

P-Glycoprotein as a Therapeutic Target in Hematological Malignancies: A Challenge to Overcome

Abstract
1. Introduction
2. P-Glycoprotein Generalities
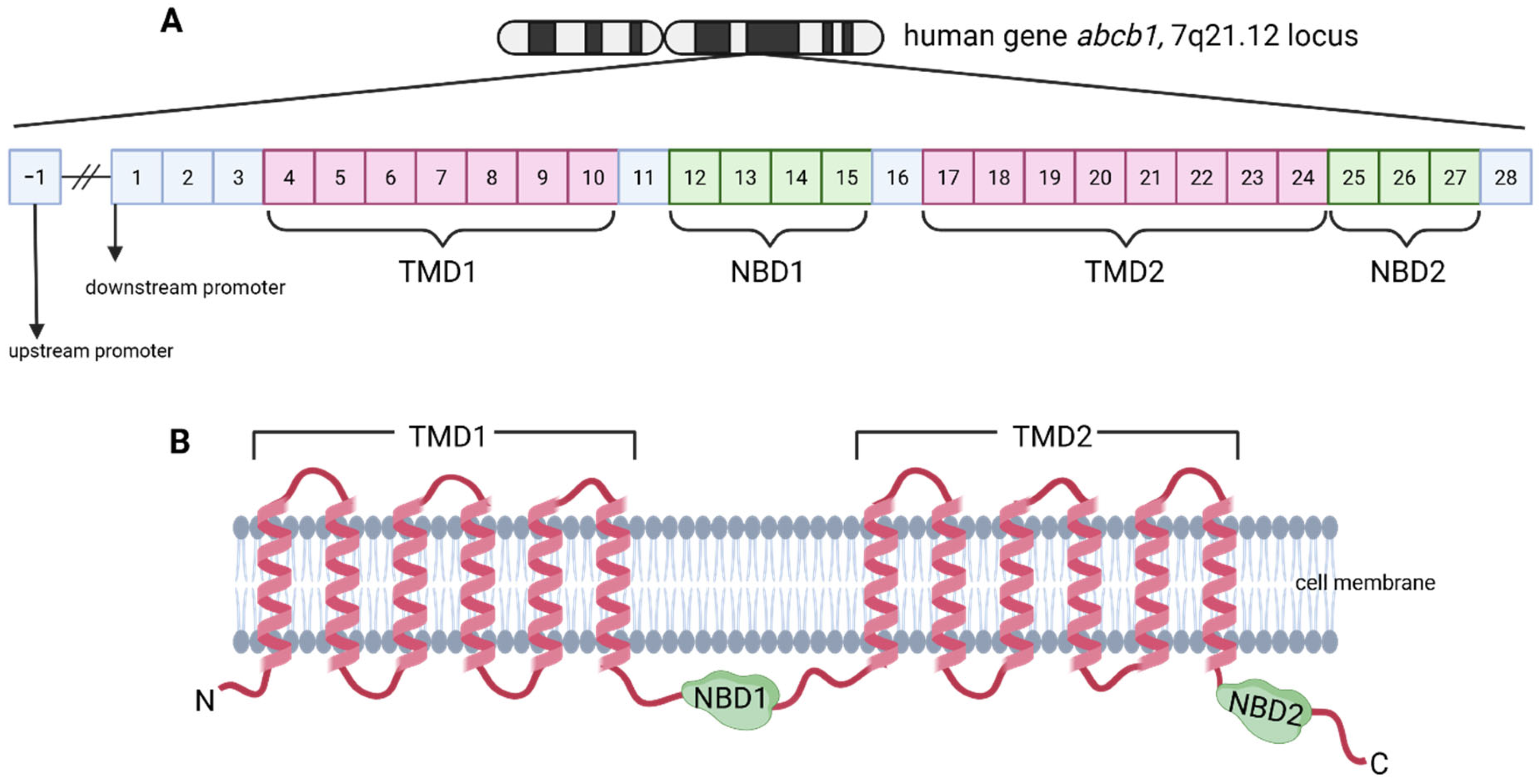
- The Main Binding Cavity (MBC), situated at the apex of P-gp, is a critical region for substrate binding prior to their efflux from the cell. This cavity plays a central role in the interaction with a diverse array of compounds, encompassing both substrates and non-substrates of P-gp. Specifically, the MBC exhibits a preferential affinity for substrates, facilitating their subsequent transportation to the extracellular environment. Compounds classified as strong substrates, defined by an efflux ratio (ER) exceeding 2, demonstrate a particularly high affinity for this binding site [18].
- Distinct from the MBC, located at the apex of P-gp, Other Binding Sites (OBS) are situated within the middle region of the protein. These sites are primarily for interacting with non-substrates, compounds that are not effluxed by P-gp, thus facilitating their entry into the cell. Non-substrates, characterized by an ER of less than 1, preferentially bind to the OBS. Compounds exhibiting intermediate ER values (between 1 and 2) demonstrate a more balanced binding affinity, distributing relatively equally between the MBC and the OBS [18].
3. ABCB1 Gene Expression
3.1. ABCB1 Regulation
3.1.1. Circadian Regulation
3.1.2. Post-Transcriptional Regulation
3.1.3. Epigenetic Regulation
3.1.4. Signaling Pathways
3.2. MDR1 Polymorphisms
4. Importance of P-Glycoprotein in Hematological Malignancies
4.1. Multiple Myeloma
4.2. Leukemias
4.3. Lymphomas
5. P-Glycoprotein Expression in Immune Cell Subsets
6. Strategies to Modulate P-gp Function
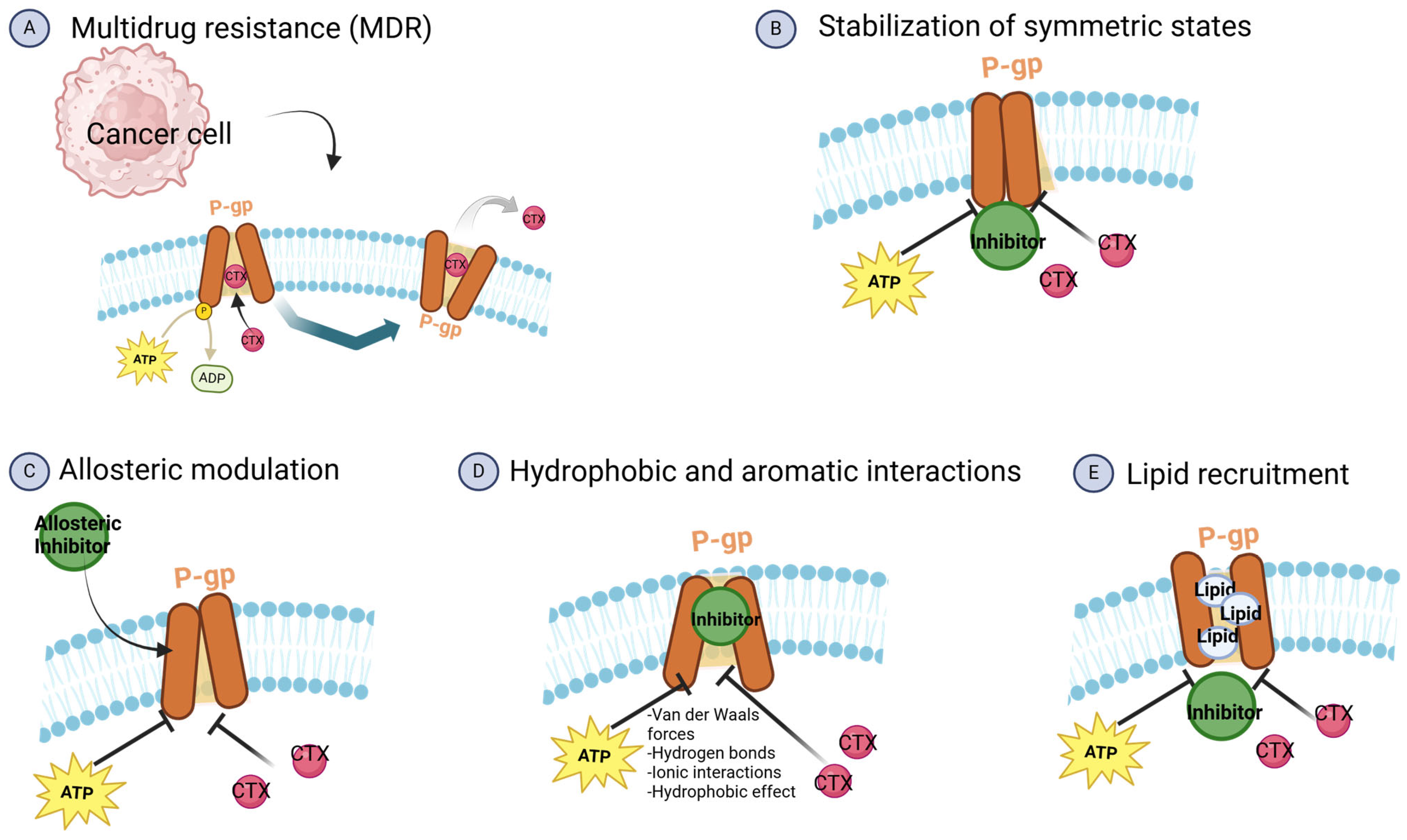
6.1. Inhibitors

{kind=link}
{kind=link}
{kind=link}
| Author | Inhibitor | Main Results |
|---|---|---|
| First generation | ||
| Dalton WS et al., 1995 [113] | Verapamil | No beneficial effect was observed from the addition of oral verapamil to the combination of vincristine, doxorubicin, and dexamethasone chemotherapy regimen for the treatment of drug-resistant myeloma patients. |
| Lum BL et al., 1992 [114] | Cyclosporine A | High doses of cyclosporine A and etoposide doses should be reduced by approximately 50% to compensate for the pharmacokinetic effects of cyclosporine A on etoposide. |
| Philip PA et al., 1992 [115] | Nifedipine | Patients with various malignancies received nifedipine at three dose levels; the cardiovascular effects of nifedipine were dose-limiting, but it did not interfere with the pharmacokinetics of etoposide. |
| Moriki Y et al., 2004 [116] | Pentazocine (PTZ) | The increment of PTZ uptake by the brain could be explained by the saturable efflux transport system involving a P-gp-mediated efflux mechanism of PTZ transport at the blood–brain barrier. |
| Cunningham C et al., 2009 [117] | Meperidine | Meperidine shows characteristics of an opioid agonist that lacks interaction with P-gp. |
| Regev R et al., 2007 [118] | Diethyl ether | Anesthetics enhance drug transport across cell membranes, thereby reducing P-gp-mediated multidrug resistance. At high concentrations, this effect is so pronounced that P-gp activity cannot be accurately measured. |
| Regev R et al.2007 [118] | Chloroform | Anesthetics enhance drug transport across cell membranes, thereby reducing P-gp-mediated multidrug resistance. At high concentrations, this effect is so pronounced that P-gp activity cannot be accurately measured. |
| Regev R et al., 2007 [118] | Propofol | Anesthetics enhance drug transport across cell membranes, thereby reducing P-gp-mediated multidrug resistance. At high concentrations, this effect is so pronounced that P-gp activity cannot be accurately measured. |
| Regev R et al., 2007 [118] | Benzyl alcohol | Anesthetics enhance drug transport across cell membranes, thereby reducing P-gp-mediated multidrug resistance. At high concentrations, this effect is so pronounced that P-gp activity cannot be accurately measured. |
| Gosland MP et al., 1996 [119] | Cefoperazone | Cefoperazone and ceftriaxone showed a remarkable ability to increase the sensitivity of cells resistant to drugs, such as doxorubicin, etoposide, and vinblastine. |
| Gosland MP et al., 1996 [119] | Ceftriaxone | Ceftriaxone and cefoperazone showed a remarkable ability to increase the sensitivity of cells resistant to drugs, such as doxorubicin, etoposide, and vinblastine. |
| Fuchs D et al., 2010 [120] | Salinomycin | Salinomycin can reverse multidrug resistance in leukemia stem cell-like cells by inhibiting ABC transporters. |
| Janneh O et al., 2010 [121] | Nigericin | Increased rh123 accumulation by 1.9-fold. |
| Asakura E et al., 2004 [122] | Azithromycin | Combination with doxorubicin in K562/ADR cell line: reversed P-gp dependent anticancer drug resistance. |
| Janneh O et al., 2010 [121] | Brefeldin A | Increase cellular accumulation of zidovudine (P-gp substrate) in the P-gp over-expressing cell line 3T3-F442A. |
| Janneh O et al., 2010 [121] | Bafilomycin | Increase cellular accumulation of zidovudine (P-gp substrate) in the P-gp over-expressing cell line 3T3-F442A. |
| Second generation | ||
| Punt CJ et al., 1997 [123] | S9788 | Multidrug-resistance-reversing agent S9788 concentrations are achieved in patients at nontoxic doses. Treatment with the combination of doxorubicin and S9788 produced a significant increase in the occurrence of grade 3–4 granulocytopenia. |
| Saeki T. et al., 2007 [124] | Dofequidar (MS-209) | In patients with advanced or recurrent breast cancer, dofequidar + fluorouracil was well tolerated and is suggested to have efficacy in patients who had not received prior therapy. |
| Warner et al., 1998 [125] | Dexverapamil | Study of dexverapamil plus anthracycline in patients with metastatic breast cancer who have progressed on the same anthracycline regimen and did not increase anthracycline toxicity. |
| Nuessler V et al. 1997 [126] | Dexniguldipine | A phase I study using dexniguldipine alone and in combination with vinblastine in patients with metastatic or locally advanced cancer found cardiovascular adverse events such as a blood drop, blood pressure, and decreased heart rate. |
| Solary E et al., 2000 [127] | Cinchonine | An i.v. infusion of cinchonine might be started 12 h before chemotherapy infusion and requires continuous cardiac monitoring, but no reduction in cytotoxic drug doses. |
| Kolitz J et al., 2010 [128] | PSC-833 (valspodar) | Randomized phase III trial to compare the effectiveness of combination chemotherapy with or without valspodar followed by interleukin-2 or no further therapy in treating older patients with acute myeloid leukemia. Grade 4 toxicities during IL-2 therapy, included thrombocytopenia and neutropenia, and grade 3 toxicities included anemia, infection, and malaise/fatigue. Low-dose IL-2 maintenance immunotherapy is not a successful strategy in older AML patients. Clinical trial: NCT00006363. |
| Gandhi L et al., 2007 [129] | VX-710 (biricodar) | Biricodar did not significantly enhance antitumor activity or survival, although minimal toxicity was reported. |
| Third generation | ||
| Cripe LD et al., 2010 [130] | LY-335979 (zosuquidar) | Erythromycin inhibited in vitro P-gp-mediated transport of both ximelagatran and melagatran and reduced biliary excretion of melagatran in the rats. |
| Chi K et al., 2005 [131] | OC144-093 (ontogen) | Inhibition of P-gp and multidrug resistance reversal at nM concentrations. No effect on paclitaxel pharmacokinetics. Well tolerated. Toxicities were mainly attributable to paclitaxel (febrile neutropenia). |
| Kelly RJ et al., 2011 [132] | XR-9576 (tariquidar) | Tariquidar, in combination either with paclitaxel and carboplatin or with vinorelbine, was tested on phase III clinical trials as the first-line therapy in non-small-cell lung cancer patients, but had to be stopped due to the high toxicity observed. Clinical trial: NCT00042302. |
| Bihorel S et al., 2007 [133] | GF120918 (elacridar) | A phase I and pharmacologic study of elacridar in combination with doxorubicin in patients with advanced solid tumors; elacridar pharmacokinetics were not influenced by coadministration of doxorubicin and produced only minimal side effects at a dose level yielding concentrations able to inhibit the action of P-gp in vitro (hematologic toxicity, namely neutropenia, somnolence, and occasional gastrointestinal complaints). Clinical trial: 2010-020759-30. |
| Fourth generation | ||
| Yan C et al., 2023 [134] | OY-101 | The excellent synergistic anticancer effect with vincristine (VCR) against drug-resistant cells of Eca109/VCR was confirmed using a reversal activity assay. |
| Yan C et al., 2015 [134] | FD18 | Flavonoid dimer FD18 is a new class of dimeric P-gp modulator that can modulate multidrug resistance toward paclitaxel, vinblastine, vincristine, doxorubicin, daunorubicin, and mitoxantrone in human breast cancer LCC6MDR in vitro. |
| Yu T et al., 2024 [135] | OY-103-B | For the VCR-resistant Eca109 cell line (Eca109/VCR), co-administration of 5.0 μM OY-103-B resulted in a reversal fold of up to 727.2, superior to the typical third-generation P-gp inhibitor tariquidar, and it reversed tumor drug resistance by inhibiting P-gp. |
| Modulation through signaling pathways | ||
| Wang H., 2016 [60] | Osthole | Reversed P-gp-mediated multidrug resistance by inhibiting the PI3K/Akt signaling cascade. |
| Hopff S., 2020 [136] | MBR-60 | It shows significant apoptotic effects and the mechanism of the compound involves the intrinsic pathway of apoptosis, which demonstrates selectivity for tumor cells over healthy leukocytes. |
| Li Z., 2024 [137] | ZIF-90@ICG | Impairs mitochondrial functions, downregulating the intracellular ATP level and inhibiting P-gp expression. |
| Qin K., 2018 [138] | MTX+4-HC | It is mediated through the inhibition of the JAK2/STAT3 signaling pathway. |
| Wang Y., 2018 [139] | FZD1 | FZD1 activation through the Wnt/β-catenin signaling pathway positively regulates the expression of MDR1; therefore, by inhibiting it, it may be possible to reduce the expression and function of P-gp. |
| Eadie L., 2014 [140] | Imatinib | This TKI is an ABCB1 inhibitor at high micromolar concentrations; however, the clinical relevance of these observations in some studies is limited. |
| Eadie L., 2014 [140] | Nilotinib | Exhibits a concentration-dependent interaction with ABCB1, acting as a substrate at lower concentrations and as an inhibitor at higher concentrations. The inhibitory effect is observed at concentrations above typical therapeutic levels. |
| Modulation from a genetic approach | ||
| Wang H., 2016 [60] | Osthole | Inhibited the cellular efflux of P-gp substrates and downregulated the expression of the MDR1 gene. |
| Tariq I. et al., 2020 [141] | Lipodendriplexes | Demonstrated enhanced cellular uptake, reduced toxicity, and increased gene knockdown efficiency. By silencing MDR1, lipodendriplexes sensitized cancer cells to chemotherapeutic drugs. |
6.2. Genetic Approach
6.3. Targeting Related Signaling Pathways
7. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Nanayakkara, A.K.; Follit, C.A.; Chen, G.; Williams, N.S.; Vogel, P.D.; Wise, J.G. Targeted Inhibitors of P-Glycoprotein Increase Chemotherapeutic-Induced Mortality of Multidrug Resistant Tumor Cells. Sci. Rep. 2018, 8, 967. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, I.S.; He, W.; Yin, L. Understanding of Human ATP Binding Cassette Superfamily and Novel Multidrug Resistance Modulators to Overcome MDR. Biomed. Pharmacother. 2018, 100, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.M.; Chen, J.W.; Chen, L.X.; Xie, X.; Mao, N. Overexpression of P-Glycoprotein on Fibroblast-like Synoviocytes in Refractory Rheumatoid Arthritis Patients: A Potential Mechanism for Multidrug Resistance in Rheumatoid Arthritis Treatment. Genet. Mol. Res. 2016, 15, 15027927. [Google Scholar] [CrossRef] [PubMed]
- García-Carrasco, M.; Mendoza-Pinto, C.; Macias Díaz, S.; Vera-Recabarren, M.; Vázquez de Lara, L.; Méndez Martínez, S.; Soto-Santillán, P.; González-Ramírez, R.; Ruiz-Arguelles, A. P-Glycoprotein in Autoimmune Rheumatic Diseases. Autoimmun. Rev. 2015, 14, 594–600. [Google Scholar] [CrossRef]
- Yoshimori, M.; Takada, H.; Imadome, K.; Kurata, M.; Yamamoto, K.; Koyama, T.; Shimizu, N.; Fujiwara, S.; Miura, O.; Arai, A. P-glycoprotein Is Expressed and Causes Resistance to Chemotherapy in EBV-positive T-cell Lymphoproliferative Diseases. Cancer Med. 2015, 4, 1494–1504. [Google Scholar] [CrossRef]
- Robinson, K.; Tiriveedhi, V. Perplexing Role of P-Glycoprotein in Tumor Microenvironment. Front. Oncol. 2020, 10, 265. [Google Scholar] [CrossRef]
- Dong, J.; Qin, Z.; Zhang, W.-D.; Cheng, G.; Yehuda, A.G.; Ashby, C.R.; Chen, Z.-S.; Cheng, X.-D.; Qin, J.-J. Medicinal Chemistry Strategies to Discover P-Glycoprotein Inhibitors: An Update. Drug Resist. Updates 2020, 49, 100681. [Google Scholar] [CrossRef]
- Karthika, C.; Sureshkumar, R.; Zehravi, M.; Akter, R.; Ali, F.; Ramproshad, S.; Mondal, B.; Tagde, P.; Ahmed, Z.; Khan, F.S.; et al. Multidrug Resistance of Cancer Cells and the Vital Role of P-Glycoprotein. Life 2022, 12, 897. [Google Scholar] [CrossRef]
- Goebel, J.; Chmielewski, J.; Hrycyna, C.A. The Roles of the Human ATP-Binding Cassette Transporters P-Glycoprotein and ABCG2 in Multidrug Resistance in Cancer and at Endogenous Sites: Future Opportunities for Structure-Based Drug Design of Inhibitors. Cancer Drug Resist. 2021, 4, 784–804. [Google Scholar] [CrossRef]
- Waghray, D.; Zhang, Q. Inhibit or Evade Multidrug Resistance P-Glycoprotein in Cancer Treatment. J. Med. Chem. 2018, 61, 5108–5121. [Google Scholar] [CrossRef]
- Wise, J.G.; Nanayakkara, A.K.; Aljowni, M.; Chen, G.; De Oliveira, M.C.; McCormick, L.A.; Olengue, K.; Lippert, A.R.; Vogel, P.D. Optimizing Targeted Inhibitors of P-Glycoprotein Using Computational and Structure-Guided Approaches. J. Med. Chem. 2019, 62, 10645–10663. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Sethi, N.; Patel, P.; Shah, S.; Patel, K. Exploring the Potential of P-Glycoprotein Inhibitors in the Targeted Delivery of Anti-Cancer Drugs: A Comprehensive Review. Eur. J. Pharm. Biopharm. 2024, 198, 114267. [Google Scholar] [CrossRef] [PubMed]
- Seelig, A. P-Glycoprotein: One Mechanism, Many Tasks and the Consequences for Pharmacotherapy of Cancers. Front. Oncol. 2020, 10, 576559. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Garg, T.; Tanmay, M.; Arora, S. Polymeric Drug-Delivery Systems: Role in P-Gp Efflux System Inhibition. Crit. Rev. Ther. Drug Carr. Syst. 2015, 32, 247–275. [Google Scholar] [CrossRef]
- Chen, C.J.; Clark, D.; Ueda, K.; Pastan, I.; Gottesman, M.M.; Roninson, I.B. Genomic Organization of the Human Multidrug Resistance (MDR1) Gene and Origin of P-Glycoproteins. J. Biol. Chem. 1990, 265, 506–514. [Google Scholar] [CrossRef]
- Skinner, K.T.; Palkar, A.M.; Hong, A.L. Genetics of ABCB1 in Cancer. Cancers 2023, 15, 4236. [Google Scholar] [CrossRef]
- Wang, L.; Sun, Y. Efflux Mechanism and Pathway of Verapamil Pumping by Human P-Glycoprotein. Arch. Biochem. Biophys. 2020, 696, 108675. [Google Scholar] [CrossRef]
- Mukhametov, A.; Raevsky, O.A. On the Mechanism of Substrate/Non-Substrate Recognition by P-Glycoprotein. J. Mol. Graph. Model. 2017, 71, 227–232. [Google Scholar] [CrossRef]
- Sharom, F.J. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 2014, 4, 41. [Google Scholar] [CrossRef]
- Chen, C.; Lee, M.-H.; Weng, C.-F.; Leong, M.K. Theoretical Prediction of the Complex P-Glycoprotein Substrate Efflux Based on the Novel Hierarchical Support Vector Regression Scheme. Molecules 2018, 23, 1820. [Google Scholar] [CrossRef]
- Kurre, D.; Dang, P.X.; Le, L.T.M.; Gadkari, V.V.; Alam, A. Structural Insights into Binding-Site Access and Ligand Recognition by Human ABCB1. EMBO J. 2025, 44, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Silva, R.; Vilas-Boas, V.; Carmo, H.; Dinis-Oliveira, R.J.; Carvalho, F.; de Lourdes Bastos, M.; Remião, F. Modulation of P-Glycoprotein Efflux Pump: Induction and Activation as a Therapeutic Strategy. Pharmacol. Ther. 2015, 149, 1–123. [Google Scholar] [CrossRef] [PubMed]
- Henrique, R.; Oliveira, A.I.; Costa, V.L.; Baptista, T.; Martins, A.T.; Morais, A.; Oliveira, J.; Jerónimo, C. Epigenetic Regulation of MDR1 Gene through Post-Translational Histone Modifications in Prostate Cancer. BMC Genom. 2013, 14, 898. [Google Scholar] [CrossRef] [PubMed]
- Pappas, J.J.; Petropoulos, S.; Suderman, M.; Iqbal, M.; Moisiadis, V.; Turecki, G.; Matthews, S.G.; Szyf, M. The Multidrug Resistance 1 Gene Abcb1 in Brain and Placenta: Comparative Analysis in Human and Guinea Pig. PLoS ONE 2014, 9, e111135. [Google Scholar] [CrossRef]
- Samuel, C.E. Adenosine Deaminase Acting on RNA (ADAR1), a Suppressor of Double-Stranded RNA–Triggered Innate Immune Responses. J. Biol. Chem. 2019, 294, 1710–1720. [Google Scholar] [CrossRef]
- Omata, Y.; Yamauchi, T.; Tsuruta, A.; Matsunaga, N.; Koyanagi, S.; Ohdo, S. RNA Editing Enzyme ADAR1 Governs the Circadian Expression of P-Glycoprotein in Human Renal Cells by Regulating Alternative Splicing of the ABCB1 Gene. J. Biol. Chem. 2021, 296, 100601. [Google Scholar] [CrossRef]
- Zhang, S.L.; Lahens, N.F.; Yue, Z.; Arnold, D.M.; Pakstis, P.P.; Schwarz, J.E.; Sehgal, A. A Circadian Clock Regulates Efflux by the Blood-Brain Barrier in Mice and Human Cells. Nat. Commun. 2021, 12, 617. [Google Scholar] [CrossRef]
- Ikemura, K.; Yamamoto, M.; Miyazaki, S.; Mizutani, H.; Iwamoto, T.; Okuda, M. MicroRNA-145 Post-Transcriptionally Regulates the Expression and Function of P-Glycoprotein in Intestinal Epithelial Cells. Mol. Pharmacol. 2013, 83, 399–405. [Google Scholar] [CrossRef]
- Thorne, J.L.; Battaglia, S.; Baxter, D.E.; Hayes, J.L.; Hutchinson, S.A.; Jana, S.; Millican-Slater, R.A.; Smith, L.; Teske, M.C.; Wastall, L.M.; et al. MiR-19b Non-Canonical Binding Is Directed by HuR and Confers Chemosensitivity through Regulation of P-Glycoprotein in Breast Cancer. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2018, 1861, 996–1006. [Google Scholar] [CrossRef]
- Huang, W.; Paul, D.; Calin, G.A.; Bayraktar, R. MiR-142: A Master Regulator in Hematological Malignancies and Therapeutic Opportunities. Cells 2023, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Peixoto da Silva, S.; Caires, H.R.; Bergantim, R.; Guimarães, J.E.; Vasconcelos, M.H. MiRNAs Mediated Drug Resistance in Hematological Malignancies. Semin. Cancer Biol. 2022, 83, 283–302. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Shan, Z.; Li, C.; Yang, L. MiR-129 Regulates Cisplatin-Resistance in Human Gastric Cancer Cells by Targeting P-Gp. Biomed. Pharmacother. 2017, 86, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Valizadeh, A.; Mir, S.M.; Asemi, Z.; Karimian, A.; Majidina, M.; Safa, A.; Yosefi, B. MiRNA-29a Reverses P-Glycoprotein-Mediated Drug Resistance and Inhibits Proliferation via up-Regulation of PTEN in Colon Cancer Cells. Eur. J. Pharmacol. 2020, 880, 173138. [Google Scholar] [CrossRef]
- Meng, Q.; Liang, C.; Hua, J.; Zhang, B.; Liu, J.; Zhang, Y.; Wei, M.; Yu, X.; Xu, J.; Shi, S. A MiR-146a-5p/TRAF6/NF-KB P65 Axis Regulates Pancreatic Cancer Chemoresistance: Functional Validation and Clinical Significance. Theranostics 2020, 10, 3967–3979. [Google Scholar] [CrossRef]
- Duan, H.; Wang, C.; Zhou, K.; Wang, T.; Li, Y.; Qiu, D.; Li, Q.; Zhang, Y.; Hua, Y. The Effect of Histone Deacetylase Inhibition on the Expression of P-Glycoprotein in Human Placental Trophoblast Cell Lines. Placenta 2017, 49, 37–47. [Google Scholar] [CrossRef]
- Jin, W.; Lu, Y.; Li, Q.; Wang, J.; Zhang, H.; Chang, G.; Lin, Y.; Pang, T. Down-Regulation of the P-Glycoprotein Relevant for Multidrug Resistance by Intracellular Acidification through the Crosstalk of MAPK Signaling Pathways. Int. J. Biochem. Cell Biol. 2014, 54, 111–121. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, M.; Aneja, R.; Chandra, R.; Lage, H.; Joshi, H.C. Reversal of P-Glycoprotein–Mediated Multidrug Resistance in Cancer Cells by the c-Jun NH2-Terminal Kinase. Cancer Res. 2006, 66, 445–452. [Google Scholar] [CrossRef]
- Antonio-Andrés, G.; Rangel-Santiago, J.; Tirado-Rodríguez, B.; Martinez-Ruiz, G.U.; Klunder-Klunder, M.; Vega, M.I.; Lopez-Martinez, B.; Jiménez-Hernández, E.; Torres Nava, J.; Medina-Sanson, A.; et al. Role of Yin Yang-1 (YY1) in the Transcription Regulation of the Multi-Drug Resistance (MDR1) Gene. Leuk. Lymphoma 2018, 59, 2628–2638. [Google Scholar] [CrossRef]
- Verheul, T.C.J.; van Hijfte, L.; Perenthaler, E.; Barakat, T.S. The Why of YY1: Mechanisms of Transcriptional Regulation by Yin Yang 1. Front. Cell Dev. Biol. 2020, 8, 592164. [Google Scholar] [CrossRef]
- Song, S.; Pei, G.; Du, Y.; Wu, J.; Ni, X.; Wang, S.; Jiang, B.; Luo, M.; Yu, J. Interaction between CD133 and PI3K-P85 Promotes Chemoresistance in Gastric Cancer Cells. Am. J. Transl. Res. 2018, 10, 304–314. [Google Scholar]
- Foley, S.E.; Dente, M.J.; Lei, X.; Sallis, B.F.; Loew, E.B.; Meza-Segura, M.; Fitzgerald, K.A.; McCormick, B.A. Microbial Metabolites Orchestrate a Distinct Multi-Tiered Regulatory Network in the Intestinal Epithelium That Directs P-Glycoprotein Expression. mBio 2022, 13, e01993-22. [Google Scholar] [CrossRef] [PubMed]
- Megías-Vericat, J.E.; Rojas, L.; Herrero, M.J.; Bosó, V.; Montesinos, P.; Moscardó, F.; Poveda, J.L.; Sanz, M.Á.; Aliño, S.F. Influence of ABCB1 Polymorphisms upon the Effectiveness of Standard Treatment for Acute Myeloid Leukemia: A Systematic Review and Meta-Analysis of Observational Studies. Pharmacogenomics J. 2015, 15, 109–118. [Google Scholar] [CrossRef]
- Hitzl, M.; Drescher, S.; van der Kuip, H.; Schäffeler, E.; Fischer, J.; Schwab, M.; Eichelbaum, M.; Fromm, M.F. The C3435T Mutation in the Human MDR1 Gene Is Associated with Altered Efflux of the P-Glycoprotein Substrate Rhodamine 123 from CD56+ Natural Killer Cells. Pharmacogenetics 2001, 11, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Buda, G.; Ricci, D.; Huang, C.C.; Favis, R.; Cohen, N.; Zhuang, S.H.; Harousseau, J.-L.; Sonneveld, P.; Bladé, J.; Orlowski, R.Z. Polymorphisms in the Multiple Drug Resistance Protein 1 and in P-Glycoprotein 1 Are Associated with Time to Event Outcomes in Patients with Advanced Multiple Myeloma Treated with Bortezomib and Pegylated Liposomal Doxorubicin. Ann. Hematol. 2010, 89, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Wu, H.; Yu, Q.; Kim, D.H.; Lipton, J.H.; Angelini, S.; Soverini, S.; Vivona, D.; Takahashi, N.; Cao, J. ABCB1 Polymorphisms Predict Imatinib Response in Chronic Myeloid Leukemia Patients: A Systematic Review and Meta-Analysis. Pharmacogenom. J. 2015, 15, 127–134. [Google Scholar] [CrossRef]
- Gerecke, C.; Fuhrmann, S.; Strifler, S.; Schmidt-Hieber, M.; Einsele, H.; Knop, S. The Diagnosis and Treatment of Multiple Myeloma. Dtsch. Arztebl. Int. 2016, 113, 470–476. [Google Scholar] [CrossRef]
- Abraham, J.; Salama, N.N.; Azab, A.K. The Role of P-Glycoprotein in Drug Resistance in Multiple Myeloma. Leuk. Lymphoma 2015, 56, 26–33. [Google Scholar] [CrossRef]
- Mynott, R.L.; Wallington-Beddoe, C.T. Inhibition of P-Glycoprotein Does Not Increase the Efficacy of Proteasome Inhibitors in Multiple Myeloma Cells. ACS Pharmacol. Transl. Sci. 2021, 4, 713–729. [Google Scholar] [CrossRef]
- Nicholson, R.; Menezes, A.C.; Azevedo, A.; Leckenby, A.; Davies, S.; Seedhouse, C.; Gilkes, A.; Knapper, S.; Tonks, A.; Darley, R.L. Protein Kinase C Epsilon Overexpression Is Associated with Poor Patient Outcomes in AML and Promotes Daunorubicin Resistance Through P-Glycoprotein-Mediated Drug Efflux. Front. Oncol. 2022, 12, 840046. [Google Scholar] [CrossRef]
- Hawley, T.S.; Riz, I.; Yang, W.; Wakabayashi, Y.; DePalma, L.; Chang, Y.; Peng, W.; Zhu, J.; Hawley, R.G. Identification of an ABCB1 (P-glycoprotein)-positive Carfilzomib-resistant Myeloma Subpopulation by the Pluripotent Stem Cell Fluorescent Dye CDy1. Am. J. Hematol. 2013, 88, 265–272. [Google Scholar] [CrossRef]
- Ilyas, M.; Aamir, K.M.; Manzoor, S.; Deriche, M. Linear Programming Based Computational Technique for Leukemia Classification Using Gene Expression Profile. PLoS ONE 2023, 18, e0292172. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Thiele, J.; Kvasnicka, H.M.; Orazi, A.; Gianelli, U.; Gangat, N.; Vannucchi, A.M.; Barbui, T.; Arber, D.A.; Tefferi, A. The International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Myeloproliferative Neoplasms. Am. J. Hematol. 2023, 98, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Abhishek, A.; Jha, R.K.; Sinha, R.; Jha, K. Automated Detection and Classification of Leukemia on a Subject-Independent Test Dataset Using Deep Transfer Learning Supported by Grad-CAM Visualization. Biomed. Signal Process Control 2023, 83, 104722. [Google Scholar] [CrossRef]
- da Silveira Júnior, L.S.; Soares, V.d.L.; Jardim da Silva, A.S.; Gil, E.A.; Pereira de Araújo, M.d.G.; Merces Gonçalves, C.A.; Paiva, A.d.S.; Moura de Oliveira, T.M.; Oliveira, G.H.d.M.; Kramer Cavacanti e Silva, D.G.; et al. P-glycoprotein and Multidrug Resistance-associated Protein-1 Expression in Acute Myeloid Leukemia: Biological and Prognosis Implications. Int. J. Lab. Hematol. 2020, 42, 594–603. [Google Scholar] [CrossRef]
- Wen, Z.; Li, H.; Zhang, J. The Expression and Clinical Significance of Murine Double Minute 2, Lysosome-Associated Membrane Protein 1, and P-Glycoprotein in Pediatric Acute Lymphoblastic Leukemia. Transl. Pediatr. 2020, 9, 677–685. [Google Scholar] [CrossRef]
- Muñoz-Pérez, M.J.; Casco, S.; Garza-González, M.d.C.; Soto-Vega, E. P-Glycoprotein Activity Correlates With Treatment Response in 2 Leukemia Child Patients. J. Pediatr. Hematol. Oncol. 2018, 40, e490–e494. [Google Scholar] [CrossRef]
- Gao, F.; Dong, W.; Yang, W.; Liu, J.; Zheng, Z.; Sun, K. Expression of P-Gp in Acute Myeloid Leukemia and the Reversal Function of As2O3 on Drug Resistance. Oncol. Lett. 2015, 9, 177–182. [Google Scholar] [CrossRef]
- Tian, Y.; Lei, Y.; Wang, Y.; Lai, J.; Wang, J.; Xia, F. Mechanism of Multidrug Resistance to Chemotherapy Mediated by P-glycoprotein (Review). Int. J. Oncol. 2023, 63, 119. [Google Scholar] [CrossRef]
- Wang, H.; Jia, X.-H.; Chen, J.-R.; Wang, J.-Y.; Li, Y.-J. Osthole Shows the Potential to Overcome P-Glycoprotein-Mediated Multidrug Resistance in Human Myelogenous Leukemia K562/ADM Cells by Inhibiting the PI3K/Akt Signaling Pathway. Oncol. Rep. 2016, 35, 3659–3668. [Google Scholar] [CrossRef]
- Muthusamy, G.; Gunaseelan, S.; Prasad, N.R. Ferulic Acid Reverses P-Glycoprotein-Mediated Multidrug Resistance via Inhibition of PI3K/Akt/NF-ΚB Signaling Pathway. J. Nutr. Biochem. 2019, 63, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Coculova, M.; Imrichova, D.; Seres, M.; Messingerova, L.; Bohacova, V.; Sulova, Z.; Breier, A. The Expression of P-Glycoprotein in Leukemia Cells Is Associated with the Upregulated Expression of Nestin, a Class 6 Filament Protein. Leuk. Res. 2016, 48, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X.; Zeidan, A.M. Epidemiology of Acute Myeloid Leukemia: Recent Progress and Enduring Challenges. Blood Rev. 2019, 36, 70–87. [Google Scholar] [CrossRef]
- Labbozzetta, M.; Poma, P.; Notarbartolo, M. Natural Inhibitors of P-Glycoprotein in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2023, 24, 4140. [Google Scholar] [CrossRef]
- Muthiah, D.; Henshaw, G.K.; DeBono, A.J.; Capuano, B.; Scammells, P.J.; Callaghan, R. Overcoming P-Glycoprotein–Mediated Drug Resistance with Noscapine Derivatives. Drug Metab. Dispos. 2019, 47, 164–172. [Google Scholar] [CrossRef]
- Prado-Carrillo, O.; Arenas-Ramírez, A.; Llaguno-Munive, M.; Jurado, R.; Pérez-Rojas, J.; Cervera-Ceballos, E.; Garcia-Lopez, P. Ketoconazole Reverses Imatinib Resistance in Human Chronic Myelogenous Leukemia K562 Cells. Int. J. Mol. Sci. 2022, 23, 7715. [Google Scholar] [CrossRef]
- Fischer, H.; Ullah, M.; de la Cruz, C.C.; Hunsaker, T.; Senn, C.; Wirz, T.; Wagner, B.; Draganov, D.; Vazvaei, F.; Donzelli, M.; et al. Entrectinib, a TRK/ROS1 Inhibitor with Anti-CNS Tumor Activity: Differentiation from Other Inhibitors in Its Class Due to Weak Interaction with P-Glycoprotein. Neuro Oncol. 2020, 22, 819–829. [Google Scholar] [CrossRef]
- Tan, S.-F.; Dunton, W.; Liu, X.; Fox, T.E.; Morad, S.A.F.; Desai, D.; Doi, K.; Conaway, M.R.; Amin, S.; Claxton, D.F.; et al. Acid Ceramidase Promotes Drug Resistance in Acute Myeloid Leukemia through NF-ΚB-Dependent P-Glycoprotein Upregulation. J. Lipid Res. 2019, 60, 1078–1086. [Google Scholar] [CrossRef]
- Prijić, S.; Ugrina, I.; Labar, B.; Nemet, D.; Batinić, J.; Zadro, R.; Ries, S.; Gjadrov-Kuvedžić, K.; Davidović, S.; Batinić, D. Prognostic Significance of Constitutive Phosphatidylinositol 3-Kinase/Akt and Mitogen-Activated Protein Kinase Phosphorylation in Acute Myeloid Leukemia. Leuk. Lymphoma 2015, 56, 2281–2288. [Google Scholar] [CrossRef]
- Ammar, M.; Louati, N.; Frikha, I.; Medhaffar, M.; Ghozzi, H.; Elloumi, M.; Menif, H.; Zeghal, K.; Ben Mahmoud, L. Overexpression of P-glycoprotein and Resistance to Imatinib in Chronic Myeloid Leukemia Patients. J. Clin. Lab. Anal. 2020, 34. [Google Scholar] [CrossRef]
- Eadie, L.N.; Hughes, T.P.; White, D.L. ABCB1 Overexpression Is a Key Initiator of Resistance to Tyrosine Kinase Inhibitors in CML Cell Lines. PLoS ONE 2016, 11, e0161470. [Google Scholar] [CrossRef] [PubMed]
- Dessilly, G.; Panin, N.; Elens, L.; Haufroid, V.; Demoulin, J.-B. Impact of ABCB1 1236C > T-2677G > T-3435C > T Polymorphisms on the Anti-Proliferative Activity of Imatinib, Nilotinib, Dasatinib and Ponatinib. Sci. Rep. 2016, 6, 29559. [Google Scholar] [CrossRef] [PubMed]
- Dulucq, S.; Bouchet, S.; Turcq, B.; Lippert, E.; Etienne, G.; Reiffers, J.; Molimard, M.; Krajinovic, M.; Mahon, F.-X. Multidrug Resistance Gene (MDR1) Polymorphisms Are Associated with Major Molecular Responses to Standard-Dose Imatinib in Chronic Myeloid Leukemia. Blood 2008, 112, 2024–2027. [Google Scholar] [CrossRef]
- Bedewy, A.M.L.; Elmaghraby, S.M.; Kandil, N.S. ABCB1 and BMI1 MRNA Expression in Patients with Chronic Myeloid Leukemia: Impact on Imatinib Efficacy. Blood Res. 2019, 54, 57–62. [Google Scholar] [CrossRef]
- Rinaldetti, S.; Pfirrmann, M.; Manz, K.; Guilhot, J.; Dietz, C.; Panagiotidis, P.; Spiess, B.; Seifarth, W.; Fabarius, A.; Müller, M.; et al. Effect of ABCG2, OCT1, and ABCB1 (MDR1) Gene Expression on Treatment-Free Remission in a EURO-SKI Subtrial. Clin. Lymphoma Myeloma Leuk. 2018, 18, 266–271. [Google Scholar] [CrossRef]
- da Cunha Vasconcelos, F.; Mauricio Scheiner, M.A.; Moellman-Coelho, A.; Mencalha, A.L.; Renault, I.Z.; Rumjanek, V.M.; Maia, R.C. Low ABCB1 and High OCT1 Levels Play a Favorable Role in the Molecular Response to Imatinib in CML Patients in the Community Clinical Practice. Leuk. Res. 2016, 51, 3–10. [Google Scholar] [CrossRef]
- Pongstaporn, W.; Pakakasama, S.; Chaksangchaichote, P.; Pongtheerat, T.; Hongeng, S.; Permitr, S. MDR1 C3435T and C1236T Polymorphisms: Association with High-Risk Childhood Acute Lymphoblastic Leukemia. Asian Pac. J. Cancer Prev. 2015, 16, 2839–2843. [Google Scholar] [CrossRef]
- Talaat, R.M.; El-Kelliny, M.Y.K.; El-Akhras, B.A.; Bakry, R.M.; Riad, K.F.; Guirgis, A.A. Association of C3435T, C1236T and C4125A Polymorphisms of the MDR-1 Gene in Egyptian Children with Acute Lymphoblastic Leukaemia. Asian Pac. J. Cancer Prev. 2018, 19, 2535–2543. [Google Scholar] [CrossRef]
- Yue, Q.; Xiong, B.; Chen, L.; Chen, Y.; Bu, F.; Liu, X.; Cheng, F. MDR1 C3435T Polymorphism and Childhood Acute Lymphoblastic Leukemia Susceptibility: An Updated Meta-Analysis. Biomed. Pharmacother. 2015, 69, 76–81. [Google Scholar] [CrossRef]
- Ramírez-Pacheco, A.; Moreno-Guerrero, S.; Alamillo, I.; Medina-Sanson, A.; Lopez, B.; Moreno-Galván, M. Mexican Childhood Acute Lymphoblastic Leukemia: A Pilot Study of the MDR1 and MTHFR Gene Polymorphisms and Their Associations with Clinical Outcomes. Genet. Test. Mol. Biomark. 2016, 20, 597–602. [Google Scholar] [CrossRef]
- Bhatia, P.; Masih, S.; Varma, N.; Bansal, D.; Trehan, A. High Expression of Lung Resistance Protein MRNA at Diagnosis Predicts Poor Early Response to Induction Chemotherapy in Childhood Acute Lymphoblastic Leukemia. Asian Pac. J. Cancer Prev. 2015, 16, 6663–6668. [Google Scholar] [CrossRef] [PubMed]
- Gregers, J.; Gréen, H.; Christensen, I.J.; Dalhoff, K.; Schroeder, H.; Carlsen, N.; Rosthoej, S.; Lausen, B.; Schmiegelow, K.; Peterson, C. Polymorphisms in the ABCB1 Gene and Effect on Outcome and Toxicity in Childhood Acute Lymphoblastic Leukemia. Pharmacogenom. J. 2015, 15, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Gurbaxani, S.; Anastasi, J.; Hyjek, E. Diffuse Large B-Cell Lymphoma—More Than a Diffuse Collection of Large B Cells: An Entity in Search of a Meaningful Classification. Arch. Pathol. Lab. Med. 2009, 133, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- de Leval, L.; Jaffe, E.S. Lymphoma Classification. Cancer J. 2020, 26, 176–185. [Google Scholar] [CrossRef]
- de Leval, L. Abstract IA02: Principles of Lymphoma Classification. Clin. Cancer Res. 2015, 21, IA02. [Google Scholar] [CrossRef]
- Pi, M.; Kuang, H.; Yue, C.; Yang, Q.; Wu, A.; Li, Y.; Assaraf, Y.G.; Yang, D.-H.; Wu, S. Targeting Metabolism to Overcome Cancer Drug Resistance: A Promising Therapeutic Strategy for Diffuse Large B Cell Lymphoma. Drug Resist. Updates 2022, 61, 100822. [Google Scholar] [CrossRef]
- Han, L.; Guo, X.; Bian, H.; Zhou, Y.; Li, T.; Yang, J. Changed Expression and Function of P-Gp in Peripheral Blood CD56 + Cells Predicting Chemoresistance in Non-Hodgkin Lymphoma Patients. Cancer Biomark. 2015, 15, 289–297. [Google Scholar] [CrossRef]
- Nam, Y.-S.; Im, K.-I.; Kim, N.; Song, Y.; Lee, J.-S.; Jeon, Y.-W.; Cho, S.-G. Down-Regulation of Intracellular Reactive Oxygen Species Attenuates P-Glycoprotein-Associated Chemoresistance in Epstein-Barr Virus-Positive NK/T-Cell Lymphoma. Am. J. Transl. Res. 2019, 11, 1359–1373. [Google Scholar]
- Hu, B.; Oki, Y. Novel Immunotherapy Options for Extranodal NK/T-Cell Lymphoma. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Miyazaki, K. Current Treatment Approaches for NK/T-Cell Lymphoma. J. Clin. Exp. Hematop. 2017, 57, 98–108. [Google Scholar] [CrossRef]
- Chen, Q.; Feng, J.; Wu, J.; Yu, Z.; Zhang, W.; Chen, Y.; Yao, P.; Zhang, H. HKDC1 C-Terminal Based Peptides Inhibit Extranodal Natural Killer/T-Cell Lymphoma by Modulation of Mitochondrial Function and EBV Suppression. Leukemia 2020, 34, 2736–2748. [Google Scholar] [CrossRef] [PubMed]
- Berg, T.; Hegelund-Myrbäck, T.; Öckinger, J.; Zhou, X.-H.; Brännström, M.; Hagemann-Jensen, M.; Werkström, V.; Seidegård, J.; Grunewald, J.; Nord, M.; et al. Expression of MATE1, P-Gp, OCTN1 and OCTN2, in Epithelial and Immune Cells in the Lung of COPD and Healthy Individuals. Respir. Res. 2018, 19, 68. [Google Scholar] [CrossRef] [PubMed]
- Arias Garcia, F.I.I.; Guillen Medina, M.R.; Perez Guerrero, E.E.; Padilla Ortega, J.A.; Landeros Perez, A.A.; Ron Magaña, A.L.; Vargas Carretero, C.J.; Chamorro Morales, N.H.; Villalobos, L.O.; Paredes Uc, J.A. Soluble and Membrane P-Glycoprotein Expression in Lymphocytes from Diffuse Large B Cell Non-Hodgkin’s Lymphoma Patients Treated with R-CHOP. Blood 2023, 142, 5798. [Google Scholar] [CrossRef]
- García-Carrasco, M.; Mendoza-Pinto, C.; Macías-Díaz, S.; Etchegaray-Morales, I.; Méndez-Martínez, S.; Soto-Santillán, P.; Pérez-Romano, B.; Jiménez-Herrera, E.A.; Guzmán-Ruiz, O.; Ruiz-Argüelles, A. Clinical Relevance of P-Glycoprotein Activity on Peripheral Blood Mononuclear Cells and Polymorphonuclear Neutrophils to Methotrexate in Systemic Lupus Erythematosus Patients. Clin. Rheumatol. 2017, 36, 2267–2272. [Google Scholar] [CrossRef]
- Wu, L.; Cai, S.; Deng, Y.; Zhang, Z.; Zhou, X.; Su, Y.; Xu, D. PD-1/PD-L1 Enhanced Cisplatin Resistance in Gastric Cancer through PI3K/AKT Mediated P-Gp Expression. Int. Immunopharmacol. 2021, 94, 107443. [Google Scholar] [CrossRef]
- Sun, W.; Hu, S.; Wang, X. Advances and Clinical Applications of Immune Checkpoint Inhibitors in Hematological Malignancies. Cancer Commun. 2024, 44, 1071–1097. [Google Scholar] [CrossRef]
- Izawa, A.; Schatton, T.; Frank, N.Y.; Ueno, T.; Yamaura, K.; Pendse, S.S.; Margaryan, A.; Grimm, M.; Gasser, M.; Waaga-Gasser, A.M.; et al. A Novel in Vivo Regulatory Role of P-Glycoprotein in Alloimmunity. Biochem. Biophys. Res. Commun. 2010, 394, 646–652. [Google Scholar] [CrossRef]
- Huang, X.; Hussain, B.; Chang, J. Peripheral Inflammation and Blood–Brain Barrier Disruption: Effects and Mechanisms. CNS Neurosci. Ther. 2021, 27, 36–47. [Google Scholar] [CrossRef]
- Egashira, M.; Kawamata, N.; Sugimoto, K.; Kaneko, T.; Oshimi, K. P-Glycoprotein Expression on Normal and Abnormally Expanded Natural Killer Cells and Inhibition of P-Glycoprotein Function by Cyclosporin A and Its Analogue, PSC833. Blood 1999, 93, 599–606. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yamashiro, T.; Nagatake, H.; Yamamoto, T.; Watanabe, N.; Tanaka, H.; Shigenobu, K.; Tsuruo, T. Expression and Function of Multidrug Resistance P-Glycoprotein in a Cultured Natural Killer Cell-Rich Population Revealed by MRK16 Monoclonal Antibody and AHC-52. Biochem. Pharmacol. 1994, 48, 1641–1646. [Google Scholar] [CrossRef]
- Yamashiro, T.; Watanabe, N.; Yokoyama, K.K.; Koga, C.; Tsuruo, T.; Kobayashi, Y. Requirement of Expression of P-Glycoprotein on Human Natural Killer Leukemia Cells for Cell-Mediated Cytotoxicity. Biochem. Pharmacol. 1998, 55, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Misawa, Y.; Watanabe, N.; Kawanishi, T.; Tanaka, H.; Shigenobu, K.; Kobayashi, Y. Role of P-Glycoprotein in Human Natural Killer-Like Cell Line-Mediated Cytotoxicity. Exp. Cell Res. 1999, 253, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Trambas, C.; Wang, Z.; Cianfriglia, M.; Woods, G. Evidence That Natural Killer Cells Express Mini P-glycoproteins but Not Classic 170 KDa P-glycoprotein. Br. J. Haematol. 2001, 114, 177–184. [Google Scholar] [CrossRef]
- Lai, J.-I.; Tseng, Y.-J.; Chen, M.-H.; Huang, C.-Y.F.; Chang, P.M.-H. Clinical Perspective of FDA Approved Drugs With P-Glycoprotein Inhibition Activities for Potential Cancer Therapeutics. Front. Oncol. 2020, 10, 561936. [Google Scholar] [CrossRef]
- Xu, W.; Chen, S.; Wang, X.; Min, J.; Tanaka, S.; Onda, K.; Sugiyama, K.; Yamada, H.; Hirano, T. Cepharanthine Synergistically Promotes Methylprednisolone Pharmacodynamics against Human Peripheral Blood Mononuclear Cells Possibly via Regulation of P-Glycoprotein/Glucocorticoid Receptor Translocation. BMC Complement. Med. Ther. 2024, 24, 186. [Google Scholar] [CrossRef]
- Huang, C.-Z.; Wang, Y.-F.; Zhang, Y.; Peng, Y.-M.; Liu, Y.-X.; Ma, F.; Jiang, J.-H.; Wang, Q.-D. Cepharanthine Hydrochloride Reverses P-Glycoprotein-Mediated Multidrug Resistance in Human Ovarian Carcinoma A2780/Taxol Cells by Inhibiting the PI3K/Akt Signaling Pathway. Oncol. Rep. 2017, 38, 2558–2564. [Google Scholar] [CrossRef]
- Wang, S.-Q.; Teng, Q.-X.; Wang, S.; Lei, Z.-N.; Hu, H.-H.; Lv, H.-F.; Chen, B.-B.; Wang, J.-Z.; Shi, X.-J.; Xu, W.-F.; et al. Preclinical Studies of the Triazolo [1,5-a]Pyrimidine Derivative WS-716 as a Highly Potent, Specific and Orally Active P-Glycoprotein (P-Gp) Inhibitor. Acta Pharm. Sin. B 2022, 12, 3263–3280. [Google Scholar] [CrossRef]
- Shah, D.; Ajazuddin; Bhattacharya, S. Role of Natural P-Gp Inhibitor in the Effective Delivery for Chemotherapeutic Agents. J. Cancer Res. Clin. Oncol. 2023, 149, 367–391. [Google Scholar] [CrossRef]
- Lee, J.; Kang, J.; Kwon, N.-Y.; Sivaraman, A.; Naik, R.; Jin, S.-Y.; Oh, A.R.; Shin, J.-H.; Na, Y.; Lee, K.; et al. Dual Inhibition of P-Gp and BCRP Improves Oral Topotecan Bioavailability in Rodents. Pharmaceutics 2021, 13, 559. [Google Scholar] [CrossRef]
- Jouan, E.; Le Vée, M.; Mayati, A.; Denizot, C.; Parmentier, Y.; Fardel, O. Evaluation of P-Glycoprotein Inhibitory Potential Using a Rhodamine 123 Accumulation Assay. Pharmaceutics 2016, 8, 12. [Google Scholar] [CrossRef]
- Bogaard, L.; Tsoi, K.; van de Steeg, B.; Brandon, E.F.A.; Geers, L.; van Herwaarden, M.; Jansman, F.; Maas, D.; Monster-Simons, M.; Ong, D.S.Y.; et al. A Practical Assessment Protocol for Clinically Relevant P-Glycoprotein-Mediated Drug-Drug Interactions. Front. Pharmacol. 2024, 15, 1412692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, H.; Ashby, C.R.; Assaraf, Y.G.; Chen, Z.-S.; Liu, H.-M. Chemical Molecular-Based Approach to Overcome Multidrug Resistance in Cancer by Targeting P-Glycoprotein (P-Gp). Med. Res. Rev. 2021, 41, 525–555. [Google Scholar] [CrossRef] [PubMed]
- Dalton, W.S.; Crowley, J.J.; Salmon, S.S.; Grogan, T.M.; Laufman, L.R.; Weiss, G.R.; Bonnet, J.D. A Phase III Randomized Study of Oral Verapamil as a Chemosensitizer to Reverse Drug Resistance in Patients with Refractory Myeloma. A Southwest Oncology Group Study. Cancer 1995, 75, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Lum, B.L.; Kaubisch, S.; Yahanda, A.M.; Adler, K.M.; Jew, L.; Ehsan, M.N.; Brophy, N.A.; Halsey, J.; Gosland, M.P.; Sikic, B.I. Alteration of Etoposide Pharmacokinetics and Pharmacodynamics by Cyclosporine in a Phase I Trial to Modulate Multidrug Resistance. J. Clin. Oncol. 1992, 10, 1635–1642. [Google Scholar] [CrossRef]
- Philip, P.; Joel, S.; Monkman, S.; Dolega-Ossowski, E.; Tonkin, K.; Carmichael, J.; Idle, J.; Harris, A. A Phase I Study on the Reversal of Multidrug Resistance (MDR) in Vivo: Nifedipine plus Etoposide. Br. J. Cancer 1992, 65, 267–270. [Google Scholar] [CrossRef]
- Moriki, Y.; Suzuki, T.; Fukami, T.; Hanano, M.; Tomono, K.; Watanabe, J. Involvement of P-Glycoprotein in Blood-Brain Barrier Transport of Pentazocine in Rats Using Brain Uptake Index Method. Biol. Pharm. Bull. 2004, 27, 932–935. [Google Scholar] [CrossRef]
- Hassan, H.E.; Mercer, S.L.; Cunningham, C.W.; Coop, A.; Eddington, N.D. Evaluation of the P-Glycoprotein (Abcb1) Affinity Status of a Series of Morphine Analogs: Comparative Study with Meperidine Analogs to Identify Opioids with Minimal P-Glycoprotein Interactions. Int. J. Pharm. 2009, 375, 48–54. [Google Scholar] [CrossRef]
- Regev, R.; Katzir, H.; Yeheskely-Hayon, D.; Eytan, G.D. Modulation of P-glycoprotein-mediated Multidrug Resistance by Acceleration of Passive Drug Permeation across the Plasma Membrane. FEBS J. 2007, 274, 6204–6214. [Google Scholar] [CrossRef]
- Gosland, M.P.; Gillespie, M.N.; Tsuboi, C.P.; Tofiq, S.; Olson, J.W.; Crooks, P.A.; Aziz, S.M. Reversal of Doxorubicin, Etoposide, Vinblastine, and Taxol Resistance in Multidrug Resistant Human Sarcoma Cells by a Polymer of Spermine. Cancer Chemother. Pharmacol. 1996, 37, 593–600. [Google Scholar] [CrossRef]
- Fuchs, D.; Daniel, V.; Sadeghi, M.; Opelz, G.; Naujokat, C. Salinomycin Overcomes ABC Transporter-Mediated Multidrug and Apoptosis Resistance in Human Leukemia Stem Cell-like KG-1a Cells. Biochem. Biophys. Res. Commun. 2010, 394, 1098–1104. [Google Scholar] [CrossRef]
- Janneh, O.; Owen, A.; Bray, P.G.; Back, D.J.; Pirmohamed, M. The Accumulation and Metabolism of Zidovudine in 3T3-F442A Pre-adipocytes. Br. J. Pharmacol. 2010, 159, 484–493. [Google Scholar] [CrossRef] [PubMed]
- Asakura, E.; Nakayama, H.; Sugie, M.; Lan Zhao, Y.; Nadai, M.; Kitaichi, K.; Shimizu, A.; Miyoshi, M.; Takagi, K.; Takagi, K.; et al. Azithromycin Reverses Anticancer Drug Resistance and Modifies Hepatobiliary Excretion of Doxorubicin in Rats. Eur. J. Pharmacol. 2004, 484, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Punt, C.; Voest, E.; Tueni, E.; Van Oosterom, A.; Backx, A.; De Mulder, P.; Hecquet, B.; Lucas, C.; Gerard, B.; Bleiberg, H. Phase IB Study of Doxorubicin in Combination with the Multidrug Resistance Reversing Agent S9788 in Advanced Colorectal and Renal Cell Cancer. Br. J. Cancer 1997, 76, 1376–1381. [Google Scholar] [CrossRef] [PubMed]
- Saeki, T.; Nomizu, T.; Toi, M.; Ito, Y.; Noguchi, S.; Kobayashi, T.; Asaga, T.; Minami, H.; Yamamoto, N.; Aogi, K.; et al. Dofequidar Fumarate (MS-209) in Combination With Cyclophosphamide, Doxorubicin, and Fluorouracil for Patients With Advanced or Recurrent Breast Cancer. J. Clin. Oncol. 2007, 25, 411–417. [Google Scholar] [CrossRef]
- Warner, E.; Hedley, D.; Andrulis, I.; Myers, R.; Trudeau, M.; Warr, D.; Pritchard, K.I.; Blackstein, M.; Goss, P.E.; Franssen, E.; et al. Phase II Study of Dexverapamil plus Anthracycline in Patients with Metastatic Breast Cancer Who Have Progressed on the Same Anthracycline Regimen. Clin. Cancer Res. 1998, 4, 1451–1457. [Google Scholar]
- Nuessler, V.; Scheulen, M.E.; Oberneder, R.; Kriegmair, M.; Goebel, K.J.; Rathgeb, F.; Wurst, W.; Zech, K.; Wilmanns, W. Phase I and Pharmacokinetic Study of the P-Glycoprotein Modulator Dexniguldipine-HCL. Eur. J. Med. Res. 1997, 2, 55–61. [Google Scholar]
- Solary, E.; Mannone, L.; Moreau, D.; Caillot, D.; Casasnovas, R.-O.; Guy, H.; Grandjean, M.; Wolf, J.-E.; André, F.; Fenaux, P.; et al. Phase I Study of Cinchonine, a Multidrug Resistance Reversing Agent, Combined with the CHVP Regimen in Relapsed and Refractory Lymphoproliferative Syndromes. Leukemia 2000, 14, 2085–2094. [Google Scholar] [CrossRef]
- Kolitz, J.E.; George, S.L.; Marcucci, G.; Vij, R.; Powell, B.L.; Allen, S.L.; DeAngelo, D.J.; Shea, T.C.; Stock, W.; Baer, M.R.; et al. P-Glycoprotein Inhibition Using Valspodar (PSC-833) Does Not Improve Outcomes for Patients Younger than Age 60 Years with Newly Diagnosed Acute Myeloid Leukemia: Cancer and Leukemia Group B Study 19808. Blood 2010, 116, 1413–1421. [Google Scholar] [CrossRef]
- Gandhi, L.; Harding, M.W.; Neubauer, M.; Langer, C.J.; Moore, M.; Ross, H.J.; Johnson, B.E.; Lynch, T.J. A Phase II Study of the Safety and Efficacy of the Multidrug Resistance Inhibitor VX-710 Combined with Doxorubicin and Vincristine in Patients with Recurrent Small Cell Lung Cancer. Cancer 2007, 109, 924–932. [Google Scholar] [CrossRef]
- Cripe, L.D.; Uno, H.; Paietta, E.M.; Litzow, M.R.; Ketterling, R.P.; Bennett, J.M.; Rowe, J.M.; Lazarus, H.M.; Luger, S.; Tallman, M.S. Zosuquidar, a Novel Modulator of P-Glycoprotein, Does Not Improve the Outcome of Older Patients with Newly Diagnosed Acute Myeloid Leukemia: A Randomized, Placebo-Controlled Trial of the Eastern Cooperative Oncology Group 3999. Blood 2010, 116, 4077–4085. [Google Scholar] [CrossRef]
- Chi, K.N.; Chia, S.K.; Dixon, R.; Newman, M.J.; Wacher, V.J.; Sikic, B.; Gelmon, K.A. A Phase I Pharmacokinetic Study of the P-Glycoprotein Inhibitor, ONT-093, in Combination with Paclitaxel in Patients with Advanced Cancer. Invest. New Drugs 2005, 23, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.J.; Draper, D.; Chen, C.C.; Robey, R.W.; Figg, W.D.; Piekarz, R.L.; Chen, X.; Gardner, E.R.; Balis, F.M.; Venkatesan, A.M.; et al. A Pharmacodynamic Study of Docetaxel in Combination with the P-Glycoprotein Antagonist Tariquidar (XR9576) in Patients with Lung, Ovarian, and Cervical Cancer. Clin. Cancer Res. 2011, 17, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Bihorel, S.; Camenisch, G.; Lemaire, M.; Scherrmann, J.-M. Modulation of the Brain Distribution of Imatinib and Its Metabolites in Mice by Valspodar, Zosuquidar and Elacridar. Pharm. Res. 2007, 24, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.S.W.; Wong, I.L.K.; Chan, K.-F.; Kan, J.W.Y.; Chong, T.C.; Law, M.C.; Zhao, Y.; Chan, S.W.; Chan, T.H.; Chow, L.M.C. A New Class of Safe, Potent, and Specific P-Gp Modulator: Flavonoid Dimer FD18 Reverses P-Gp-Mediated Multidrug Resistance in Human Breast Xenograft in Vivo. Mol. Pharm. 2015, 12, 3507–3517. [Google Scholar] [CrossRef]
- Yu, T.; Zeng, R.; Guan, Y.; Pan, B.; Li, H.-W.; Gu, J.; Zheng, P.-F.; Qian, Y.; Ouyang, Q. Discovery of New Tricyclic Spiroindole Derivatives as Potent P-Glycoprotein Inhibitors for Reversing Multidrug Resistance Enabled by a Synthetic Methodology-Based Library. RSC Med. Chem. 2024, 15, 1675–1685. [Google Scholar] [CrossRef]
- Hopff, S.M.; Onambele, L.A.; Brandenburg, M.; Berkessel, A.; Prokop, A. Discovery of a Cobalt (III) Salen Complex That Induces Apoptosis in Burkitt like Lymphoma and Leukemia Cells, Overcoming Multidrug Resistance in Vitro. Bioorg Chem. 2020, 104, 104193. [Google Scholar] [CrossRef]
- Li, Z.; Ma, X.; Yang, Y.; Wang, Y.; Zhu, W.; Deng, X.; Chen, T.; Gao, C.; Zhang, Y.; Yang, W.; et al. Crizotinib Resistance Reversal in ALK-Positive Lung Cancer through Zeolitic Imidazolate Framework-Based Mitochondrial Damage. Acta Biomater. 2024, 185, 381–395. [Google Scholar] [CrossRef]
- Qin, K.; Chen, K.; Zhao, W.; Zhao, X.; Luo, J.; Wang, Q.; Gao, C.; Li, X.; Wang, C. Methotrexate Combined with 4-Hydroperoxycyclophosphamide Downregulates Multidrug-Resistance P-Glycoprotein Expression Induced by Methotrexate in Rheumatoid Arthritis Fibroblast-Like Synoviocytes via the JAK2/STAT3 Pathway. J. Immunol. Res. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Imai, Y.; Shiseki, M.; Tanaka, J.; Motoji, T. Knockdown of the Wnt Receptor Frizzled-1 (FZD1) Reduces MDR1/P-Glycoprotein Expression in Multidrug Resistant Leukemic Cells and Inhibits Leukemic Cell Proliferation. Leuk. Res. 2018, 67, 99–108. [Google Scholar] [CrossRef]
- Eadie, L.N.; Hughes, T.P.; White, D.L. Interaction of the Efflux Transporters ABCB1 and ABCG2 With Imatinib, Nilotinib, and Dasatinib. Clin. Pharmacol. Ther. 2014, 95, 294–306. [Google Scholar] [CrossRef]
- Tariq, I.; Ali, M.Y.; Janga, H.; Ali, S.; Amin, M.U.; Ambreen, G.; Ali, U.; Pinnapireddy, S.R.; Schäfer, J.; Schulte, L.N.; et al. Downregulation of MDR 1 Gene Contributes to Tyrosine Kinase Inhibitor Induce Apoptosis and Reduction in Tumor Metastasis: A Gravity to Space Investigation. Int. J. Pharm. 2020, 591, 119993. [Google Scholar] [CrossRef] [PubMed]
- Mohseni, M.; Samadi, N.; Ghanbari, P.; Yousefi, B.; Tabasinezhad, M.; Sharifi, S.; Nazemiyeh, H. Co-Treatment by Docetaxel and Vinblastine Breaks down P-Glycoprotein Mediated Chemo-Resistance. Iran. J. Basic. Med. Sci. 2016, 19, 300–309. [Google Scholar]
- Kim, J.Y.; Park, Y.J.; Lee, B.-M.; Yoon, S. Co-Treatment With HIV Protease Inhibitor Nelfinavir Greatly Increases Late-Phase Apoptosis of Drug-Resistant KBV20C Cancer Cells Independently of P-Glycoprotein Inhibition. Anticancer. Res. 2019, 39, 3757–3765. [Google Scholar] [CrossRef] [PubMed]
- Dastvan, R.; Mishra, S.; Peskova, Y.B.; Nakamoto, R.K.; Mchaourab, H.S. Mechanism of Allosteric Modulation of P-Glycoprotein by Transport Substrates and Inhibitors. Science 2019, 364, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Mollazadeh, S.; Hadizadeh, F.; Ferreira, R.J. Theoretical Studies on 1,4-Dihydropyridine Derivatives as P-Glycoprotein Allosteric Inhibitors: Insights on Symmetry and Stereochemistry. J. Biomol. Struct. Dyn. 2021, 39, 4752–4763. [Google Scholar] [CrossRef]
- Mollazadeh, S.; Sahebkar, A.; Hadizadeh, F.; Behravan, J.; Arabzadeh, S. Structural and Functional Aspects of P-Glycoprotein and Its Inhibitors. Life Sci. 2018, 214, 118–123. [Google Scholar] [CrossRef]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef]
- Ekins, S.; Kim, R.B.; Leake, B.F.; Dantzig, A.H.; Schuetz, E.G.; Lan, L.-B.; Yasuda, K.; Shepard, R.L.; Winter, M.A.; Schuetz, J.D.; et al. Three-Dimensional Quantitative Structure-Activity Relationships of Inhibitors of P-Glycoprotein. Mol. Pharmacol. 2002, 61, 964–973. [Google Scholar] [CrossRef]
- Srivastava, S.; Choudhary, B.S.; Sharma, M.; Malik, R. Pharmacophore Modeling and 3D-QSAR Studies of Galloyl Benzamides as Potent P-Gp Inhibitors. Med. Chem. Res. 2016, 25, 1140–1147. [Google Scholar] [CrossRef]
- Kapoor, K.; Pant, S.; Tajkhorshid, E. Active Participation of Membrane Lipids in Inhibition of Multidrug Transporter P-Glycoprotein. Chem. Sci. 2021, 12, 6293–6306. [Google Scholar] [CrossRef]
- Gao, Y.; Wei, C.; Luo, L.; Tang, Y.; Yu, Y.; Li, Y.; Xing, J.; Pan, X. Membrane-Assisted Tariquidar Access and Binding Mechanisms of Human ATP-Binding Cassette Transporter P-Glycoprotein. Front. Mol. Biosci. 2024, 11. [Google Scholar] [CrossRef] [PubMed]
- Ooko, E.; Alsalim, T.; Saeed, B.; Saeed, M.E.M.; Kadioglu, O.; Abbo, H.S.; Titinchi, S.J.J.; Efferth, T. Modulation of P-Glycoprotein Activity by Novel Synthetic Curcumin Derivatives in Sensitive and Multidrug-Resistant T-Cell Acute Lymphoblastic Leukemia Cell Lines. Toxicol. Appl. Pharmacol. 2016, 305, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Reis, M.A.; Paterna, A.; Ferreira, R.J.; Lage, H.; Ferreira, M.-J.U. Macrocyclic Diterpenes Resensitizing Multidrug Resistant Phenotypes. Bioorg. Med. Chem. 2014, 22, 3696–3702. [Google Scholar] [CrossRef]
- Norouzi-Barough, L.; Sarookhani, M.; Salehi, R.; Sharifi, M.; Moghbelinejad, S. CRISPR/Cas9, a New Approach to Successful Knockdown of ABCB1/P-Glycoprotein and Reversal of Chemosensitivity in Human Epithelial Ovarian Cancer Cell Line. Iran. J. Basic. Med. Sci. 2018, 21, 181–187. [Google Scholar] [CrossRef]
- Liu, T.; Li, Z.; Zhang, Q.; Bernstein, K.D.A.; Lozano-Calderon, S.; Choy, E.; Hornicek, F.J.; Duan, Z. Targeting ABCB1 (MDR1) in Multi-Drug Resistant Osteosarcoma Cells Using the CRISPR-Cas9 System to Reverse Drug Resistance. Oncotarget 2016, 7, 83502–83513. [Google Scholar] [CrossRef]
- Chufan, E.E.; Kapoor, K.; Ambudkar, S.V. Drug–Protein Hydrogen Bonds Govern the Inhibition of the ATP Hydrolysis of the Multidrug Transporter P-Glycoprotein. Biochem. Pharmacol. 2016, 101, 40–53. [Google Scholar] [CrossRef]
- Lyu, J.H.; Huang, B.; Park, D.W.; Baek, S.H. Regulation of PHLDA1 Expression by JAK2-ERK1/2-STAT3 Signaling Pathway. J. Cell Biochem. 2016, 117, 483–490. [Google Scholar] [CrossRef]
- Ageeva, T.; Rizvanov, A.; Mukhamedshina, Y. NF-ΚB and JAK/STAT Signaling Pathways as Crucial Regulators of Neuroinflammation and Astrocyte Modulation in Spinal Cord Injury. Cells 2024, 13, 581. [Google Scholar] [CrossRef]
- Wang, H.; Wei, W.; Zhang, J.-P.; Song, Z.; Li, Y.; Xiao, W.; Liu, Y.; Zeng, M.-S.; Petrus, M.N.; Thomas, C.J.; et al. A Novel Model of Alternative NF-ΚB Pathway Activation in Anaplastic Large Cell Lymphoma. Leukemia 2021, 35, 1976–1989. [Google Scholar] [CrossRef]
- Shiraiwa, K.; Matsuse, M.; Nakazawa, Y.; Ogi, T.; Suzuki, K.; Saenko, V.; Xu, S.; Umezawa, K.; Yamashita, S.; Tsukamoto, K.; et al. JAK/STAT3 and NF-ΚB Signaling Pathways Regulate Cancer Stem-Cell Properties in Anaplastic Thyroid Cancer Cells. Thyroid. 2019, 29, 674–682. [Google Scholar] [CrossRef]
- Bharathiraja, P.; Balamurugan, K.; Govindasamy, C.; Prasad, N.R.; Pore, P.M. Solasodine Targets NF-ΚB Signaling to Overcome P-Glycoprotein Mediated Multidrug Resistance in Cancer. Exp. Cell Res. 2024, 441, 114153. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Dutta, P.; Mukherjee, P.; Datta, E.R.; Efferth, T.; Bhattacharya, S.; Choudhuri, S.K. Reversal of Drug Resistance in P-Glycoprotein-Expressing T-Cell Acute Lymphoblastic CEM Leukemia Cells by Copper N-(2-Hydroxy Acetophenone) Glycinate and Oxalyl Bis (N-Phenyl) Hydroxamic Acid. Cancer Lett. 2006, 244, 16–23. [Google Scholar] [CrossRef]
- Peters, W.; Wobbes, T.; Roelofs, H.; Jansen, J. Glutathione, Glutathione-S-Transferase And P-170 Glycoprotein in Metastases of Malignant Melanomas. Int. J. Oncol. 1994, 4, 1323–1327. [Google Scholar] [CrossRef]
- Mickisch, G.; Bier, H.; Bergler, W.; Bak, M.; Tschada, R.; Alken, P. P-170 Glycoprotein, Glutathione and Associated Enzymes in Relation to Chemoresistance of Primary Human Renal Cell Carcinomas. Urol. Int. 1990, 45, 170–176. [Google Scholar] [CrossRef]
- Terrier, P.; Townsend, A.J.; Coindre, J.M.; Triche, T.J.; Cowan, K.H. An Immunohistochemical Study of Pi Class Glutathione S-Transferase Expression in Normal Human Tissue. Am. J. Pathol. 1990, 137, 845–853. [Google Scholar]
- Chen, X.; Zheng, X.; Zhou, X.; Wang, W.; Liu, Q.; Xia, X. Clinical Analysis of Multidrug Resistance-Related Protein Expression in Gastric Cancer Cells. J. Food Drug Saf. Res. 2024, 1, 1–10. [Google Scholar] [CrossRef]
- Werle, M.; Hoffer, M. Glutathione and Thiolated Chitosan Inhibit Multidrug Resistance P-Glycoprotein Activity in Excised Small Intestine. J. Control. Release 2006, 111, 41–46. [Google Scholar] [CrossRef]
- Miller, T.P.; Grogan, T.M.; Dalton, W.S.; Spier, C.M.; Scheper, R.J.; Salmon, S.E. P-Glycoprotein Expression in Malignant Lymphoma and Reversal of Clinical Drug Resistance with Chemotherapy plus High-Dose Verapamil. J. Clin. Oncol. 1991, 9, 17–24. [Google Scholar] [CrossRef]
- Salmon, S.E.; Grogan, T.M.; Miller, T.; Scheper, R.; Dalton, W.S. Prediction of Doxorubicin Resistance in Vitro in Myeloma, Lymphoma, and Breast Cancer by P-Glycoprotein Staining. J. Natl. Cancer Inst. 1989, 81, 696–701. [Google Scholar] [CrossRef]
- Chen, J.; Du, Y.; Hou, H.; Li, W.; Sun, C.; Liang, F.; Wang, H. Unveiling the Correlation Between the Membrane Assembly of P-Gp and Drug Resistance in Multiple Myeloma Using Super-Resolution Fluorescence Imaging. Anal. Chem. 2024, 96, 11673–11681. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, B.; Wang, Z.; Cui, Y.; Ge, Z. P-Glycoprotein Detection for Multidrug Resistance of Leukemia Using SERS Immunoassay. Blood 2018, 132, 2218. [Google Scholar] [CrossRef]
- Wang, Y.; Zong, S.; Wu, L.; Zhang, Y.; Wang, Z.; Wang, Z.; Chen, B.; Cui, Y. Evaluation of Multidrug Resistance of Leukemia Using Surface-Enhanced Raman Scattering Method for Clinical Applications. ACS Appl. Mater. Interfaces 2018, 10, 24999–25005. [Google Scholar] [CrossRef] [PubMed]
- Quarti, J.; Torres, D.N.M.; Ferreira, E.; Vidal, R.S.; Casanova, F.; Chiarini, L.B.; Fialho, E.; Rumjanek, V.M. Selective Cytotoxicity of Piperine Over Multidrug Resistance Leukemic Cells. Molecules 2021, 26, 934. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Álvarez-Carrasco, P.; Morales-Villamil, F.; Maldonado-Bernal, C. P-Glycoprotein as a Therapeutic Target in Hematological Malignancies: A Challenge to Overcome. Int. J. Mol. Sci. 2025, 26, 4701. https://doi.org/10.3390/ijms26104701
Álvarez-Carrasco P, Morales-Villamil F, Maldonado-Bernal C. P-Glycoprotein as a Therapeutic Target in Hematological Malignancies: A Challenge to Overcome. International Journal of Molecular Sciences. 2025; 26(10):4701. https://doi.org/10.3390/ijms26104701
Chicago/Turabian StyleÁlvarez-Carrasco, Pablo, Fernanda Morales-Villamil, and Carmen Maldonado-Bernal. 2025. "P-Glycoprotein as a Therapeutic Target in Hematological Malignancies: A Challenge to Overcome" International Journal of Molecular Sciences 26, no. 10: 4701. https://doi.org/10.3390/ijms26104701
APA StyleÁlvarez-Carrasco, P., Morales-Villamil, F., & Maldonado-Bernal, C. (2025). P-Glycoprotein as a Therapeutic Target in Hematological Malignancies: A Challenge to Overcome. International Journal of Molecular Sciences, 26(10), 4701. https://doi.org/10.3390/ijms26104701