
Fingolimod Prevents Neuroinflammation but Has a Limited Effect on the Development of Ataxia in a Mouse Model for SCA1


 and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
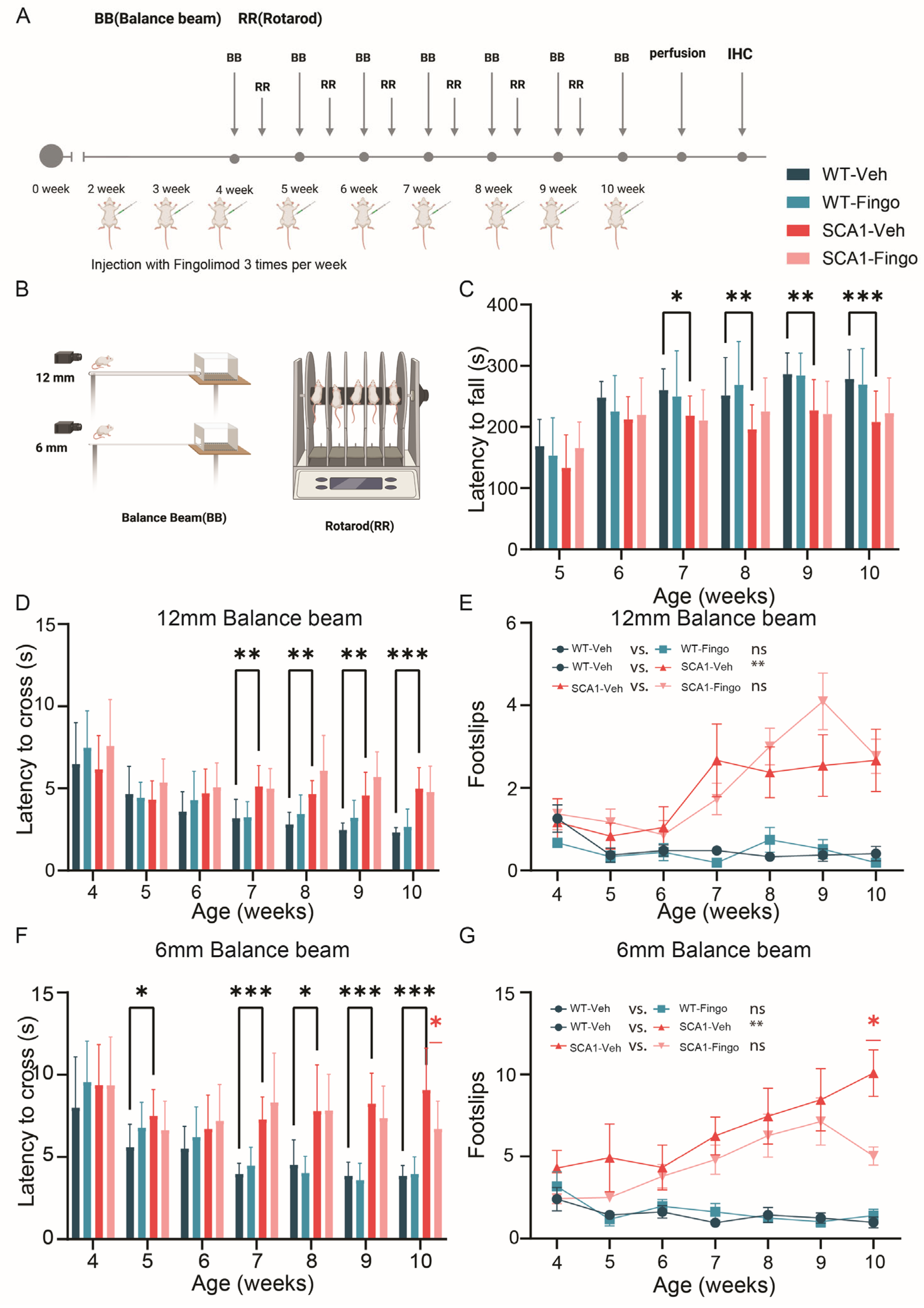
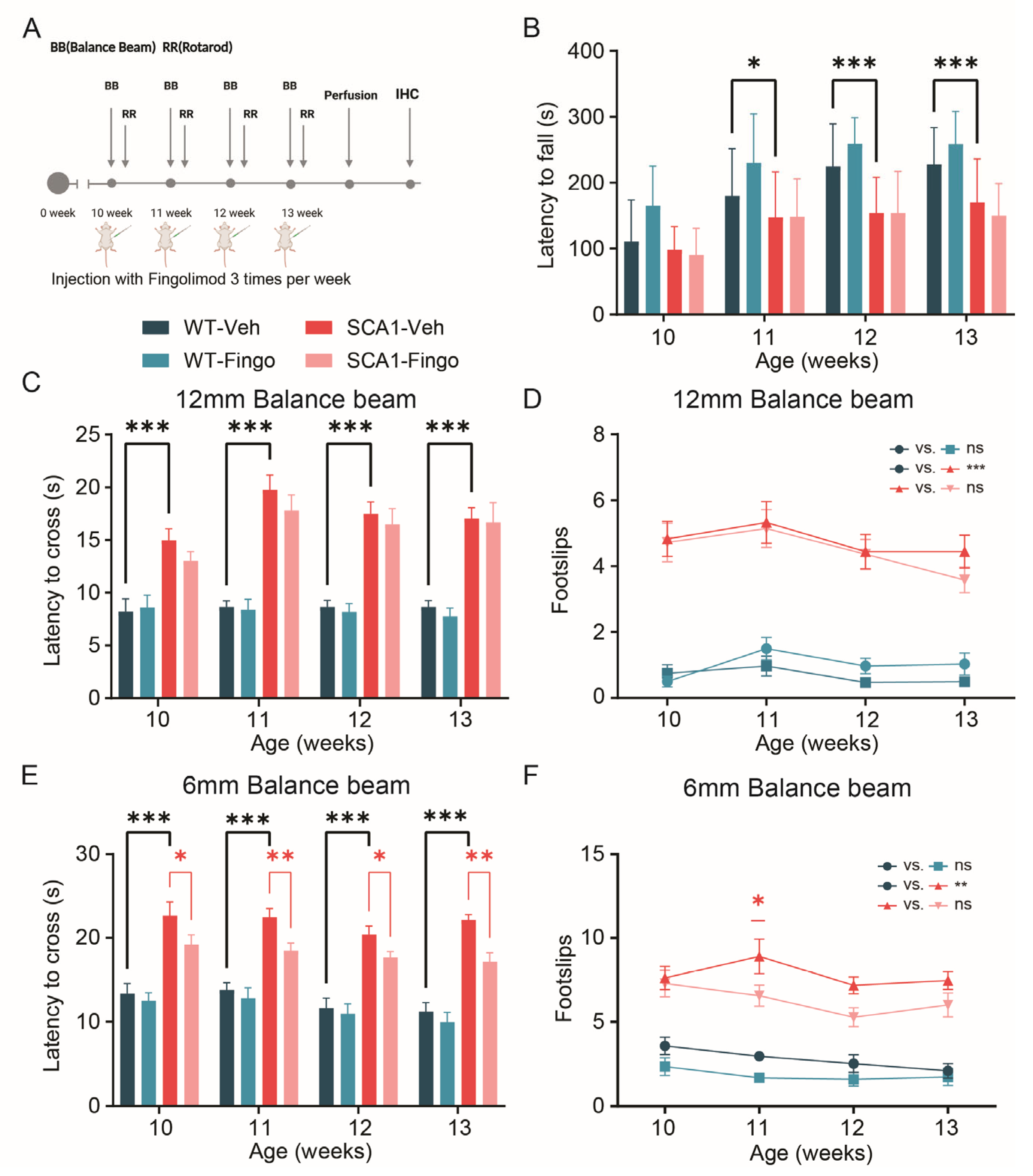
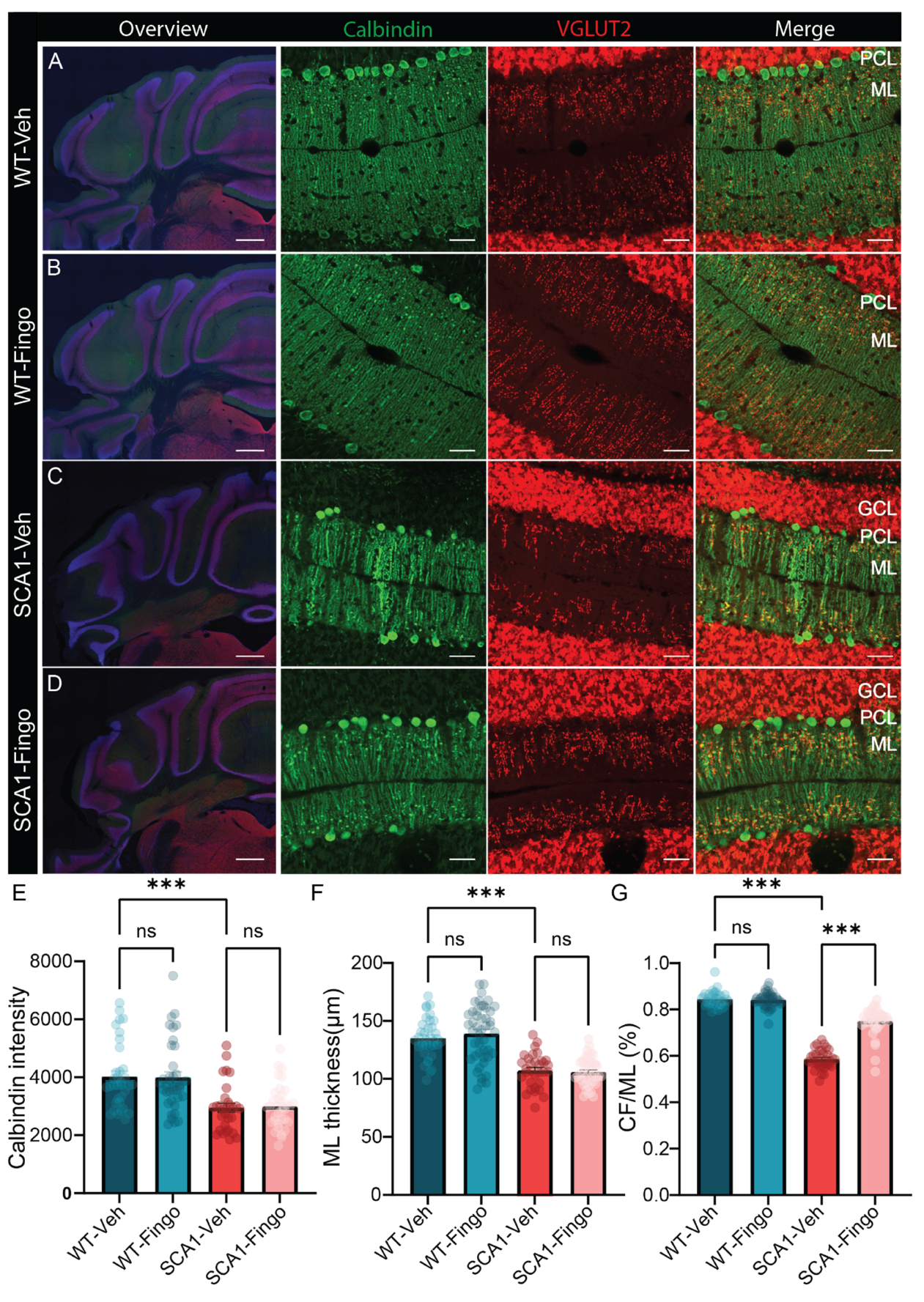
2.1. Minimal Improvement in Behavioral Deficits of SCA1 Mice Following Fingolimod Treatment
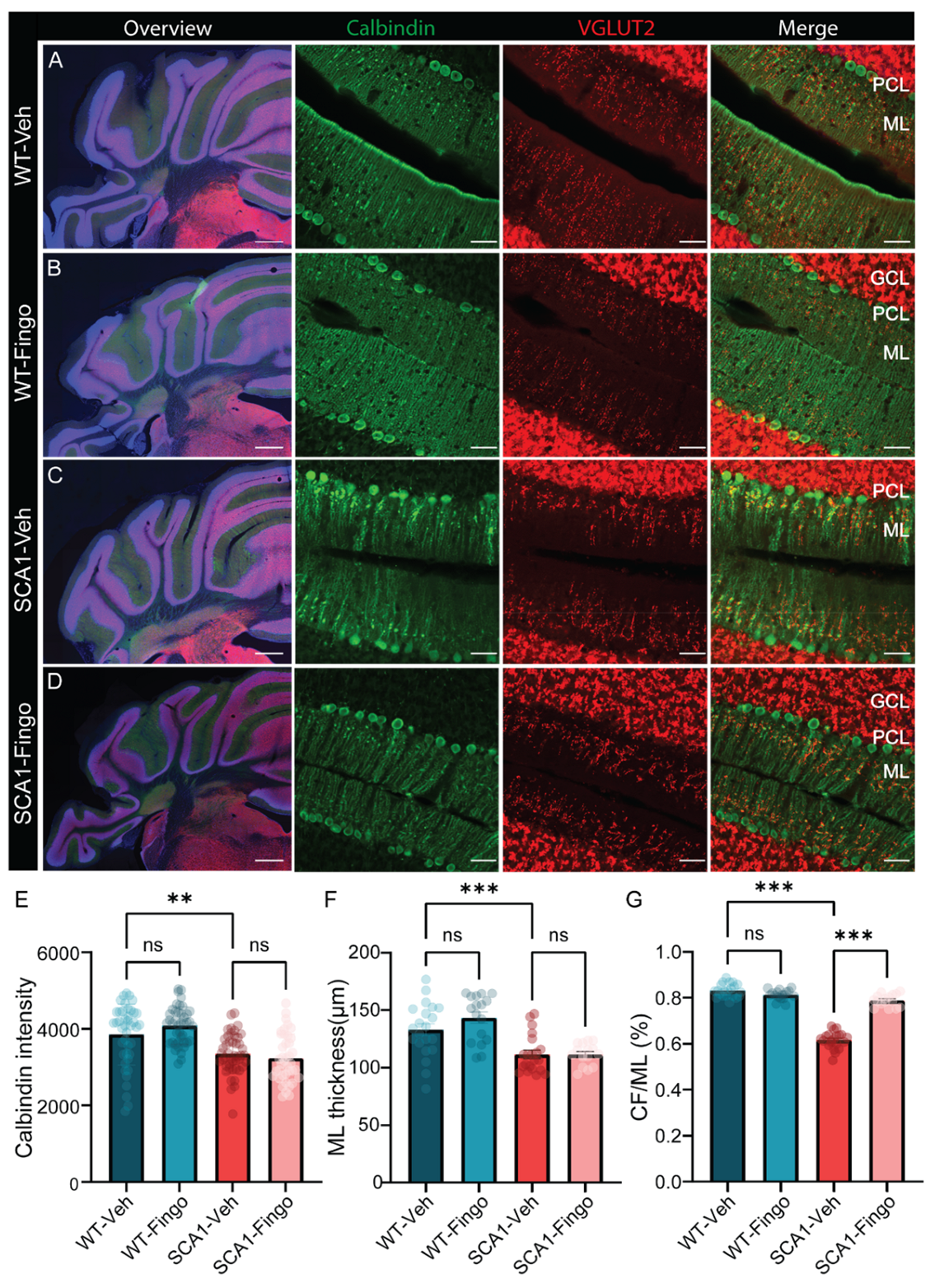
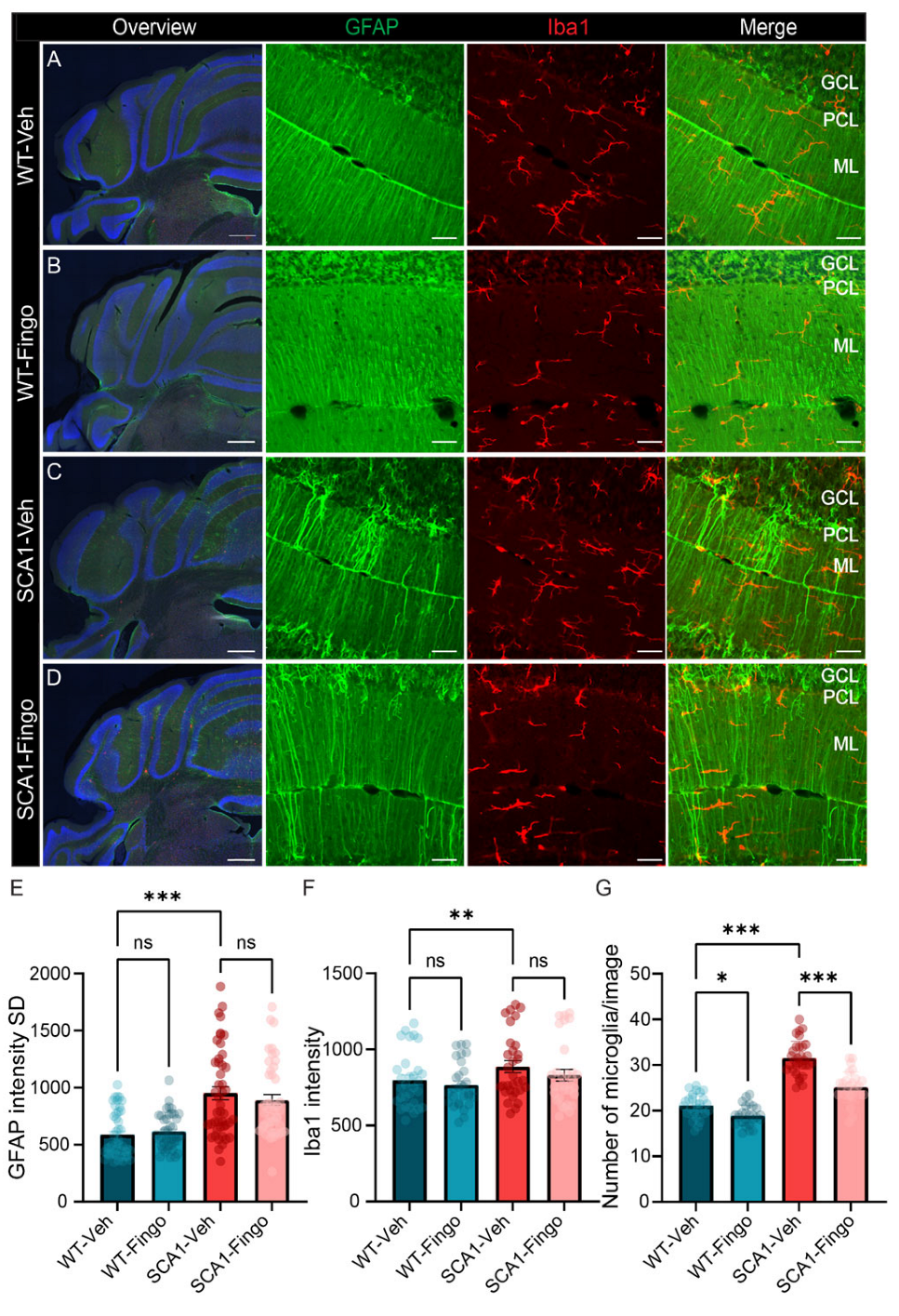
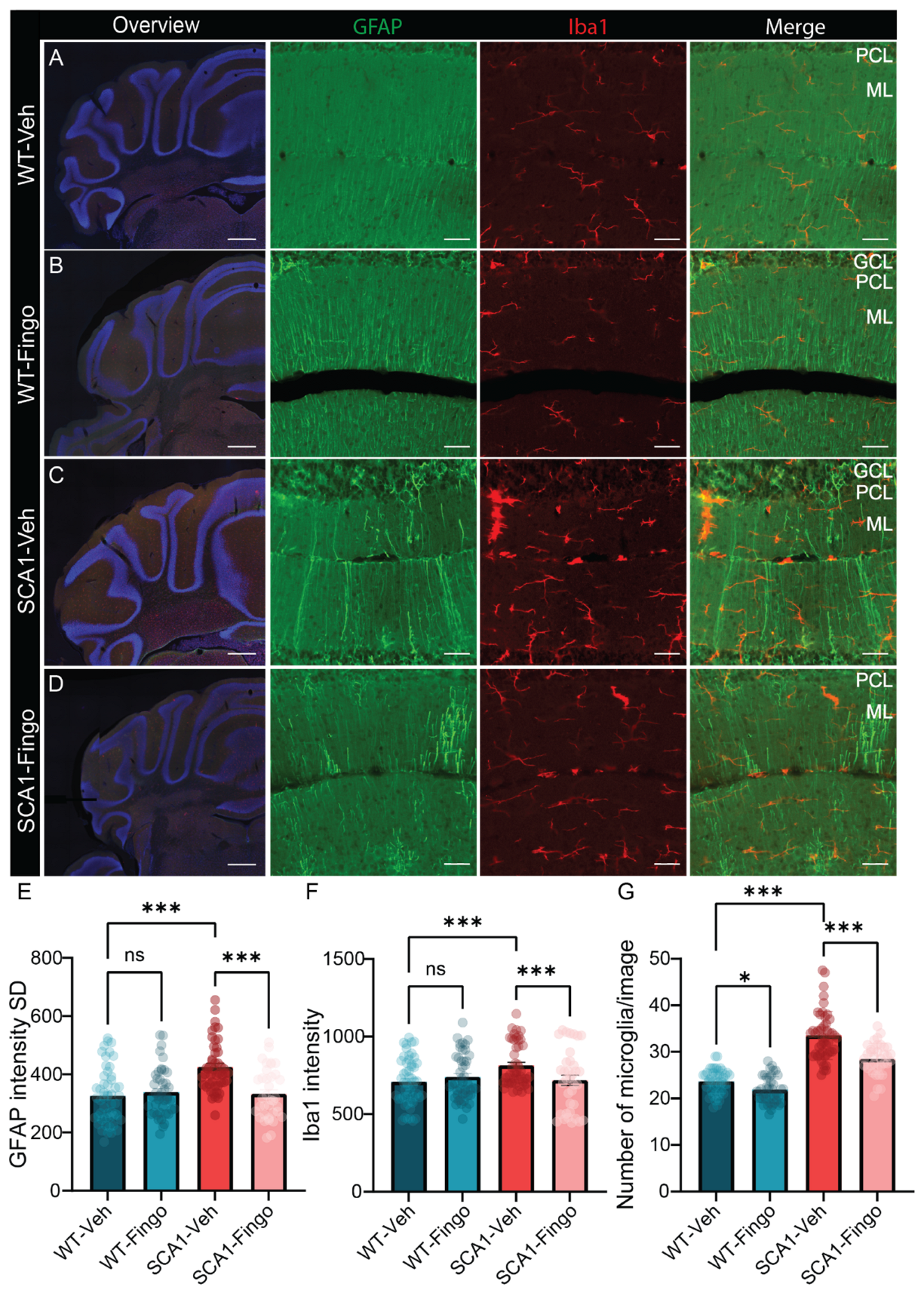
2.2. Fingolimod Attenuates the Inflammatory Responses in ATXN1[82Q] Mice
2.3. Delaying the Start of Treatment with Fingolimod Has a Minimal Positive Effect on the Behavioral Impairment in SCA1 Mice
2.4. Long-Term Use of Fingolimod Primarily Decreases Astrocyte Expression in SCA1 Mice in the Mid to Late Stage of the Disease
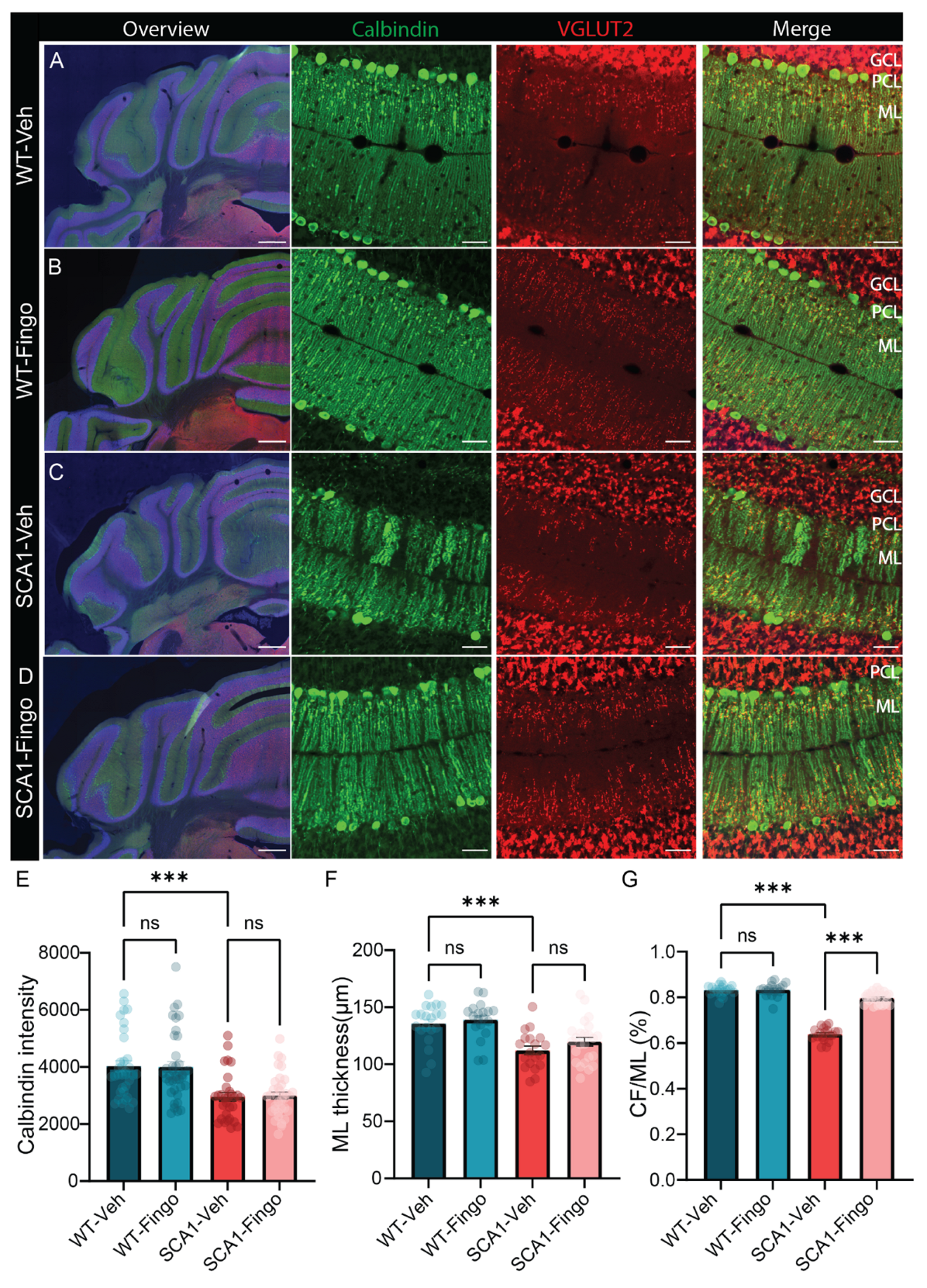
2.5. Short-Term Administration of Fingolimod Decreases Behavioral Deficits in SCA1 Mice in the Late Stages of the Disease
2.6. Short-Term Treatment with Fingolimod Is Also Capable of Reducing the Inflammatory Responses in SCA1 Mice
3. Discussion
4. Methods and Materials
4.1. Animals
4.2. Fingolimod Administration
- Regimen 1. Treatment for 8 weeks, starting week 2, behavioral analysis from week 4;
- Regimen 2. Treatment for 8 weeks, starting week 5, behavioral analysis from week 5;
- Regimen 3. Treatment for 4 weeks, starting week 10, behavioral analysis from week 10.
4.3. Balance Beam and Rotarod Tests
4.4. Tissue Collection
4.5. Immunohistochemistry
4.6. Imaging and Quantification
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar Ataxia. Nat. Rev. Dis. Prim. 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Horber, V.; Andersen, G.L.; Arnaud, C.; De La Cruz, J.; Dakovic, I.; Greitane, A.; Hensey, O.; Himmelmann, K.; Hollody, K.; Horridge, K.; et al. Prevalence, Clinical Features, Neuroimaging, and Genetic Findings in Children with Ataxic Cerebral Palsy in Europe. Neurology 2023, 101, e2509–e2521. [Google Scholar] [CrossRef] [PubMed]
- Tejwani, L.; Lim, J. Pathogenic Mechanisms Underlying Spinocerebellar Ataxia Type 1. Cell. Mol. Life Sci. 2020, 77, 4015–4029. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lukowicz, A.; Qu, W.; Johnson, A.; Cvetanovic, M. Astroglia Contribute to the Pathogenesis of Spinocerebellar Ataxia Type 1 (SCA1) in a Biphasic, Stage-of-Disease Specific Manner. Glia 2018, 66, 1972–1987. [Google Scholar] [CrossRef]
- Zühlke, C.; Dalski, A.; Hellenbroich, Y.; Bubel, S.; Schwinger, E.; Bürk, K. Spinocerebellar Ataxia Type 1 (SCA1): Phenotype-Genotype Correlation Studies in Intermediate Alleles. Eur. J. Hum. Genet. 2002, 10, 204–209. [Google Scholar] [CrossRef]
- Kerkhof, L.M.C.; van de Warrenburg, B.P.C.; van Roon-Mom, W.M.C.; Buijsen, R.A.M. Therapeutic Strategies for Spinocerebellar Ataxia Type 1. Biomolecules 2023, 13, 788. [Google Scholar] [CrossRef]
- Lieberman, A.P.; Shakkottai, V.G.; Albin, R.L. Polyglutamine Repeats in Neurodegenerative Diseases. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 1–27. [Google Scholar] [CrossRef]
- Paulson, H.L. The Spinocerebellar Ataxias. J. Neuro-Ophthalmol. 2009, 29, 227. [Google Scholar] [CrossRef]
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide Repeat Disorders. Annu. Rev. Neurosci. 2007, 30, 575–621. [Google Scholar] [CrossRef]
- Zoghbi, H.Y.; Orr, H.T. Pathogenic Mechanisms of a Polyglutamine-Mediated Neurodegenerative Disease, Spinocerebellar Ataxia Type 1. J Biol. Chem. 2009, 284, 7425–7429. [Google Scholar] [CrossRef]
- White, J.J.; Bosman, L.W.J.; Blot, F.G.C.; Osório, C.; Kuppens, B.W.; Krijnen, W.H.J.J.; Andriessen, C.; De Zeeuw, C.I.; Jaarsma, D.; Schonewille, M. Region-Specific Preservation of Purkinje Cell Morphology and Motor Behavior in the ATXN1[82Q] Mouse Model of Spinocerebellar Ataxia 1. Brain Pathol. 2021, 31, e12946. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetti, D.; Watase, K.; Xu, B.; Matzuk, M.M.; Orr, H.T.; Zoghbi, H.Y. Repeat Instability and Motor Incoordination in Mice with a Targeted Expanded CAG Repeat in the Sca1 Locus. Hum. Mol. Genet. 2000, 9, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Burright, E.N.; Brent Clark, H.; Servadio, A.; Matilla, T.; Feddersen, R.M.; Yunis, W.S.; Duvick, L.A.; Zoghbi, H.Y.; Orr, H.T. SCA1 Transgenic Mice: A Model for Neurodegeneration Caused by an Expanded CAG Trinucleotide Repeat. Cell 1995, 82, 937–948. [Google Scholar] [CrossRef]
- Hourez, R.; Servais, L.; Orduz, D.; Gall, D.; Millard, I.; de Kerchove d’Exaerde, A.; Cheron, G.; Orr, H.T.; Pandolfo, M.; Schiffmann, S.N. Aminopyridines Correct Early Dysfunction and Delay Neurodegeneration in a Mouse Model of Spinocerebellar Ataxia Type 1. J. Neurosci. 2011, 31, 11795–11807. [Google Scholar] [CrossRef]
- Rosa, J.-G.; Hamel, K.; Soles, A.; Sheeler, C.; Borgenheimer, E.; Gilliat, S.; Sbrocco, K.; Ghanoum, F.; Handler, H.; Forster, C.; et al. BDNF Is Altered in a Brain-Region Specific Manner and Rescues Deficits in Spinocerebellar Ataxia Type 1. Neurobiol. Dis. 2023, 178, 106023. [Google Scholar] [CrossRef]
- Asher, M.; Rosa, J.-G.; Cvetanovic, M. Mood Alterations in Mouse Models of Spinocerebellar Ataxia Type 1. Sci. Rep. 2021, 11, 713. [Google Scholar] [CrossRef]
- Osório, C.; White, J.J.; Lu, H.; Beekhof, G.C.; Fiocchi, F.R.; Andriessen, C.A.; Dijkhuizen, S.; Post, L.; Schonewille, M. Pre-Ataxic Loss of Intrinsic Plasticity and Motor Learning in a Mouse Model of SCA1. Brain 2023, 146, 2332–2345. [Google Scholar] [CrossRef]
- Ibrahim, M.F.; Power, E.M.; Potapov, K.; Empson, R.M. Motor and Cerebellar Architectural Abnormalities during the Early Progression of Ataxia in a Mouse Model of SCA1 and How Early Prevention Leads to a Better Outcome Later in Life. Front. Cell. Neurosci. 2017, 11, 292. [Google Scholar] [CrossRef]
- Sucha, M.; Benediktova, S.; Tichanek, F.; Jedlicka, J.; Kapl, S.; Jelinkova, D.; Purkartova, Z.; Tuma, J.; Kuncova, J.; Cendelin, J. Experimental Treatment with Edaravone in a Mouse Model of Spinocerebellar Ataxia 1. Int. J. Mol. Sci. 2023, 24, 10689. [Google Scholar] [CrossRef]
- Notartomaso, S.; Zappulla, C.; Biagioni, F.; Cannella, M.; Bucci, D.; Mascio, G.; Scarselli, P.; Fazio, F.; Weisz, F.; Lionetto, L.; et al. Pharmacological Enhancement of mGlu1 Metabotropic Glutamate Receptors Causes a Prolonged Symptomatic Benefit in a Mouse Model of Spinocerebellar Ataxia Type 1. Mol. Brain 2013, 6, 48. [Google Scholar] [CrossRef]
- Bushart, D.D.; Huang, H.; Man, L.J.; Morrison, L.M.; Shakkottai, V.G. A Chlorzoxazone-Baclofen Combination Improves Cerebellar Impairment in Spinocerebellar Ataxia Type 1. Mov. Disord. 2021, 36, 622–631. [Google Scholar] [CrossRef] [PubMed]
- Belozor, O.S.; Vasilev, A.; Mileiko, A.G.; Mosina, L.D.; Mikhailov, I.G.; Ox, D.A.; Boitsova, E.B.; Shuvaev, A.N.; Teschemacher, A.G.; Kasparov, S.; et al. Memantine Suppresses the Excitotoxicity but Fails to Rescue the Ataxic Phenotype in SCA1 Model Mice. Biomed. Pharmacother. 2024, 174, 116526. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.S.; Monteys, A.M.; Corbau, R.; Gonzalez-Alegre, P.; Davidson, B.L. RNAi Prevents and Reverses Phenotypes Induced by Mutant Human Ataxin-1. Ann. Neurol. 2016, 80, 754–765. [Google Scholar] [CrossRef]
- Keiser, M.S.; Kordower, J.H.; Gonzalez-Alegre, P.; Davidson, B.L. Broad Distribution of Ataxin 1 Silencing in Rhesus Cerebella for Spinocerebellar Ataxia Type 1 Therapy. Brain 2015, 138, 3555–3566. [Google Scholar] [CrossRef]
- Qu, W.; Johnson, A.; Kim, J.H.; Lukowicz, A.; Svedberg, D.; Cvetanovic, M. Inhibition of Colony-Stimulating Factor 1 Receptor Early in Disease Ameliorates Motor Deficits in SCA1 Mice. J. Neuroinflamm. 2017, 14, 107. [Google Scholar] [CrossRef]
- Heneka, M.T.; van der Flier, W.M.; Jessen, F.; Hoozemanns, J.; Thal, D.R.; Boche, D.; Brosseron, F.; Teunissen, C.; Zetterberg, H.; Jacobs, A.H.; et al. Neuroinflammation in Alzheimer disease. Nat. Rev. Immunol. 2025, 25, 321–352. [Google Scholar] [CrossRef]
- Edamakanti, C.R.; Mohan, V.; Opal, P. Reactive Bergmann Glia Play a Central Role in Spinocerebellar Ataxia Inflammation via the JNK Pathway. J. Neuroinflamm. 2023, 20, 126. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Zhao, J.-W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of Microglial Phagocytosis Is Sufficient to Prevent Inflammatory Neuronal Death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef]
- Kappos, L.; Antel, J.; Comi, G.; Montalban, X.; O’Connor, P.; Polman, C.H.; Haas, T.; Korn, A.A.; Karlsson, G.; Radue, E.W. Oral Fingolimod (FTY720) for Relapsing Multiple Sclerosis. N. Engl. J. Med. 2006, 355, 1124–1140. [Google Scholar] [CrossRef]
- Blot, F.G.C.; Krijnen, W.H.J.J.; Den Hoedt, S.; Osório, C.; White, J.J.; Mulder, M.T.; Schonewille, M. Sphingolipid Metabolism Governs Purkinje Cell Patterned Degeneration in ATXN1[82Q]/+ Mice. Proc. Natl. Acad. Sci. USA 2021, 118, e2016969118. [Google Scholar] [CrossRef]
- Nijnik, A.; Clare, S.; Hale, C.; Chen, J.; Raisen, C.; Mottram, L.; Lucas, M.; Estabel, J.; Ryder, E.; Adissu, H.; et al. The Role of Sphingosine-1-Phosphate Transporter Spns2 in Immune System Function. J. Immunol. 2012, 189, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Hartung, H.-P. Mechanism of Action of Oral Fingolimod (FTY720) in Multiple Sclerosis. Clin. Neuropharmacol. 2010, 33, 91. [Google Scholar] [CrossRef] [PubMed]
- Salas-Perdomo, A.; Miró-Mur, F.; Gallizioli, M.; Brait, V.H.; Justicia, C.; Meissner, A.; Urra, X.; Chamorro, A.; Planas, A.M. Role of the S1P Pathway and Inhibition by Fingolimod in Preventing Hemorrhagic Transformation after Stroke. Sci. Rep. 2019, 9, 8309. [Google Scholar] [CrossRef] [PubMed]
- Bravo, G.Á.; Cedeño, R.R.; Casadevall, M.P.; Ramió-Torrentà, L. Sphingosine-1-Phosphate (S1P) and S1P Signaling Pathway Modulators, from Current Insights to Future Perspectives. Cells 2022, 11, 2058. [Google Scholar] [CrossRef]
- Kappos, L.; Radue, E.-W.; O’Connor, P.; Polman, C.; Hohlfeld, R.; Calabresi, P.; Selmaj, K.; Agoropoulou, C.; Leyk, M.; Zhang-Auberson, L.; et al. A Placebo-Controlled Trial of Oral Fingolimod in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2010, 362, 387–401. [Google Scholar] [CrossRef]
- Noteboom, S.; Seiler, M.; Chien, C.; Rane, R.P.; Barkhof, F.; Strijbis, E.M.M.; Paul, F.; Schoonheim, M.M.; Ritter, K. Evaluation of Machine Learning-Based Classification of Clinical Impairment and Prediction of Clinical Worsening in Multiple Sclerosis. J. Neurol. 2024, 271, 5577–5589. [Google Scholar] [CrossRef]
- Carreras, I.; Aytan, N.; Choi, J.-K.; Tognoni, C.M.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. Dual Dose-Dependent Effects of Fingolimod in a Mouse Model of Alzheimer’s Disease. Sci. Rep. 2019, 9, 10972. [Google Scholar] [CrossRef]
- Ferro, A.; Sheeler, C.; Rosa, J.-G.; Cvetanovic, M. Role of Microglia in Ataxias. J. Mol. Biol. 2019, 431, 1792–1804. [Google Scholar] [CrossRef]
- Lee, J.Y.; Han, S.H.; Park, M.H.; Baek, B.; Song, I.-S.; Choi, M.-K.; Takuwa, Y.; Ryu, H.; Kim, S.H.; He, X.; et al. Neuronal SphK1 Acetylates COX2 and Contributes to Pathogenesis in a Model of Alzheimer’s Disease. Nat. Commun. 2018, 9, 1479. [Google Scholar] [CrossRef]
- Standoli, S.; Rapino, C.; Di Meo, C.; Rudowski, A.; Kämpfer-Kolb, N.; Volk, L.M.; Thomas, D.; Trautmann, S.; Schreiber, Y.; Meyer zu Heringdorf, D.; et al. Sphingosine Kinases at the Intersection of Pro-Inflammatory LPS and Anti-Inflammatory Endocannabinoid Signaling in BV2 Mouse Microglia Cells. Int. J. Mol. Sci. 2023, 24, 8508. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M. Astrocyte Reactivity and Reactive Astrogliosis: Costs and Benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Kuo, J.R.; Wang, S.J. Fingolimod Inhibits Glutamate Release through Activation of S1P1 Receptors and the G Protein Βγ Subunit-Dependent Pathway in Rat Cerebrocortical Nerve Terminals. Neuropharmacology 2021, 185, 108451. [Google Scholar] [CrossRef] [PubMed]
- Barra, G.; Lepore, A.; Gagliardi, M.; Somma, D.; Matarazzo, M.R.; Costabile, F.; Pasquale, G.; Mazzoni, A.; Gallo, C.; Nuzzo, G.; et al. Sphingosine Kinases Promote IL-17 Expression in Human T Lymphocytes. Sci. Rep. 2018, 8, 13233. [Google Scholar] [CrossRef]
- Avni, D.; Harikumar, K.B.; Sanyal, A.J.; Spiegel, S. Deletion or Inhibition of SphK1 Mitigates Fulminant Hepatic Failure by Suppressing TNFα-Dependent Inflammation and Apoptosis. FASEB J. 2021, 35, e21415. [Google Scholar] [CrossRef]
- Nayak, D.; Huo, Y.; Kwang, W.X.T.; Pushparaj, P.N.; Kumar, S.D.; Ling, E.-A.; Dheen, S.T. Sphingosine Kinase 1 Regulates the Expression of Proinflammatory Cytokines and Nitric Oxide in Activated Microglia. Neuroscience 2010, 166, 132–144. [Google Scholar] [CrossRef]
- Sapkota, A.; Gaire, B.P.; Kang, M.-G.; Choi, J.W. S1P2 Contributes to Microglial Activation and M1 Polarization Following Cerebral Ischemia through ERK1/2 and JNK. Sci. Rep. 2019, 9, 12106. [Google Scholar] [CrossRef]
- Dong, Y.-F.; Guo, R.-B.; Ji, J.; Cao, L.-L.; Zhang, L.; Chen, Z.-Z.; Huang, J.-Y.; Wu, J.; Lu, J.; Sun, X.-L. S1PR3 Is Essential for Phosphorylated Fingolimod to Protect Astrocytes against Oxygen-Glucose Deprivation-Induced Neuroinflammation via Inhibiting TLR2/4-NFκB Signalling. J. Cell Mol. Med. 2018, 22, 3159–3166. [Google Scholar] [CrossRef]
- Kuo, S.-H.; Lin, C.-Y.; Wang, J.; Sims, P.A.; Pan, M.-K.; Liou, J.; Lee, D.; Tate, W.J.; Kelly, G.C.; Louis, E.D.; et al. Climbing Fiber-Purkinje Cell Synaptic Pathology in Tremor and Cerebellar Degenerative Diseases. Acta Neuropathol. 2017, 133, 121–138. [Google Scholar] [CrossRef]
- Nanclares, C.; Noriega Prieto, J.; Labrada-Moncada, E.; Cvetanovic, M.; Araque, A.; Kofuji, P. Altered Calcium Signaling in Bergmann Glia Contributes to Spinocerebellar Ataxia Type-1 in a Mouse Model of SCA1. Neurobiol. Dis. 2023, 187, 106318. [Google Scholar] [CrossRef]
- Stellwagen, D.; Malenka, R.C. Synaptic Scaling Mediated by Glial TNF-α. Nature 2006, 440, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Ferro, A.; Qu, W.; Lukowicz, A.; Svedberg, D.; Johnson, A.; Cvetanovic, M. Inhibition of NF-κB Signaling in IKKβF/F;LysM Cre Mice Causes Motor Deficits but Does Not Alter Pathogenesis of Spinocerebellar Ataxia Type 1. PLoS ONE 2018, 13, e0200013. [Google Scholar] [CrossRef]
- Billich, A.; Bornancin, F.; Dévay, P.; Mechtcheriakova, D.; Urtz, N.; Baumruker, T. Phosphorylation of the Immunomodulatory Drug FTY720 by Sphingosine Kinases. J. Biol. Chem. 2003, 278, 47408–47415. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Zangemeister-Wittke, U. The Sphingosine 1-Phosphate Receptor Modulator Fingolimod as a Therapeutic Agent: Recent Findings and New Perspectives. Pharmacol. Ther. 2018, 185, 34–49. [Google Scholar] [CrossRef]
- Lee, J.; Ryu, J.; Yoon, G.; Jeon, H.; Cho, Y.; Choi, J.; Song, S.Y.; Do, I.; Lee, Y.; Kim, T.; et al. Sphingosine Kinase 1 as a Potential Therapeutic Target in Epithelial Ovarian Cancer. Int. J. Cancer 2014, 137, 221–229. [Google Scholar] [CrossRef]
- Nagahashi, M.; Abe, M.; Sakimura, K.; Takabe, K.; Wakai, T. The Role of Sphingosine-1-Phosphate in Inflammation and Cancer Progression. Cancer Sci. 2018, 109, 3671–3678. [Google Scholar] [CrossRef]
- Yi, X.; Tang, X.; Li, T.; Chen, L.; He, H.; Wu, X.; Xiang, C.; Cao, M.; Wang, Z.; Wang, Y.; et al. Therapeutic Potential of the Sphingosine Kinase 1 Inhibitor, PF-543. Biomed. Pharmacother. 2023, 163, 114401. [Google Scholar] [CrossRef]
- Bu, Y.; Wu, H.; Deng, R.; Wang, Y. Therapeutic Potential of SphK1 Inhibitors Based on Abnormal Expression of SphK1 in Inflammatory Immune Related-Diseases. Front. Pharmacol. 2021, 12, 733387. [Google Scholar] [CrossRef]
- L’Italien, G.J.; Oikonomou, E.K.; Khera, R.; Potashman, M.H.; Beiner, M.W.; Maclaine, G.D.H.; Schmahmann, J.D.; Perlman, S.; Coric, V. Video-Based Kinematic Analysis of Movement Quality in a Phase 3 Clinical Trial of Troriluzole in Adults with Spinocerebellar Ataxia: A Post Hoc Analysis. Neurol. Ther. 2024, 13, 1287–1301. [Google Scholar] [CrossRef]
- Potashman, M.; Popoff, E.; Powell, L.; Mackenzie, A.; Beiner, M.W.; Coric, V.; Schmahmann, J.; L’Italien, G. Psychometric Validation of the Modified Functional Scale for the Assessment and Rating of Ataxia (f-SARA) in Patients with Spinocerebellar Ataxia. Cerebellum 2024, 23, 2095–2108. [Google Scholar] [CrossRef]
- Osório, C.; White, J.J.; Solé, P.T.; Mandemaker, N.; Olivero, F.; Kirwan, F.; de Winter, F.; Regolo, E.; Fiocchi, F.R.; Serra, I.; et al. Purkinje Cell Intrinsic Activity Shapes Cerebellar Development and Function. biorxiv 2024. [Google Scholar] [CrossRef]
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.-M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A Central Element in Neurological Diseases. Acta Neuropathol. 2016, 131, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Gleixner, A.M.; Posimo, J.M.; Pant, D.B.; Henderson, M.P.; Leak, R.K. Astrocytes Surviving Severe Stress Can Still Protect Neighboring Neurons from Proteotoxic Injury. Mol. Neurobiol. 2016, 53, 4939–4960. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, C.; Gravendeel, N.; On, A.C.; Post, L.; van Bergen, R.; Osorio, C.; Schonewille, M. Fingolimod Prevents Neuroinflammation but Has a Limited Effect on the Development of Ataxia in a Mouse Model for SCA1. Int. J. Mol. Sci. 2025, 26, 4698. https://doi.org/10.3390/ijms26104698
Yang C, Gravendeel N, On AC, Post L, van Bergen R, Osorio C, Schonewille M. Fingolimod Prevents Neuroinflammation but Has a Limited Effect on the Development of Ataxia in a Mouse Model for SCA1. International Journal of Molecular Sciences. 2025; 26(10):4698. https://doi.org/10.3390/ijms26104698
Chicago/Turabian StyleYang, Chen, Nienke Gravendeel, Amy Chin On, Laura Post, Ryan van Bergen, Catarina Osorio, and Martijn Schonewille. 2025. "Fingolimod Prevents Neuroinflammation but Has a Limited Effect on the Development of Ataxia in a Mouse Model for SCA1" International Journal of Molecular Sciences 26, no. 10: 4698. https://doi.org/10.3390/ijms26104698
APA StyleYang, C., Gravendeel, N., On, A. C., Post, L., van Bergen, R., Osorio, C., & Schonewille, M. (2025). Fingolimod Prevents Neuroinflammation but Has a Limited Effect on the Development of Ataxia in a Mouse Model for SCA1. International Journal of Molecular Sciences, 26(10), 4698. https://doi.org/10.3390/ijms26104698