.; Koh, K.S.; Rhee, B.D. EGCG and quercetin protected INS-1 cells in oxidative stress via different mechanisms. Front. Biosci. (Elite Ed.) 2010, 2, 810–817. [Google Scholar] [CrossRef]
- Sun, Z.-J.; Chen, G.; Hu, X.; Zhang, W.; Liu, Y.; Zhu, L.-X.; Zhou, Q.; Zhao, Y.-F. Activation of PI3K/Akt/IKK-α/NF-κB signaling pathway is required for the apoptosis-evasion in human salivary adenoid cystic carcinoma: Its inhibition by quercetin. Apoptosis 2010, 15, 850–863. [Google Scholar] [CrossRef]
- Guo, Y.; Zhao, Y.; Nan, Y.; Wang, X.; Chen, Y.; Wang, S. (−)-Epigallocatechin-3-gallate ameliorates memory impairment and rescues the abnormal synaptic protein levels in the frontal cortex and hippocampus in a mouse model of Alzheimer’s disease. Neuroreport 2017, 28, 590–597. [Google Scholar] [CrossRef]
- Chang, X.; Rong, C.; Chen, Y.; Yang, C.; Hu, Q.; Mo, Y.; Zhang, C.; Gu, X.; Zhang, L.; He, W. (−)-Epigallocatechin-3-gallate attenuates cognitive deterioration in Alzheimer’s disease model mice by upregulating neprilysin expression. Exp. Cell Res. 2015, 334, 136–145. [Google Scholar] [CrossRef]
- Adhami, V.M.; Ahmad, N.; Mukhtar, H. Molecular targets for green tea in prostate cancer prevention. J. Nutr. 2003, 133, 2417S–2424S. [Google Scholar] [CrossRef]
- Ahmad, K.A.; Harris, N.H.; Johnson, A.D.; Lindvall, H.C.; Wang, G.; Ahmed, K. Protein kinase CK2 modulates apoptosis induced by resveratrol and epigallocatechin-3-gallate in prostate cancer cells. Mol. Cancer Ther. 2007, 6, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Gupta, S.; Mukhtar, H. Green tea polyphenol epigallocatechin-3-gallate differentially modulates nuclear factor κB in cancer cells versus normal cells. Arch. Biochem. Biophys. 2000, 376, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Kamble, S.; Deshkar, S.; Kothapalli, L.; Chitlange, S. Bioavailability of berberine: Challenges and solutions. İstanbul J. Pharm. 2021, 51, 141–153. [Google Scholar] [CrossRef]
- Behl, T.; Singh, S.; Sharma, N.; Zahoor, I.; Albarrati, A.; Albratty, M.; Meraya, A.M.; Najmi, A.; Bungau, S. Expatiating the pharmacological and nanotechnological aspects of the alkaloidal drug berberine: Current and future trends. Molecules 2022, 27, 3705. [Google Scholar] [CrossRef]
- Kumar, A.; Chopra, K.; Mukherjee, M.; Pottabathini, R.; Dhull, D.K. Current knowledge and pharmacological profile of berberine: An update. Eur. J. Pharmacol. 2015, 761, 288–297. [Google Scholar] [CrossRef]
- Jeong, H.W.; Hsu, K.C.; Lee, J.-W.; Ham, M.; Huh, J.Y.; Shin, H.J.; Kim, W.S.; Kim, J.B. Berberine suppresses proinflammatory responses through AMPK activation in macrophages. Am. J. Physiol.-Endocrinol. Metab. 2009, 296, E955–E964. [Google Scholar] [CrossRef]
- Sajeev, A.; Sailo, B.; Unnikrishnan, J.; Talukdar, A.; Alqahtani, M.S.; Abbas, M.; Alqahtani, A.; Sethi, G.; Kunnumakara, A.B. Unlocking the Potential of Berberine: Advancing Cancer Therapy through Chemosensitization and Combination Treatments. Cancer Lett. 2024, 597, 217019. [Google Scholar] [CrossRef]
- Chen, W.; Miao, Y.-Q.; Fan, D.-J.; Yang, S.-S.; Lin, X.; Meng, L.-K.; Tang, X. Bioavailability study of berberine and the enhancing effects of TPGS on intestinal absorption in rats. Aaps Pharmscitech 2011, 12, 705–711. [Google Scholar] [CrossRef]
- Liu, Y.-T.; Hao, H.-P.; Xie, H.-G.; Lai, L.; Wang, Q.; Liu, C.-X.; Wang, G.-J. Extensive intestinal first-pass elimination and predominant hepatic distribution of berberine explain its low plasma levels in rats. Drug Metab. Dispos. 2010, 38, 1779–1784. [Google Scholar] [CrossRef]
- Darbandi, A.; Elahi, Z.; Dadgar-Zankbar, L.; Ghasemi, F.; Kakavandi, N.; Jafari, S.; Darbandi, T.; Ghanavati, R. Application of microbial enzymes in medicine and industry: Current status and future perspectives. Future Microbiol. 2024, 19, 1419–1437. [Google Scholar] [CrossRef]
- West, S. Industrial Enzymology; Stockton Press: NewYork, NY, USA, 1996. [Google Scholar]
- Rao, M.B.; Tanksale, A.M.; Ghatge, M.S.; Deshpande, V.V. Molecular and biotechnological aspects of microbial proteases. Microbiol. Mol. Biol. Rev. 1998, 62, 597–635. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Barrett, A.J. Evolutionary families of peptidases. Biochem. J. 1993, 290, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Malve, H. Exploring the ocean for new drug developments: Marine pharmacology. J. Pharm. Bioallied Sci. 2016, 8, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Cheung, R.C.F.; Wong, J.H.; Pan, W.; Chan, Y.S.; Yin, C.; Dan, X.; Ng, T.B. Marine lectins and their medicinal applications. Appl. Microbiol. Biotechnol. 2015, 99, 3755–3773. [Google Scholar] [CrossRef]
- Morlighem, J.-É.R.; Radis-Baptista, G. The place for enzymes and biologically active peptides from marine organisms for application in industrial and pharmaceutical biotechnology. Curr. Protein Pept. Sci. 2019, 20, 334–355. [Google Scholar] [CrossRef]
- Bakunina, I.; Likhatskaya, G.; Slepchenko, L.; Balabanova, L.; Tekutyeva, L.; Son, O.; Shubina, L.; Makarieva, T. Effect of pentacyclic guanidine alkaloids from the sponge Monanchora pulchra on activity of α-glycosidases from marine bacteria. Mar. Drugs 2019, 17, 22. [Google Scholar] [CrossRef]
- Kobayashi, M.; Aoki, S.; Ohyabu, N.; Kurosu, M.; Wang, W.; Kitagawa, I. Arenastatin A, a potent cytotoxic depsipeptide from the Okinawan marine sponge Dysidea arenaria. Tetrahedron Lett. 1994, 35, 7969–7972. [Google Scholar] [CrossRef]
- Magarvey, N.A.; Beck, Z.Q.; Golakoti, T.; Ding, Y.; Huber, U.; Hemscheidt, T.K.; Abelson, D.; Moore, R.E.; Sherman, D.H. Biosynthetic characterization and chemoenzymatic assembly of the cryptophycins. Potent anticancer agents from Nostoc cyanobionts. ACS Chem. Biol. 2006, 1, 766–779. [Google Scholar] [CrossRef]
- Boyer, P.D.; Krebs, E.G. The enzymes; Academic Press: Cambridge, MA, USA, 1986. [Google Scholar]
- Hoffman, T. Food related enzymes. Adv. Chem. Ser. 1974, 136, 146–185. [Google Scholar]
- Famutimi, O.G.; Adebiyi, V.G.; Akinmolu, B.G.; Dada, O.V.; Adewale, I.O. Trypsin, chymotrypsin and elastase in health and disease. Future J. Pharm. Sci. 2024, 10, 126. [Google Scholar] [CrossRef]
- Shah, D.; Mital, K. The role of trypsin: Chymotrypsin in tissue repair. Adv. Ther. 2018, 35, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, D.; Rizvi, S.M.D.; Rehman, M.T.; Khan, M.S.; Bin Dukhyil, A.; AlAjmi, M.F.; Alshehri, B.M.; Banawas, S.; Zia, Q.; Alsaweed, M. Soyasapogenol-B as a potential multitarget therapeutic agent for neurodegenerative disorders: Molecular docking and dynamics study. Entropy 2022, 24, 593. [Google Scholar] [CrossRef] [PubMed]
- Dara, T.; Vatanara, A.; Meybodi, M.N.; Vakilinezhad, M.A.; Malvajerd, S.S.; Vakhshiteh, F.; Shamsian, A.; Sharifzadeh, M.; Kaghazian, H.; Mosaddegh, M.H. Erythropoietin-loaded solid lipid nanoparticles: Preparation, optimization, and in vivo evaluation. Colloids Surf. B Biointerfaces 2019, 178, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.; Khunt, D.; Bhatt, H.; Misra, M.; Padh, H. Application of quality by design approach for intranasal delivery of rivastigmine loaded solid lipid nanoparticles: Effect on formulation and characterization parameters. Eur. J. Pharm. Sci. 2015, 78, 54–66. [Google Scholar] [CrossRef]
- Poovaiah, N.; Davoudi, Z.; Peng, H.; Schlichtmann, B.; Mallapragada, S.; Narasimhan, B.; Wang, Q. Treatment of neurodegenerative disorders through the blood–brain barrier using nanocarriers. Nanoscale 2018, 10, 16962–16983. [Google Scholar] [CrossRef]
- Zhang, C.; Chen, J.; Feng, C.; Shao, X.; Liu, Q.; Zhang, Q.; Pang, Z.; Jiang, X. Intranasal nanoparticles of basic fibroblast growth factor for brain delivery to treat Alzheimer’s disease. Int. J. Pharm. 2014, 461, 192–202. [Google Scholar] [CrossRef]
- Riedel, G.; Kang, S.; Choi, D.; Platt, B. Scopolamine-induced deficits in social memory in mice: Reversal by donepezil. Behav. Brain Res. 2009, 204, 217–225. [Google Scholar] [CrossRef]
- Fowler, S.C.; Wang, G. Chronic haloperidol produces a time-and dose-related slowing of lick rhythm in rats: Implications for rodent models of tardive dyskinesia and neuroleptic-induced parkinsonism. Psychopharmacology 1998, 137, 50–60. [Google Scholar] [CrossRef]
- Izurieta-Sánchez, P.; Sarre, S.; Ebinger, G.; Michotte, Y. Effect of trihexyphenidyl, a non-selective antimuscarinic drug, on decarboxylation of L-dopa in hemi-Parkinson rats. Eur. J. Pharmacol. 1998, 353, 33–42. [Google Scholar] [CrossRef]
- Aracava, Y.; Albuquerque, E.X.; Pereira, E.F. (R, S)-trihexyphenidyl, acting via a muscarinic receptor-independent mechanism, inhibits hippocampal glutamatergic and GABAergic synaptic transmissions: Potential relevance for treatment of organophosphorus intoxication. Neuropharmacology 2023, 239, 109684. [Google Scholar] [CrossRef]
- May, P.C.; Willis, B.A.; Lowe, S.L.; Dean, R.A.; Monk, S.A.; Cocke, P.J.; Audia, J.E.; Boggs, L.N.; Borders, A.R.; Brier, R.A. The potent BACE1 inhibitor LY2886721 elicits robust central Aβ pharmacodynamic responses in mice, dogs, and humans. J. Neurosci. 2015, 35, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Hyman, D.M.; Davids, M.S.; Siu, L.L. Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet 2020, 395, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Chen, T.; Zhang, X.; Ma, X.L.; Shi, H.S. Small molecule inhibitors targeting the cancers. MedComm 2022, 3, e181. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Attwood, M.M.; Fabbro, D.; Sokolov, A.V.; Knapp, S.; Schiöth, H.B. Trends in kinase drug discovery: Targets, indications and inhibitor design. Nat. Rev. Drug Discov. 2021, 20, 839–861. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Gray, N.S. Kinase inhibitors: The road ahead. Nat. Rev. Drug Discov. 2018, 17, 353–377. [Google Scholar] [CrossRef]
- Case, L.B. Membranes regulate biomolecular condensates. Nat. Cell Biol. 2022, 24, 404–405. [Google Scholar] [CrossRef]
- Kurreck, A.; Stintzing, S.; Modest, D.P. Efficacy, molecular biology, quality of life, or economic aspects: What do we really focus on? J. Clin. Oncol. 2022, 40, 1260–1262. [Google Scholar] [CrossRef]
- Katoh, M. Fibroblast growth factor receptors as treatment targets in clinical oncology. Nat. Rev. Clin. Oncol. 2019, 16, 105–122. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, L.; Liu, B.; Zhang, Y.; Zhu, H.; Cui, J.; Sun, A.; Hu, Y.; Jin, J.; Jiang, H. Flumatinib versus imatinib for newly diagnosed chronic phase chronic myeloid leukemia: A phase III, randomized, open-label, multi-center FESTnd study. Clin. Cancer Res. 2021, 27, 70–77. [Google Scholar] [CrossRef]
- Kwak, J.-Y.; Kim, S.-H.; Oh, S.J.; Zang, D.Y.; Kim, H.; Kim, J.-A.; Do, Y.R.; Kim, H.J.; Park, J.S.; Choi, C.W. Phase III clinical trial (RERISE study) results of efficacy and safety of radotinib compared with imatinib in newly diagnosed chronic phase chronic myeloid leukemia. Clin. Cancer Res. 2017, 23, 7180–7188. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C.; Tan, D.S. Targeted therapies for lung cancer patients with oncogenic driver molecular alterations. J. Clin. Oncol. 2022, 40, 611–625. [Google Scholar] [CrossRef] [PubMed]
- George, S.; von Mehren, M.; Fletcher, J.A.; Sun, J.; Zhang, S.; Pritchard, J.R.; Hodgson, J.G.; Kerstein, D.; Rivera, V.M.; Haluska, F.G. Phase II study of ponatinib in advanced gastrointestinal stromal tumors: Efficacy, safety, and impact of liquid biopsy and other biomarkers. Clin. Cancer Res. 2022, 28, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.-Y.; Shen, L.; Kang, Y.-K.; Rutkowski, P.; Qin, S.; Nosov, D.; Wan, D.; Trent, J.; Srimuninnimit, V.; Pápai, Z. Nilotinib versus imatinib as first-line therapy for patients with unresectable or metastatic gastrointestinal stromal tumours (ENESTg1): A randomised phase 3 trial. Lancet Oncol. 2015, 16, 550–560. [Google Scholar] [CrossRef]
- Hodi, F.S.; Corless, C.L.; Giobbie-Hurder, A.; Fletcher, J.A.; Zhu, M.; Marino-Enriquez, A.; Friedlander, P.; Gonzalez, R.; Weber, J.S.; Gajewski, T.F. Imatinib for melanomas harboring mutationally activated or amplified KIT arising on mucosal, acral, and chronically sun-damaged skin. J. Clin. Oncol. 2013, 31, 3182–3190. [Google Scholar] [CrossRef]
- Guo, J.; Si, L.; Kong, Y.; Flaherty, K.T.; Xu, X.; Zhu, Y.; Corless, C.L.; Li, L.; Li, H.; Sheng, X. Phase II, open-label, single-arm trial of imatinib mesylate in patients with metastatic melanoma harboring c-Kit mutation or amplification. J. Clin. Oncol. 2011, 29, 2904–2909. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Antonescu, C.R.; Wolchok, J.D.; Chapman, P.B.; Roman, R.-A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J. KIT as a therapeutic target in metastatic melanoma. Jama 2011, 305, 2327–2334. [Google Scholar] [CrossRef]
- Le Cesne, A.; Blay, J.-Y.; Bui, B.N.; Bouché, O.; Adenis, A.; Domont, J.; Cioffi, A.; Ray-Coquard, I.; Lassau, N.; Bonvalot, S. Phase II study of oral masitinib mesilate in imatinib-naive patients with locally advanced or metastatic gastro-intestinal stromal tumour (GIST). Eur. J. Cancer 2010, 46, 1344–1351. [Google Scholar] [CrossRef]
- Kettle, J.G.; Anjum, R.; Barry, E.; Bhavsar, D.; Brown, C.; Boyd, S.; Campbell, A.; Goldberg, K.; Grondine, M.; Guichard, S. Discovery of N-(4-{[5-Fluoro-7-(2-methoxyethoxy) quinazolin-4-yl] amino} phenyl)-2-[4-(propan-2-yl)-1 H-1, 2, 3-triazol-1-yl] acetamide (AZD3229), a Potent Pan-KIT Mutant Inhibitor for the Treatment of Gastrointestinal Stromal Tumors. J. Med. Chem. 2018, 61, 8797–8810. [Google Scholar] [CrossRef]
- Wang, J.; Hu, C.; Zhang, L. Effect of Efavirenz on the Pharmacokinetics of SHR6390 in Healthy Volunteers. Drug Des. Dev. Ther. 2024, 18, 3113–3119. [Google Scholar] [CrossRef]
- Archer, S.; Ferreira, D.; Ferreira, A.T.; Ponte, S.; Caetano, C.; Salgado, M.; Lago, P.; Pedroto, I.; Sara, A.; Daniela, F. Tofacitinib-induced eosinophilia. Rev. Esp. De Enfermadades Dig. (REED) 2023, 115, 742–743. [Google Scholar]
- Xu, F.; Xiao, C.; Sun, W.; He, Y.; Chalela, R.; Masuda, K.; Ulivi, P.; Shen, K.; Shao, Q.; Xu, J. A lung adenocarcinoma patient with ROS1 fusion and NBN germline mutation achieves long progression-free survival from sintilimab combined with niraparib after failure of ROS1 inhibitors: A case report. Ann. Transl. Med. 2022, 10, 912. [Google Scholar] [CrossRef] [PubMed]
- Tasdemiroglu, Y.; Gourdie, R.G.; He, J.-Q. In vivo degradation forms, anti-degradation strategies, and clinical applications of therapeutic peptides in non-infectious chronic diseases. Eur. J. Pharmacol. 2022, 932, 175192. [Google Scholar] [CrossRef] [PubMed]
- Yeo, X.Y.; Cunliffe, G.; Ho, R.C.; Lee, S.S.; Jung, S. Potentials of neuropeptides as therapeutic agents for neurological diseases. Biomedicines 2022, 10, 343. [Google Scholar] [CrossRef]
- Brinker, T.; Spader, H. A translational view of peptide treatment of neurological disorders. Curr. Med. Chem. 2014, 21, 2583–2590. [Google Scholar] [CrossRef]
- Parrasia, S.; Szabò, I.; Zoratti, M.; Biasutto, L. Peptides as pharmacological carriers to the brain: Promises, shortcomings and challenges. Mol. Pharm. 2022, 19, 3700–3729. [Google Scholar] [CrossRef]
- Lalatsa, A.; Schatzlein, A.G.; Uchegbu, I.F. Strategies to deliver peptide drugs to the brain. Mol. Pharm. 2014, 11, 1081–1093. [Google Scholar] [CrossRef]
- Trapani, G.; Satriano, C.; La Mendola, D. Peptides and their metal complexes in neurodegenerative diseases: From structural studies to nanomedicine prospects. Curr. Med. Chem. 2018, 25, 715–747. [Google Scholar] [CrossRef]
- Tiwari, S.K.; Chaturvedi, R.K. Peptide therapeutics in neurodegenerative disorders. Curr. Med. Chem. 2014, 21, 2610–2631. [Google Scholar] [CrossRef]
- McGonigle, P. Peptide therapeutics for CNS indications. Biochem. Pharmacol. 2012, 83, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Paolino, D.; Sinha, P.; Fresta, M.; Ferrari, M. Drug delivery systems. Encycl. Med. Devices Instrum. 2006. [Google Scholar] [CrossRef]
- Siddiqui, I.A.; Adhami, V.M.; Bharali, D.J.; Hafeez, B.B.; Asim, M.; Khwaja, S.I.; Ahmad, N.; Cui, H.; Mousa, S.A.; Mukhtar, H. Introducing nanochemoprevention as a novel approach for cancer control: Proof of principle with green tea polyphenol epigallocatechin-3-gallate. Cancer Res. 2009, 69, 1712–1716. [Google Scholar] [CrossRef] [PubMed]
- Sonaje, K.; Italia, J.; Sharma, G.; Bhardwaj, V.; Tikoo, K.; Kumar, M.R. Development of biodegradable nanoparticles for oral delivery of ellagic acid and evaluation of their antioxidant efficacy against cyclosporine A-induced nephrotoxicity in rats. Pharm. Res. 2007, 24, 899–908. [Google Scholar] [CrossRef]
- Shutava, T.G.; Balkundi, S.S.; Lvov, Y.M. (−)-Epigallocatechin gallate/gelatin layer-by-layer assembled films and microcapsules. J. Colloid Interface Sci. 2009, 330, 276–283. [Google Scholar] [CrossRef]
- Fang, J.-Y.; Hung, C.-F.; Hwang, T.-L.; Huang, Y.-L. Physicochemical characteristics and in vivo deposition of liposome-encapsulated tea catechins by topical and intratumor administrations. J. Drug Target. 2005, 13, 19–27. [Google Scholar] [CrossRef]
- Narayanan, N.K.; Nargi, D.; Randolph, C.; Narayanan, B.A. Liposome encapsulation of curcumin and resveratrol in combination reduces prostate cancer incidence in PTEN knockout mice. Int. J. Cancer 2009, 125, 1–8. [Google Scholar] [CrossRef]
- Bansal, S.S.; Vadhanam, M.V.; Gupta, R.C. Development and in vitro-in vivo evaluation of polymeric implants for continuous systemic delivery of curcumin. Pharm. Res. 2011, 28, 1121–1130. [Google Scholar] [CrossRef]
- Sakaeda, T.; Okamura, N.; Nagata, S.; Yagami, T.; Horinouchi, M.; Okumura, K.; Yamashita, F.; Hashida, M. Molecular and pharmacokinetic properties of 222 commercially available oral drugs in humans. Biol. Pharm. Bull. 2001, 24, 935–940. [Google Scholar] [CrossRef]
- Wenlock, M.C.; Austin, R.P.; Barton, P.; Davis, A.M.; Leeson, P.D. A comparison of physiochemical property profiles of development and marketed oral drugs. J. Med. Chem. 2003, 46, 1250–1256. [Google Scholar] [CrossRef]
- Vieth, M.; Siegel, M.G.; Higgs, R.E.; Watson, I.A.; Robertson, D.H.; Savin, K.A.; Durst, G.L.; Hipskind, P.A. Characteristic physical properties and structural fragments of marketed oral drugs. J. Med. Chem. 2004, 47, 224–232. [Google Scholar] [CrossRef]
- Gomez-Orellana, I. Strategies to improve oral drug bioavailability. Expert Opin. Drug Deliv. 2005, 2, 419–433. [Google Scholar] [CrossRef] [PubMed]
- Ettmayer, P.; Amidon, G.L.; Clement, B.; Testa, B. Lessons learned from marketed and investigational prodrugs. J. Med. Chem. 2004, 47, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Y.; Zhao, Y.-G.; Zhang, Y. Enhancing Drug Solubility, Bioavailability, and Targeted Therapeutic Applications through Magnetic Nanoparticles. Molecules 2024, 29, 4854. [Google Scholar] [CrossRef] [PubMed]
- Bourang, S.; Asadian, S.; Noruzpour, M.; Mansuryar, A.; Azizi, S.; Ebrahimi, H.A.; Amani Hooshyar, V. PLA-HA/Fe3O4 magnetic nanoparticles loaded with curcumin: Physicochemical characterization and toxicity evaluation in HCT116 colorectal cancer cells. Discov. Appl. Sci. 2024, 6, 186. [Google Scholar] [CrossRef]
- Sisubalan, N.; Shalini, R.; Ramya, S.; Sivamaruthi, B.S.; Chaiyasut, C. Recent advances in nanomaterials for neural applications: Opportunities and challenges. Nanomedicine 2023, 18, 1979–1994. [Google Scholar] [CrossRef]
- Liu, C.; Helsper, S.; Marzano, M.; Chen, X.; Muok, L.; Esmonde, C.; Zeng, C.; Sun, L.; Grant, S.C.; Li, Y. Human forebrain organoid-derived extracellular vesicle labeling with iron oxides for in vitro magnetic resonance imaging. Biomedicines 2022, 10, 3060. [Google Scholar] [CrossRef]
- Dahan, A.; Miller, J.M.; Amidon, G.L. Prediction of solubility and permeability class membership: Provisional BCS classification of the world’s top oral drugs. AAPS J. 2009, 11, 740–746. [Google Scholar] [CrossRef]
- Marschütz, M.K.; Bernkop-Schnürch, A. Oral peptide drug delivery: Polymer–inhibitor conjugates protecting insulin from enzymatic degradation in vitro. Biomaterials 2000, 21, 1499–1507. [Google Scholar] [CrossRef]
- Patel, A.; Cholkar, K.; Mitra, A.K. Recent developments in protein and peptide parenteral delivery approaches. Ther. Deliv. 2014, 5, 337–365. [Google Scholar] [CrossRef]
- Raj, G.M.; Raveendran, R. Introduction to Basics of Pharmacology and Toxicology: Volume 1: General and Molecular Pharmacology: Principles of Drug Action; Springer Nature: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- McDonald, T.A.; Zepeda, M.L.; Tomlinson, M.J.; Bee, W.H.; Ivens, I.A. Subcutaneous administration of biotherapeutics: Current experience in animal models. Curr. Opin. Mol. Ther. 2010, 12, 461–470. [Google Scholar]
- Prausnitz, M.R.; Langer, R. Transdermal drug delivery. Nat. Biotechnol. 2008, 26, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, N.; Andrade, F.; Segovia, N.; Ferrer-Tasies, L.; Sala, S.; Veciana, J.; Ventosa, N. Lipid-based nanovesicles for nanomedicine. Chem. Soc. Rev. 2016, 45, 6520–6545. [Google Scholar] [CrossRef] [PubMed]
- Ramadon, D.; McCrudden, M.T.; Courtenay, A.J.; Donnelly, R.F. Enhancement strategies for transdermal drug delivery systems: Current trends and applications. Drug Deliv. Transl. Res. 2021, 12, 758–791. [Google Scholar] [CrossRef]
- Martínez, A. Synthesis, in vitro activity, and molecular docking of caffeic acid and resveratrol derivatives against Alzheimer’s disease-related enzymes. Med. Chem. Res. 2024, 33, 1681–1697. [Google Scholar] [CrossRef]
- Yetisgin, A.A.; Cetinel, S.; Zuvin, M.; Kosar, A.; Kutlu, O. Therapeutic nanoparticles and their targeted delivery applications. Molecules 2020, 25, 2193. [Google Scholar] [CrossRef]
- Wakaskar, R.R. Promising effects of nanomedicine in cancer drug delivery. J. Drug Target. 2018, 26, 319–324. [Google Scholar] [CrossRef]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Afzal, O.; Altamimi, A.S.; Nadeem, M.S.; Alzarea, S.I.; Almalki, W.H.; Tariq, A.; Mubeen, B.; Murtaza, B.N.; Iftikhar, S.; Riaz, N. Nanoparticles in drug delivery: From history to therapeutic applications. Nanomaterials 2022, 12, 4494. [Google Scholar] [CrossRef]
- Khalid, M.; El-Sawy, H.S. Polymeric nanoparticles: Promising platform for drug delivery. Int. J. Pharm. 2017, 528, 675–691. [Google Scholar]
- Ramzy, L.; Nasr, M.; Metwally, A.A.; Awad, G.A. Cancer nanotheranostics: A review of the role of conjugated ligands for overexpressed receptors. Eur. J. Pharm. Sci. 2017, 104, 273–292. [Google Scholar] [CrossRef]
- Yao, Y.; Zhou, Y.; Liu, L.; Xu, Y.; Chen, Q.; Wang, Y.; Wu, S.; Deng, Y.; Zhang, J.; Shao, A. Nanoparticle-based drug delivery in cancer therapy and its role in overcoming drug resistance. Front. Mol. Biosci. 2020, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Waheed, S.; Li, Z.; Zhang, F.; Chiarini, A.; Armato, U.; Wu, J. Engineering nano-drug biointerface to overcome biological barriers toward precision drug delivery. J. Nanobiotechnol. 2022, 20, 395. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, H.; Nakara, A.; Shanmugam, V.K. Anti-inflammatory mechanism of various metal and metal oxide nanoparticles synthesized using plant extracts: A review. Biomed. Pharmacother. 2019, 109, 2561–2572. [Google Scholar] [CrossRef]
- Martano, S.; De Matteis, V.; Cascione, M.; Rinaldi, R. Inorganic nanomaterials versus polymer-based nanoparticles for overcoming neurodegeneration. Nanomaterials 2022, 12, 2337. [Google Scholar] [CrossRef]
- Ali, T.; Kim, M.J.; Rehman, S.U.; Ahmad, A.; Kim, M.O. Anthocyanin-loaded PEG-gold nanoparticles enhanced the neuroprotection of anthocyanins in an Aβ 1–42 mouse model of Alzheimer’s disease. Mol. Neurobiol. 2017, 54, 6490–6506. [Google Scholar] [CrossRef]
- Anadozie, S.O.; Effiom, D.O.; Adewale, O.B.; Jude, J.; Zosela, I.; Akawa, O.B.; Olayinka, J.N.; Roux, S. Hibiscus sabdariffa synthesized gold nanoparticles ameliorate aluminum chloride induced memory deficits through inhibition of COX-2/BACE-1 mRNA expression in rats. Arab. J. Chem. 2023, 16, 104604. [Google Scholar] [CrossRef]
- Li, L.; Tan, L.; Zhang, Q.; Cheng, Y.; Liu, Y.; Li, R.; Hou, S. Nose-to-brain delivery of self-assembled curcumin-lactoferrin nanoparticles: Characterization, neuroprotective effect and in vivo pharmacokinetic study. Front. Bioeng. Biotechnol. 2023, 11, 1168408. [Google Scholar] [CrossRef]
- Yusuf, M. Formulation and cognitive evaluation of self-assembled phosphatidylserine-chitosan nanoparticles of lycopene, an innovative technique to lessen STZ-induced oxidative stress: A vital persuader of major neurological diseases. J. Drug Deliv. Sci. Technol. 2021, 63, 102534. [Google Scholar] [CrossRef]
- Srivastav, S.; Anand, B.G.; Fatima, M.; Prajapati, K.P.; Yadav, S.S.; Kar, K.; Mondal, A.C. Piperine-coated gold nanoparticles alleviate paraquat-induced neurotoxicity in Drosophila melanogaster. ACS Chem. Neurosci. 2020, 11, 3772–3785. [Google Scholar] [CrossRef]
- Xue, J.; Liu, T.; Liu, Y.; Jiang, Y.; Seshadri, V.D.D.; Mohan, S.K.; Ling, L. Neuroprotective effect of biosynthesised gold nanoparticles synthesised from root extract of Paeonia moutan against Parkinson disease–In vitro & In vivo model. J. Photochem. Photobiol. B Biol. 2019, 200, 111635. [Google Scholar]
- Ling, L.; Jiang, Y.; Liu, Y.; Li, H.; Bari, A.; Ullah, R.; Xue, J. Role of gold nanoparticle from Cinnamomum verum against 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) induced mice model. J. Photochem. Photobiol. B Biol. 2019, 201, 111657. [Google Scholar] [CrossRef] [PubMed]
- Fakhri, S.; Abdian, S.; Zarneshan, S.N.; Moradi, S.Z.; Farzaei, M.H.; Abdollahi, M. Nanoparticles in combating neuronal dysregulated signaling pathways: Recent approaches to the nanoformulations of phytochemicals and synthetic drugs against neurodegenerative diseases. Int. J. Nanomed. 2022, 17, 299–331. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Bhardwaj, K.; Nepovimova, E.; Kuča, K.; Singh Dhanjal, D.; Bhardwaj, S.; Bhatia, S.K.; Verma, R.; Kumar, D. Antioxidant functionalized nanoparticles: A combat against oxidative stress. Nanomaterials 2020, 10, 1334. [Google Scholar] [CrossRef]
- Eriksson, P.; Tal, A.A.; Skallberg, A.; Brommesson, C.; Hu, Z.; Boyd, R.D.; Olovsson, W.; Fairley, N.; Abrikosov, I.A.; Zhang, X. Cerium oxide nanoparticles with antioxidant capabilities and gadolinium integration for MRI contrast enhancement. Sci. Rep. 2018, 8, 6999. [Google Scholar] [CrossRef]
- Gil, D.; Rodriguez, J.; Ward, B.; Vertegel, A.; Ivanov, V.; Reukov, V. Antioxidant activity of SOD and catalase conjugated with nanocrystalline ceria. Bioengineering 2017, 4, 18. [Google Scholar] [CrossRef]
- Kim, M.-H.; Jeong, H.-J. Zinc oxide nanoparticles suppress LPS-induced NF-κB activation by inducing A20, a negative regulator of NF-κB, in RAW 264.7 macrophages. J. Nanosci. Nanotechnol. 2015, 15, 6509–6515. [Google Scholar] [CrossRef]
- Kim, M.-H.; Seo, J.-H.; Kim, H.-M.; Jeong, H.-J. Aluminum-doped zinc oxide nanoparticles attenuate the TSLP levels via suppressing caspase-1 in activated mast cells. J. Biomater. Appl. 2016, 30, 1407–1416. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-brain barrier: From physiology to disease and back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Wohlfart, S.; Gelperina, S.; Kreuter, J. Transport of drugs across the blood–brain barrier by nanoparticles. J. Control. Release 2012, 161, 264–273. [Google Scholar] [CrossRef]
- Moura, R.P.; Martins, C.; Pinto, S.; Sousa, F.; Sarmento, B. Blood-brain barrier receptors and transporters: An insight on their function and how to exploit them through nanotechnology. Expert Opin. Drug Deliv. 2019, 16, 271–285. [Google Scholar] [CrossRef]
- Tanjore, H.; Kalluri, R. The role of type IV collagen and basement membranes in cancer progression and metastasis. Am. J. Pathol. 2006, 168, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Aumailley, M.; Smyth, N. The role of laminins in basement membrane function. J. Anat. 1998, 193, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Yi, C.-X. Cell type-targeting nanoparticles in treating central nervous system diseases: Challenges and hopes. Nanotechnol. Rev. 2023, 12, 20230158. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Molecules | Sources | Chemical Structure | Active Concentration | Target Enzymes | Name of ND | Mechanism of Action | In Vivo Test | References |
|---|---|---|---|---|---|---|---|---|
| Curcumin | Turmeric (Curcuma longa) | 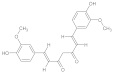 | 0 mg/kg body weight (i.p.) or 2.0 g/kg diet | Caspase-3 and caspase-9 | Cerebral ischemia/reperfusion injury | Reduces oxidative stress, decreases lipid peroxidation, restores mitochondrial function, and inhibits apoptosis by reducing caspase-3 activation | Tested in Mongolian gerbils (global cerebral ischemia model) | [173] |
| Resveratrol | Grapes, red wine, and certain berries | 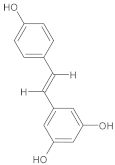 | 30 mg/kg/day | Calcium-dependent enzymes, xanthine oxidase, nitric oxide synthase (NOS), phospholipase A2 (PLA2) | Kainic acid-induced neurotoxicity (excitotoxicity in the hippocampus) | Acts as a free radical scavenger; reduces oxidative stress, neuronal death, and glial activation caused by kainic acid | Tested in adult Sprague-Dawley male rats treated with kainic acid (8 mg/kg) for 5 days | [174] |
| Galanthamine (GAL) | Amaryllidaceae plants |  | 3 mg/kg (1/10 of LD50) | Acetylcholinesterase (AChE) | Alzheimer’s disease (AD) | AChE inhibition, antioxidant activity | Male and female ICR mice (6 weeks old, 25–35 g, behavioral analysis, hematological and biochemical tests | [175] |
| 4b (Hybrid) | GAL and CU hybrid | [Structure of 4b] | 5 mg/kg (1/10 of LD50) | Acetylcholinesterase (AChE) | Alzheimer’s disease (AD) | AChE inhibition, antioxidant, BBB permeable | Male and female ICR mice (6 weeks old, 25–35 g, acute toxicity, short-term toxicity, behavioral studies | [175] |
| Apigenin | Found in plants such as parsley, celery, and chamomile | 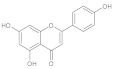 | 20 mg/kg (oral administration) | iNOS, COX-2, MAPK (p38, JNK) | Neurodegenerative diseases (Alzheimer’s disease, Parkinson’s disease) | Anti-inflammatory effect, inhibition of iNOS expression, reduction of NO production, modulation of microglial activation, inhibition of MAPK signaling pathways (p38, JNK) | MCAO mice model, TTC staining, OX-42 immunohistochemistry | [176] |
| α-Bisabolol | Found in the essential oil of Matricaria chamomilla (chamomile) | 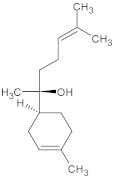 | 100 mg/kg (low dose), 200 mg/kg (high dose) | Acetylcholinesterase (AChE), α-Amylase | Diabetic Alzheimer’s disease (Type 2 Diabetes with cognitive decline) | Neuroprotective, improves spatial memory, reduces blood glucose, inhibits AChE, antioxidant activity, and anti-inflammatory | Morris water maze, open field test, acetylcholinesterase activity, and enzymatic antioxidants in rat brain | [177] |
| Lycopene | Found in tomatoes, watermelon, and red fruits | 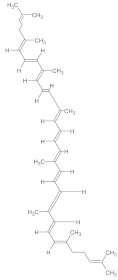 | Varies (commonly 5–10 mg/kg) | NF-κB, caspase-3, inflammatory cytokines (TNF-α, IL-1β, TGF-β) | Alzheimer’s disease (AD), neuroinflammation | Reduces pro-inflammatory cytokines (TNF-α, IL-1β, TGF-β), inhibits NF-κB and caspase-3, prevents Aβ-induced mitochondrial dysfunction, improves learning and memory functions | ICV Aβ1–42-induced rats (Morris water maze, elevated plus maze) | [178] |
| Ruscogenin | Derived from Ophiopogon japonicus (a traditional Chinese medicinal plant) |  | 10–20 mg/kg | Inflammatory cytokines, oxidative stress markers | Parkinson’s disease (PD) | Reduces oxidative stress and inflammation, improves motor function, protects dopaminergic neurons in the substantia nigra | MPTP-induced PD mice (footprint test, grip strength, rota rod, histopathology) | [179] |
| Name of Molecules | Chemical Structure | Active Concentration | Target Enzymes | Name of ND | Mechanism of Action | In Vivo Test | References |
|---|---|---|---|---|---|---|---|
| Diazepam |  | Dose range: 0.1–1 mg/kg. | Targets GABA-A receptors (enhances inhibitory neurotransmission). | Alzheimer’s disease (AD). | Enhances GABAergic neurotransmission, reducing anxiety but impairing cognitive processes like memory due to over-inhibition. | Animal Model: C57Bl/6 female mice. | [232] |
| Scopolamine | 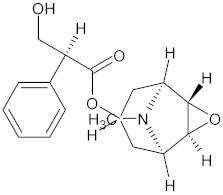 | Dose range: 0.1–1 mg/kg. | Targets muscarinic acetylcholine receptors (blocks cholinergic signaling). | Schizophrenia and autism. | Antagonizes muscarinic acetylcholine receptors, disrupting cholinergic signaling critical for memory formation. | Animal Model: C57Bl/6 female mice. | [232] |
| Donepezil |  | Dose: 1 mg/kg. | Targets acetylcholinesterase (inhibits breakdown of acetylcholine, enhancing cholinergic signaling). | Altered sociability is linked to neurotransmitter imbalances. | Inhibits acetylcholinesterase, increasing synaptic acetylcholine levels, thereby reversing memory deficits caused by cholinergic antagonism. | Animal Model: C57Bl/6 female mice. | [232] |
| Haloperidol |  | Doses: 0.06, 0.12, 0.24 mg/kg (chronic treatment for 102 days). | Target: Dopamine D2 receptors (antagonist). | Parkinson’s disease (PD): Parkinsonian-like motor impairments (e.g., bradykinesia). | Chronic D2 receptor blockade reduces dopamine signaling in the basal ganglia, leading to motor impairments characteristic of Parkinsonian effects and tardive dyskinesia. | Rats. Duration: 102 days (chronic haloperidol treatment), with drug testing conducted every second or third day. | [233] |
| Trihexyphenidyl |  | Doses: 0.15–1.0 mg/kg. | Target: Muscarinic acetylcholine receptors (antagonist). | Effect: Reduced some of haloperidol’s effects on licking behavior, Parkinson’s disease. | Mechanism: Modulate serotonin receptor activity to influence motor and behavioral outcomes linked to haloperidol-induced effects. | Rats. Duration: 102 days (chronic haloperidol treatment), with drug testing conducted every second or third day. | [233] |
| Trihexyphenidyl |  | 1 mM (local via microdialysis), 1.5 mg/kg (systemic, i.p.). L-dopa: 2 μM for 20 min (local application). | Trihexyphenidyl: Muscarinic acetylcholine receptors (non-selective antagonist). - L-dopa: Aromatic L-amino acid decarboxylase (AADC) for dopamine synthesis. | Parkinson’s disease (PD). | Trihexyphenidyl: Blocks muscarinic acetylcholine receptors, reducing cholinergic overactivity. Attenuates L-dopa-induced dopamine release systemically in intact striatum. - L-dopa: Precursor to dopamine, converted by AADC in dopaminergic neurons. | Hemi-Parkinson rat model with unilateral 6-hydroxydopamine lesion of nigrostriatal pathway. | [234] |
| (R,S)-trihexyphenidyl | 1–30 μM. | THP more effectively counters cholinergic crisis, seizures, and neuropathology triggered by OP-induced AChE inhibition. - THP blocks mAChRs and NMDARs in the brain, and inhibits α7 nAChRs. | Parkinson’s disease (PD). | - Suppresses glutamatergic synaptic transmission via an action potential-dependent mechanism. - Independent of NMDAR, mAChR, and α7 nAChR inhibition. | The study used primary hippocampal cultures derived from rats to investigate the effects of (R,S)-trihexyphenidyl (THP) on synaptic transmission. | [235] | |
| LY2886721 |  | EC50: ~10 nM in PDAPP neuronal cultures, 18.5–19.7 nM in HEK293Swe cells. | BACE1 | Alzheimer’s disease (AD). | Inhibition of BACE1 to reduce Aβ production and lower amyloid levels. | PDAPP transgenic mice (APPV717F mutation). | [236] |
| Nanomaterials | Natural Products Used in the Synthesis of NPs | Active Concentration | Target Enzymes | Name of ND | Mechanism of Action | In Vivo Test | References |
|---|---|---|---|---|---|---|---|
| Selenium NPs (SeNPs) | Aloe vera, Prunus amygdalus, Vitis vinifera, Allium sativum, Dillenia indica, Roselle plant, Cinnamomum zeylanicum, fresh citrus, lemons | 5–10 μg (non-toxic), 20–25 μg (toxic effects) | Superoxide dismutase (SOD), Glutathione peroxidase (GSH-PX | Parkinson’s disease (PD), Alzheimer’s disease (AD) | Antioxidant activity, ROS reduction, inhibition of tau hyperphosphorylation, interaction with Aβ aggregates | MPTP-induced PD model | [300] |
| PEG-AuNPs (polyethylene glycol–gold nanoparticles) | Anthocyanins | 12 μg/g/day for 14 days | p-PI3K, p-Akt, p-GSK3β | Alzheimer’s disease | Prevented tau hyperphosphorylation, protected synaptic proteins, inhibited apoptosis, and reduced neurodegeneration | Aβ1–42 mouse model of AD | [301] |
| HS-AuNPs (Hibiscus sabdariffa–synthesized gold nanoparticles) | Hibiscus sabdariffa extract | 5 mg/kg and 10 mg/kg b.w. for 14 days | Acetylcholinesterase, monoamine oxidase, adenosine deaminase, COX-2, BACE-1 | Alzheimer’s disease | Reduced oxidative stress, improved antioxidant enzyme activities (SOD, GPx, GSH), decreased inflammatory markers (COX-2, BACE-1), and ameliorated memory and learning deficit | AlCl3-induced AD model in Wistar rats | [302] |
| CUR-LF NPs (Curcumin–Lactoferrin Nanoparticles) | Curcumin (CUR), Lactoferrin (LF) | 10 mg/kg | Aβ25–35-induced oxidative stress | Alzheimer’s disease | Antioxidant, anti-inflammatory, and neuroprotective effects. CUR-LF NPs protect neurons from oxidative damage and apoptosis and improve bioavailability | Rats were administered CUR-LF NPs via the IN and IV routes. Pharmacokinetic studies were performed to evaluate brain accumulation | [303] |
| P-80-LYC-PSCNP (Polysorbate-80– Lycopene– Phosphatidylserine–Chitosan Nanoparticles) | Lycopene (LYC), Polysorbate-80 (P-80), Phosphatidylserine, Chitosan | 5 mg/kg LYC-equivalent dose | Catalase (CAT), Superoxide dismutase (SOD), Glutathione peroxidase (GPx) | Neurodegenerative diseases (oxidative stress-related | Antioxidant properties through improved enzymatic activity, alleviates oxidative stress by delivering Lycopene across the blood–brain barrier (BBB), reducing cognitive and behavioral impairments | Streptozotocin-induced oxidative stress model (SOSM), behavioral despair test, biochemical assays for enzymatic activity | [304] |
| AuNPs-piperine | Piperine | 10 μM | Likely oxidative stress-related enzymes (e.g., SOD, CAT) | Parkinson’s disease (PD) | Neuroprotection against PQ-induced oxidative stress | in Drosophila | [305] |
| PM-AuNPs | Paeonia moutan root extract | 20 mg/kg (in vivo) | iNOS, COX-2, Tyrosine hydroxylase (TH) | Parkinson’s disease (PD) | Reduces neuroinflammation and improves motor coordination | In Parkinson-induced C57BL/6 mice | [306] |
| Gold Nanoparticles (AuNPs) | Cinnamomum verum (Cinnamon) extract | 5 mg/kg, 10 mg/kg | TLR2, TLR4, NF-κB | Parkinson’s disease (PD) | Anti-inflammatory, ROS scavenging, neuroprotection | (MPTP-induced PD model in rats) | [307] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, A.A.; Khan, F.; Song, M. Alleviation of Neurological Disorders by Targeting Neurodegenerative-Associated Enzymes: Natural and Synthetic Molecules. Int. J. Mol. Sci. 2025, 26, 4707. https://doi.org/10.3390/ijms26104707
Singh AA, Khan F, Song M. Alleviation of Neurological Disorders by Targeting Neurodegenerative-Associated Enzymes: Natural and Synthetic Molecules. International Journal of Molecular Sciences. 2025; 26(10):4707. https://doi.org/10.3390/ijms26104707
Chicago/Turabian StyleSingh, Alka Ashok, Fazlurrahman Khan, and Minseok Song. 2025. "Alleviation of Neurological Disorders by Targeting Neurodegenerative-Associated Enzymes: Natural and Synthetic Molecules" International Journal of Molecular Sciences 26, no. 10: 4707. https://doi.org/10.3390/ijms26104707
APA StyleSingh, A. A., Khan, F., & Song, M. (2025). Alleviation of Neurological Disorders by Targeting Neurodegenerative-Associated Enzymes: Natural and Synthetic Molecules. International Journal of Molecular Sciences, 26(10), 4707. https://doi.org/10.3390/ijms26104707