
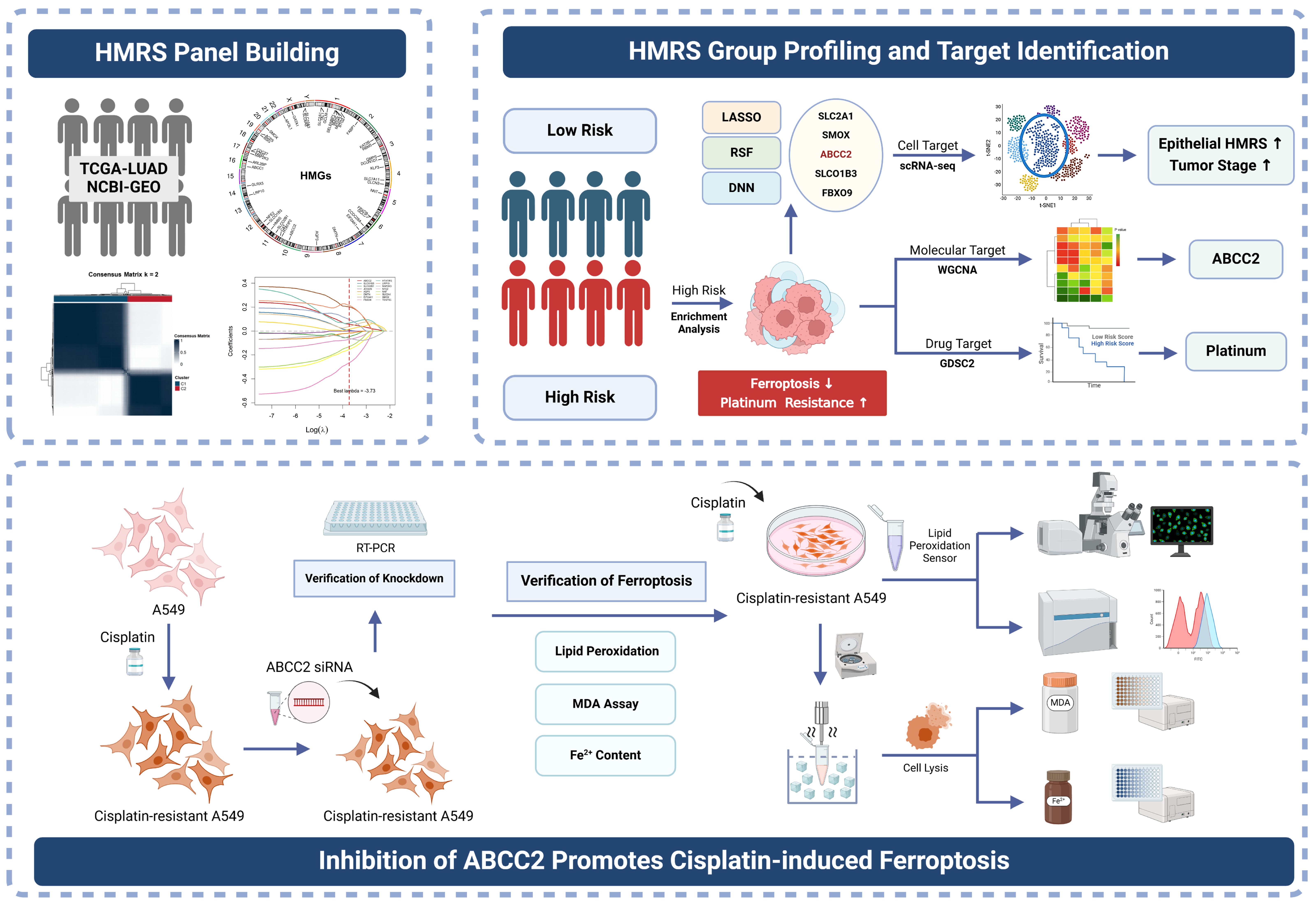
Integrating Bulk and Single-Cell Transcriptomics with Machine Learning Reveals a Heme Metabolism-Based Panel for Lung Adenocarcinoma Chemotherapy Resistance

 , and
, and
Abstract
1. Introduction
2. Results
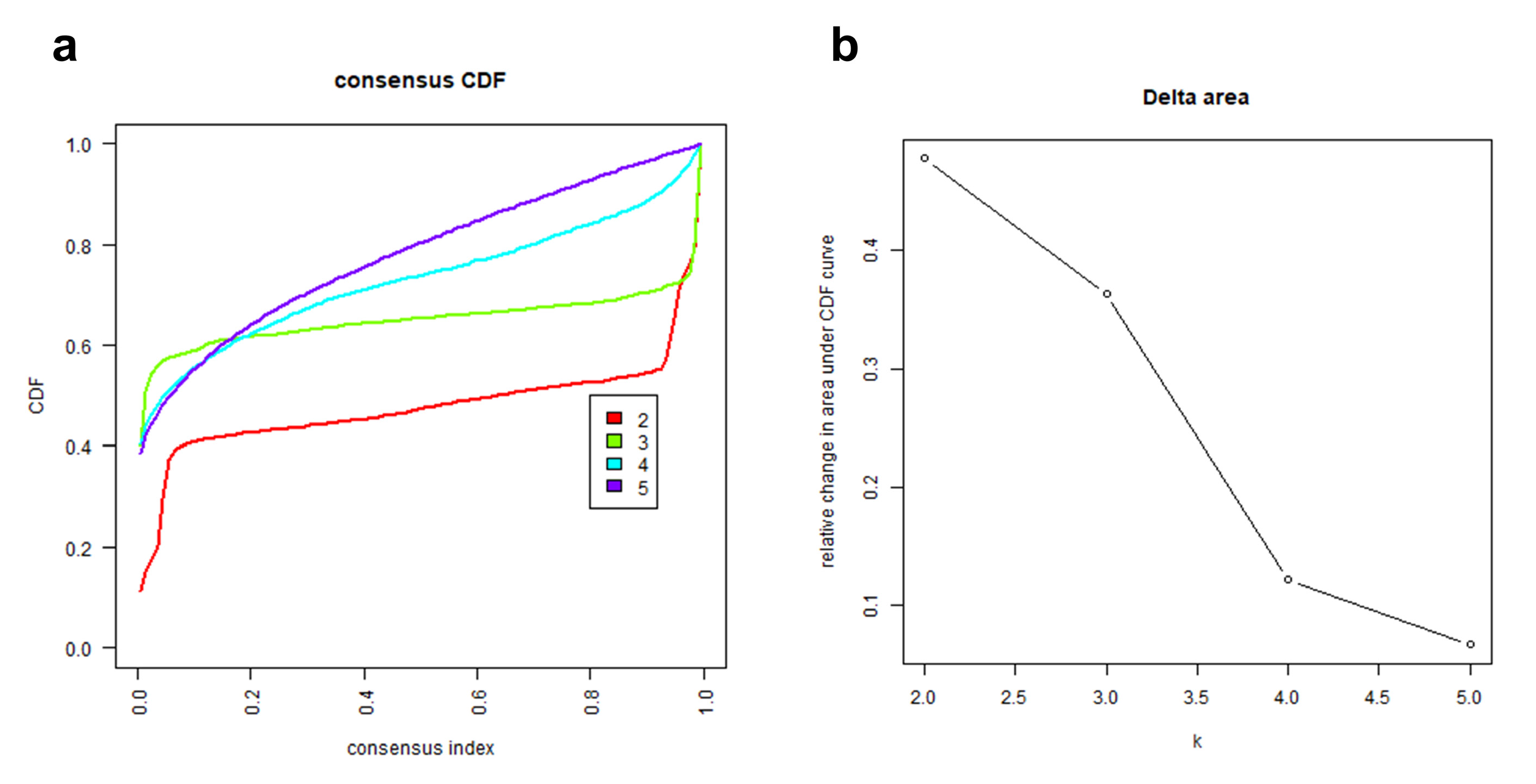
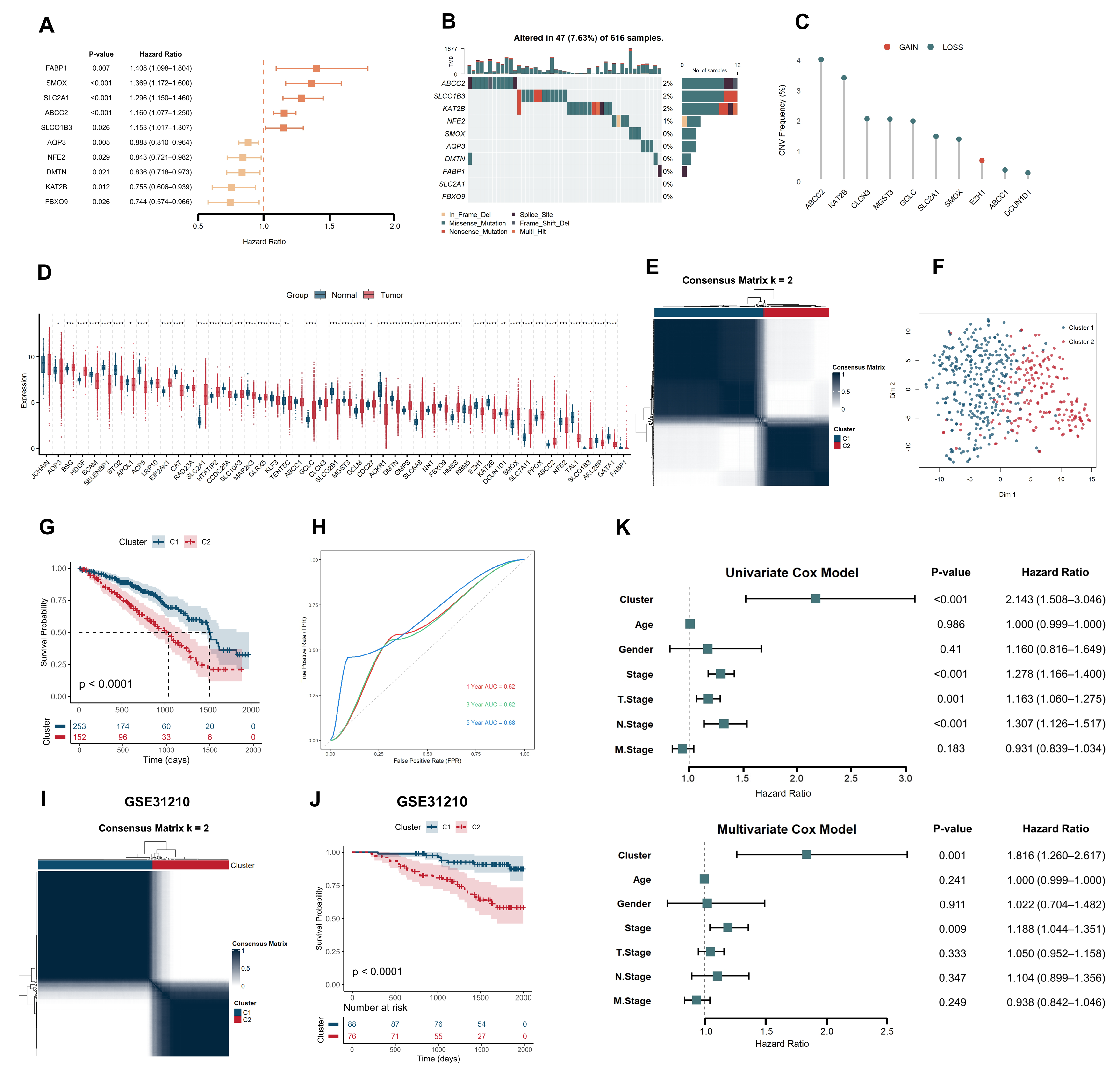
2.1. Heme Metabolism-Based Clusters Predict Prognosis in LUAD
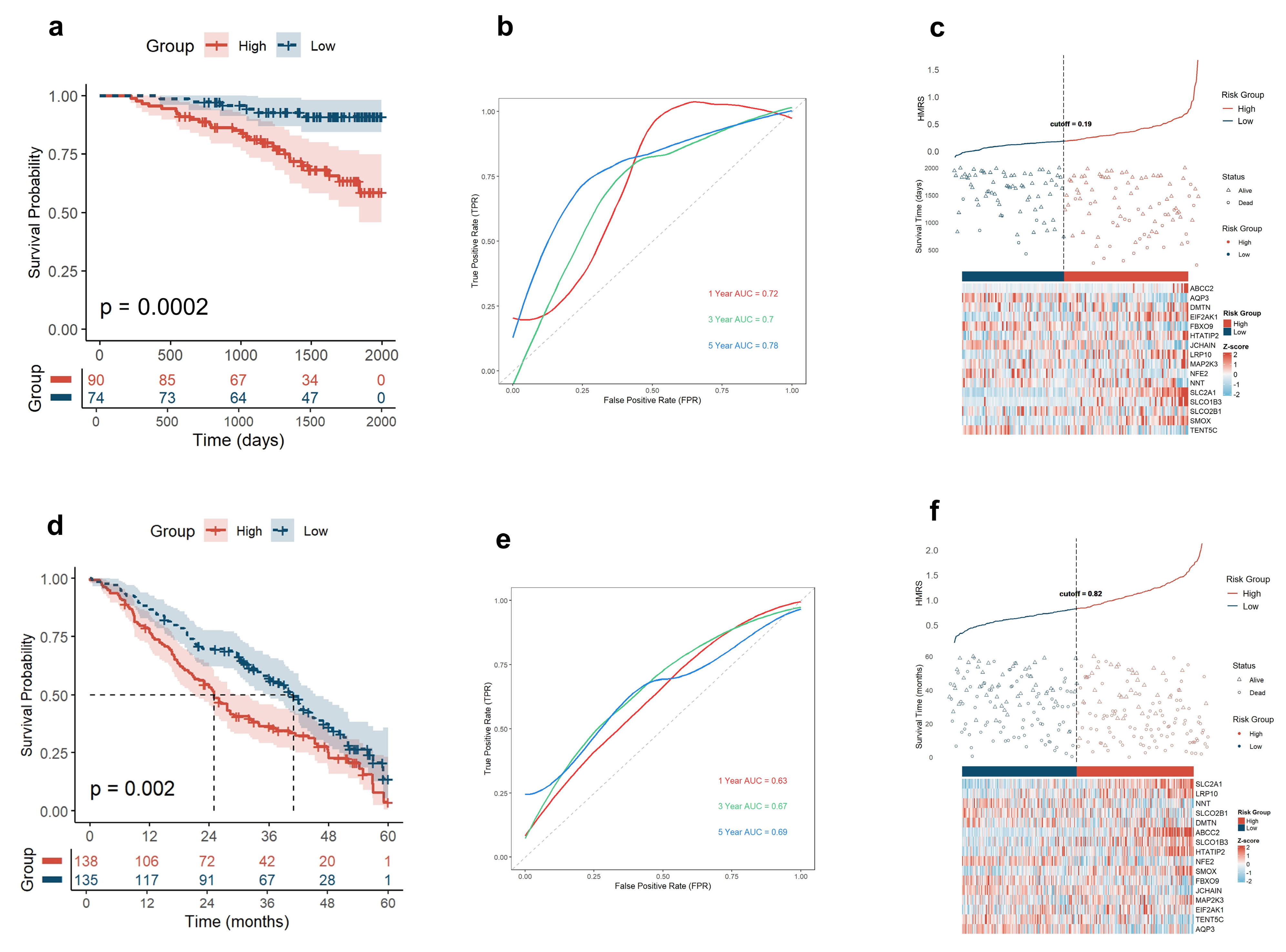
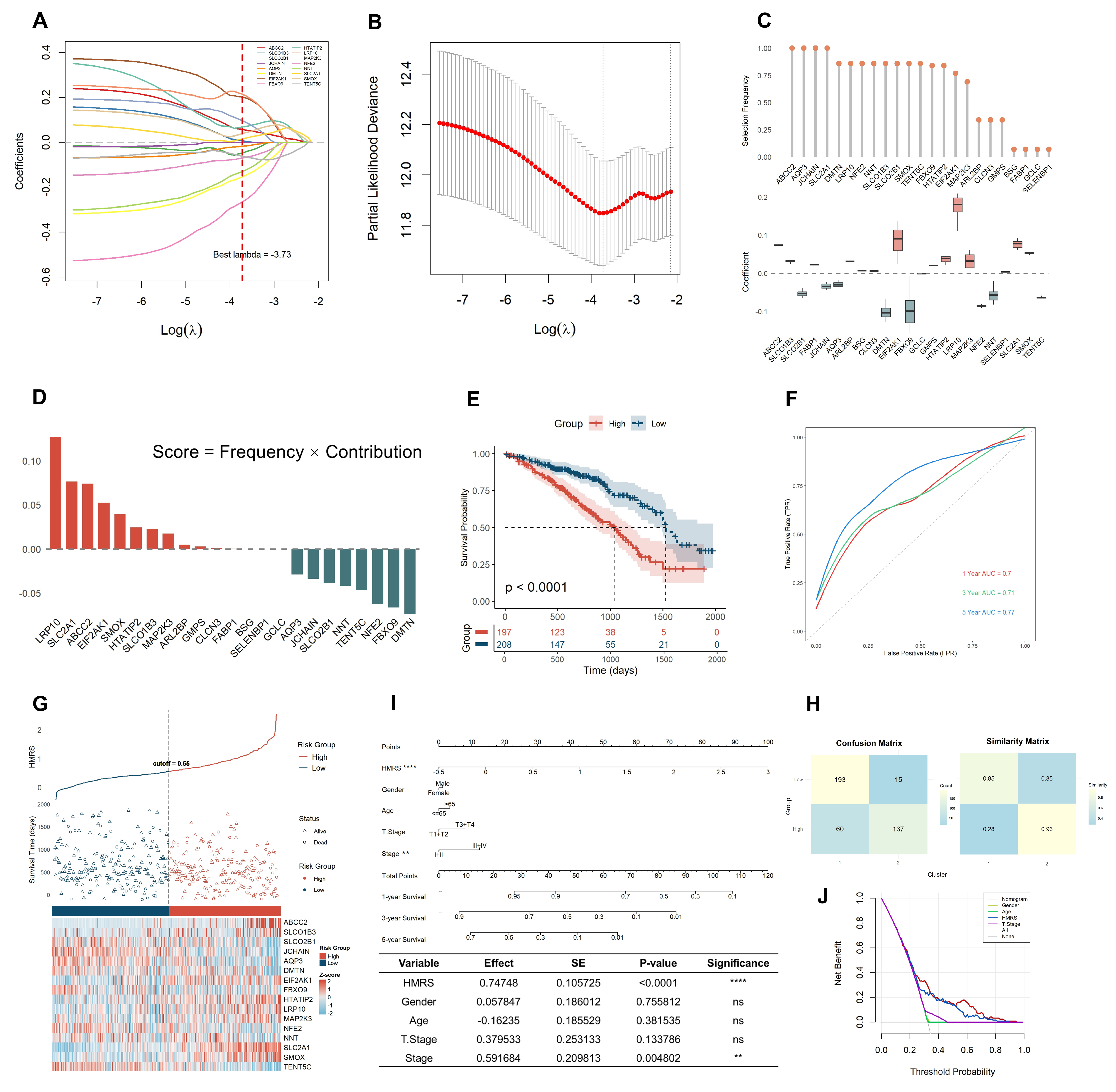
2.2. HMRS Panel Demonstrates Robust Prognostic Utility in LUAD Risk Stratification
2.3. Metabolic Reprogramming and Ferroptosis Regulation Are Key Differences in HMRS-Based Groups
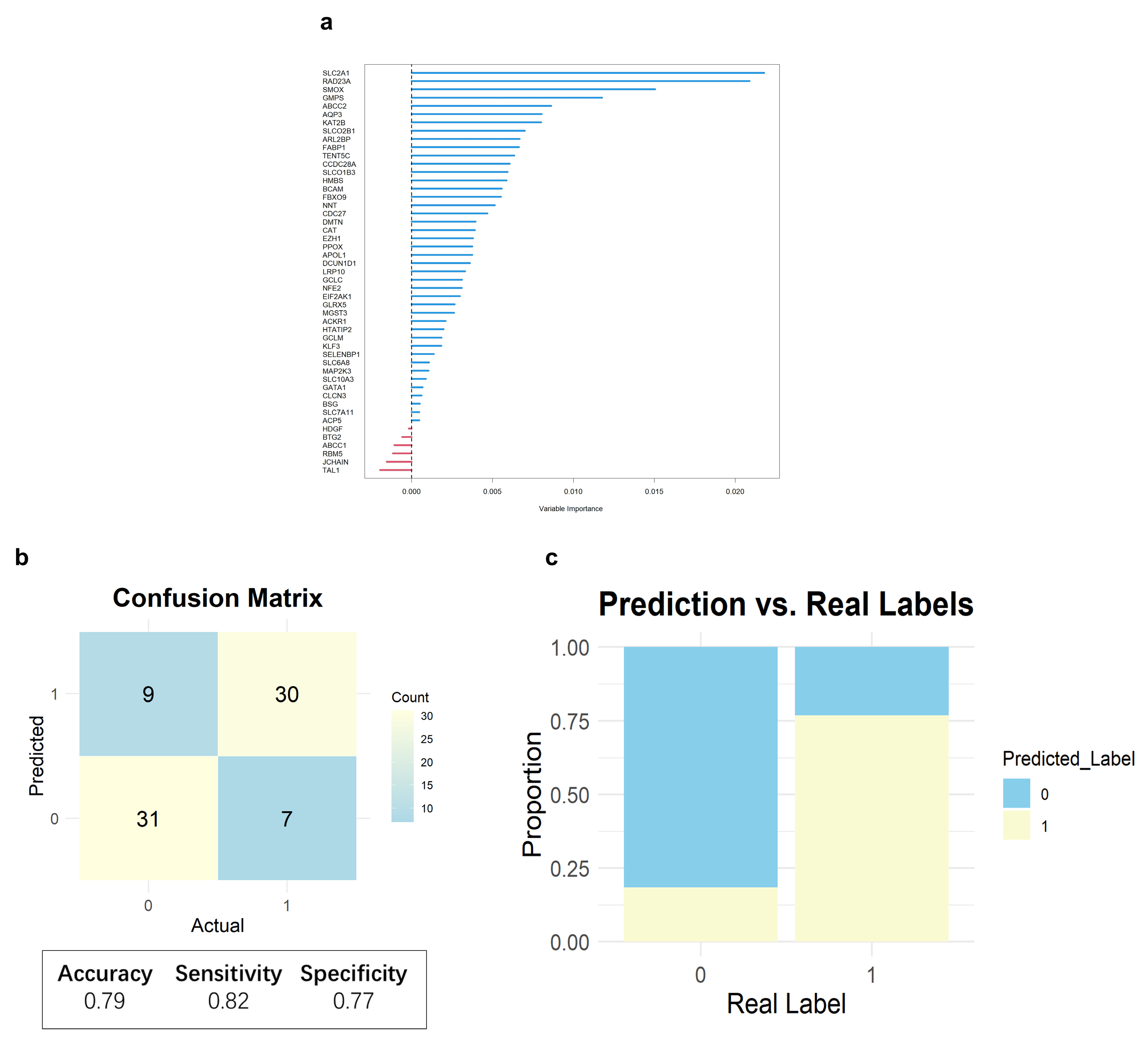
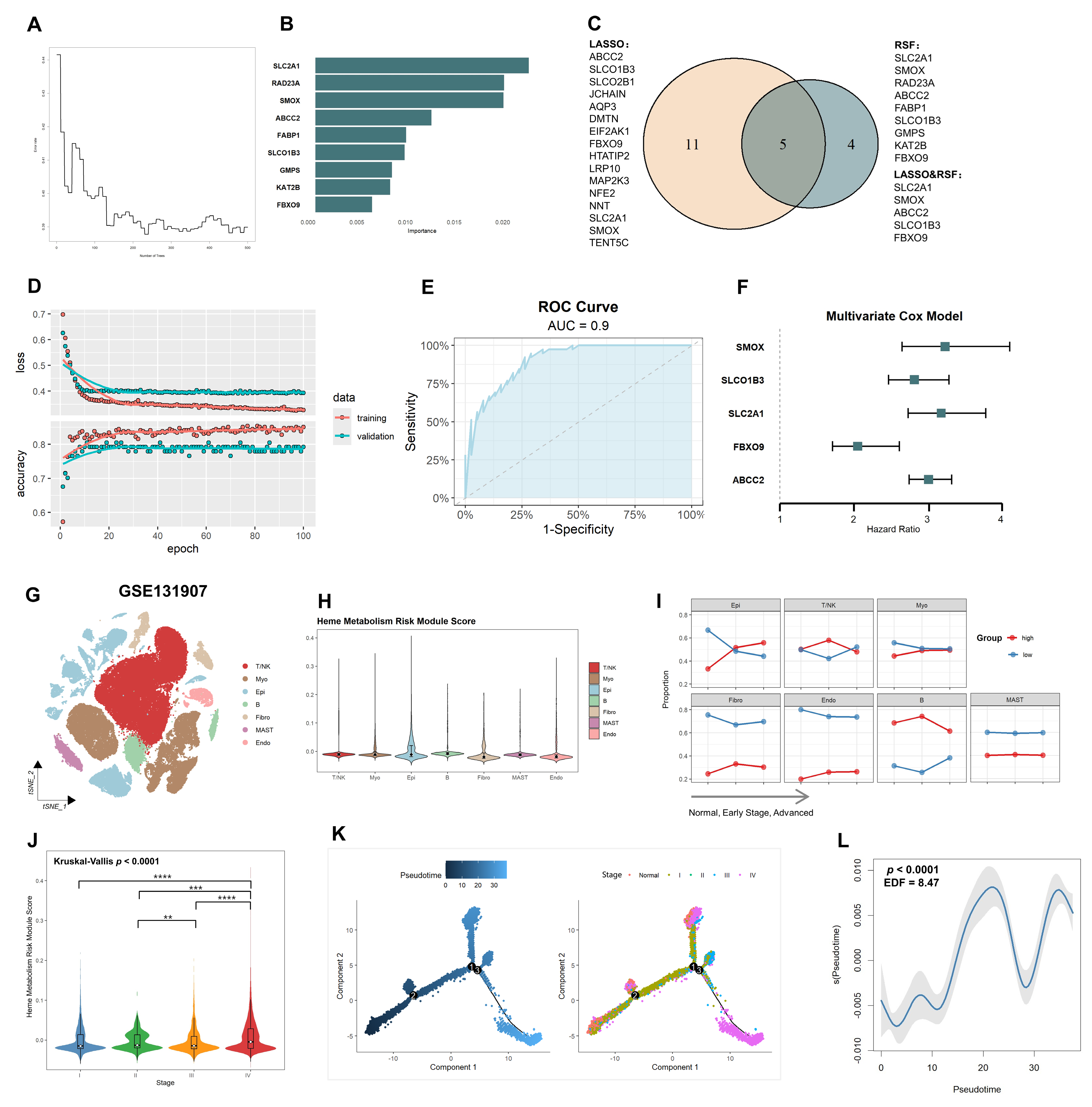
2.4. Core Heme Metabolism Genes Validation by a Deep Learning Model
2.5. Single-Cell Level Reveals Elevated Risk of Heme Metabolism in Epithelial Cells Driving Tumor Progression
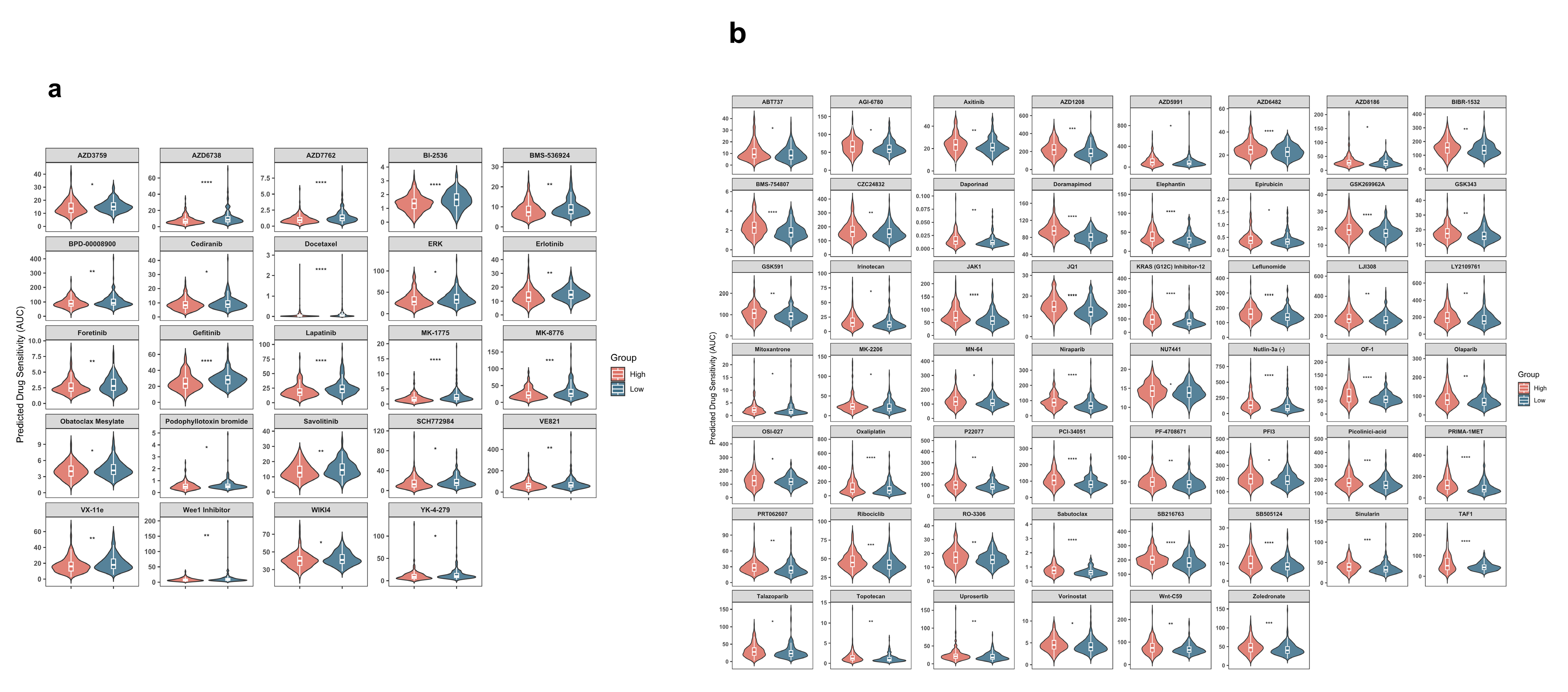
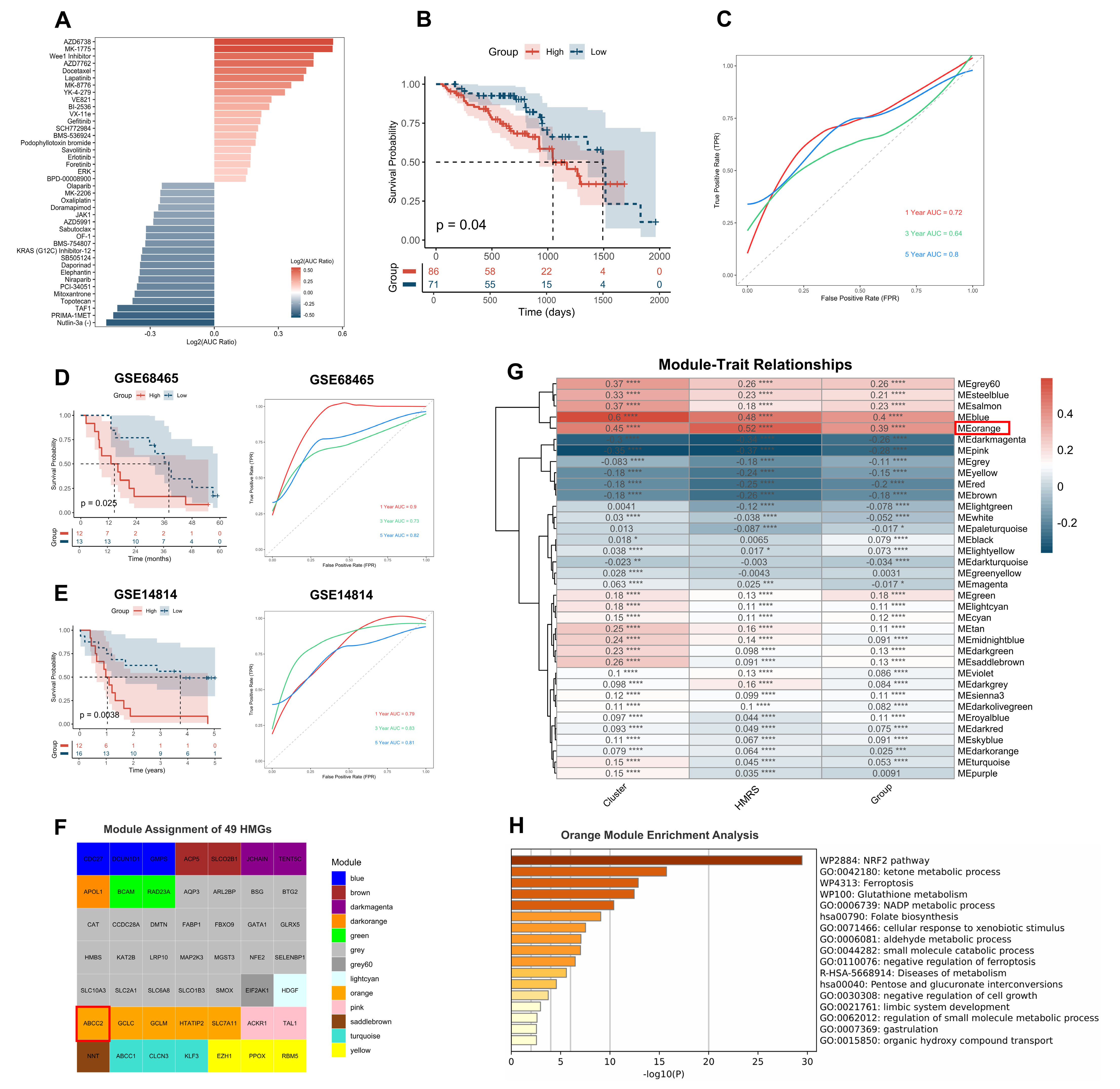
2.6. HMRS Panel as a Predictive Biomarker for Chemotherapy Sensitivity
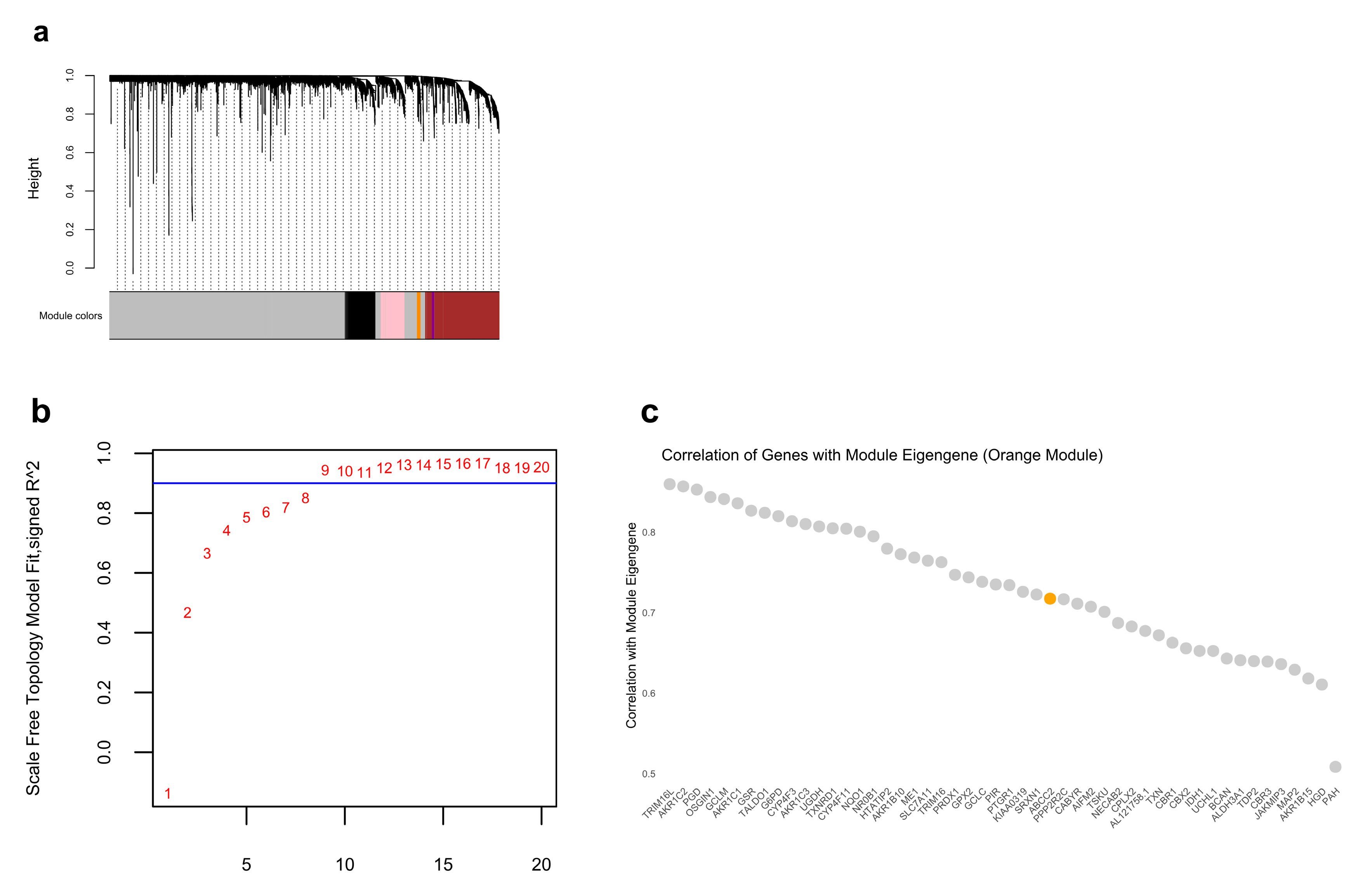
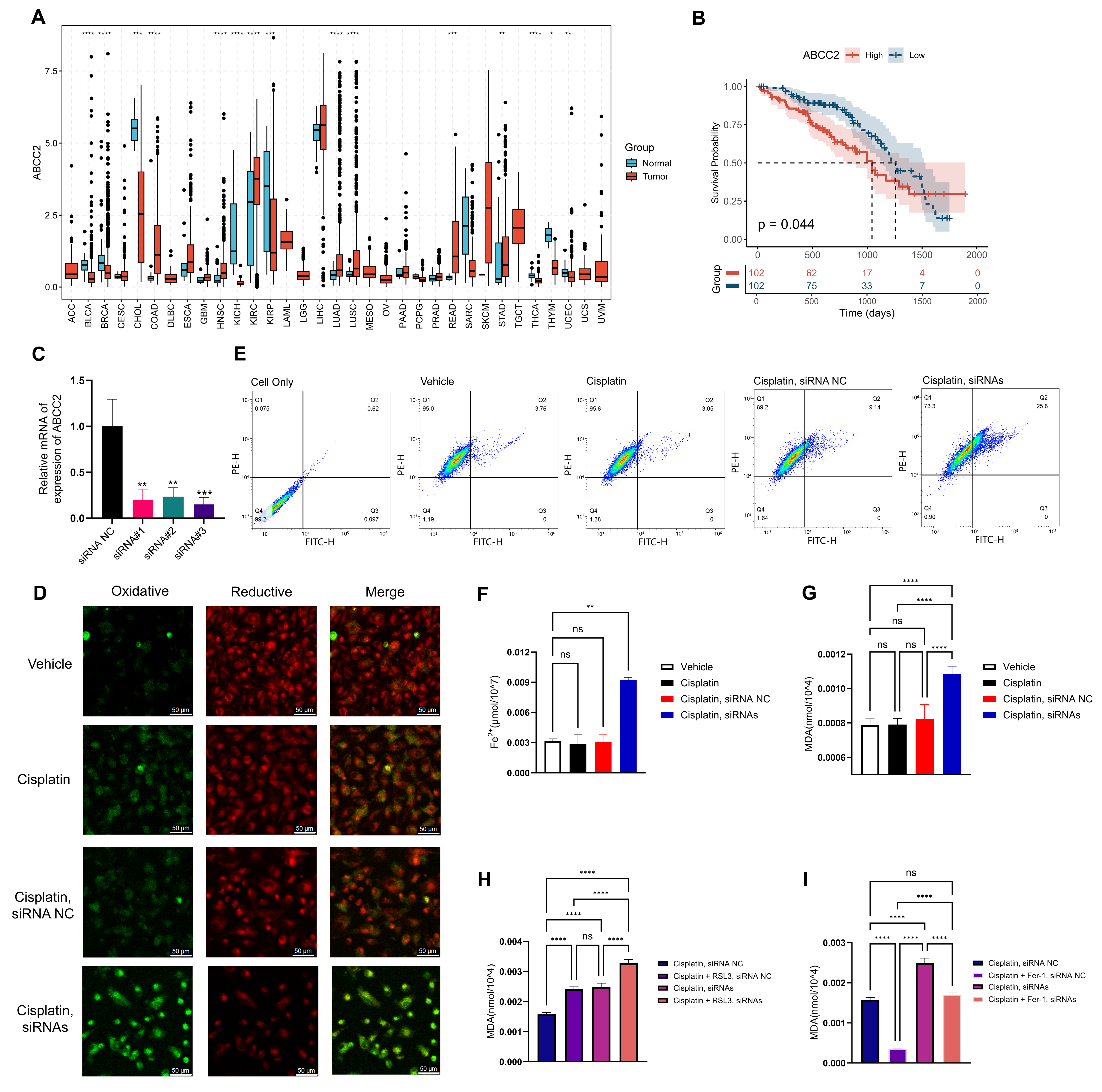
2.7. ABCC2 Is the Core Gene Identified by WGCNA
2.8. Inhibition of ABCC2 Significantly Promotes Cisplatin-Induced Ferroptosis
3. Discussion
4. Materials and Methods
4.1. Data Collection and Processing
4.2. Consensus Clustering
4.3. Construction of the HMRS Panel
4.4. Decision Curve Analysis for Evaluating the HMRS Panel
4.5. Comparative Analysis of Cluster-Based Subtyping and HMRS Risk Groups
4.6. Core Heme Metabolism Genes Identification
4.7. Deep Learning Model Construction
4.8. Functional Enrichment Analysis
4.9. Single-Cell RNA-Seq Analysis Data Collection and Processing
4.10. Drug Sensitivity Prediction
4.11. Weighted Gene Coexpression Network Analysis (WGCNA)
4.12. Cell Culture and Transfection
4.13. Cellular Lipid Peroxidation Assay
4.14. Detection of Cellular Fe2+ Content
4.15. Cellular Malondialdehyde (MDA) Content
4.16. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HMGs | Heme Metabolism-related Genes |
| ROS | Reactive Oxygen Species |
| OS | Overall Survival |
| CNV | Copy Number Variation |
| CDF | Cumulative Distribution Function |
| PCA | Principal Component Analysis |
| ROC | Receiver Operating Characteristic |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| HMRS | Heme Metabolism Risk Score |
| DCA | Decision Curve Analysis |
| GSVA | Gene Set Variation Analysis |
| GSEA | Gene Set Enrichment Analysis |
| EMT | Epithelial–Mesenchymal Transition |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| NESs | Normalized Enrichment Scores |
| RSF | Random Survival Forest |
| DNN | Deep Neural Network |
| t-SNE | t-Distributed Stochastic Neighbor Embedding |
| TCGA | The Cancer Genome Atlas |
| GEO | Gene Expression Omnibus |
| GDSC | Genomics of Drug Sensitivity in Cancer |
| LUAD | Lung adenocarcinoma |
| NSCLC | Non-Small-Cell Lung Cancer |
| MSigDB | Molecular Signatures Database |
| AUC | Areas Under the Curve |
| GC | Gastric Cancer |
| GSH | Glutathione |
| GAM | Generalized additive model |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| COX10 | CD163 | IGKV1-5 | IGLV3-27 | BTRC | EZH1 | ICAM4 | NFE2 | SELENBP1 | TRAK2 |
| COX15 | HBA1 | IGKV1D-12 | IGLV6-57 | C3 | FBXO34 | IGSF3 | NFE2L1 | SIDT2 | TRIM10 |
| ALAS1 | HBA2 | IGKV1D-16 | IGLV7-43 | CA1 | FBXO7 | ISCA1 | NNT | SLC10A3 | TRIM58 |
| FECH | HBB | IGKV1D-33 | JCHAIN | CA2 | FBXO9 | KAT2B | NR3C1 | SLC11A2 | TSPAN5 |
| CPOX | HP | IGKV1D-39 | LRP1 | CAST | FN3K | KDM7A | NUDT4 | SLC22A4 | TSPO2 |
| ABCG2 | HPR | IGKV2-28 | ABCB6 | CAT | FOXJ2 | KEL | OPTN | SLC25A37 | TYR |
| UROD | HPX | IGKV2-30 | ACKR1 | CCDC28A | FOXO3 | KHNYN | OSBP2 | SLC25A38 | UBAC1 |
| PPOX | IGHA1 | IGKV2D-28 | ACP5 | CCND3 | FTCD | KLF1 | P4HA2 | SLC2A1 | UCP2 |
| ALAD | IGHA2 | IGKV2D-30 | ACSL6 | CDC27 | GAPVD1 | KLF3 | PC | SLC30A1 | USP15 |
| ALAS2 | IGHV1-2 | IGKV2D-40 | ADD1 | CDR2 | GATA1 | LAMP2 | PDZK1IP1 | SLC30A10 | VEZF1 |
| FLVCR1 | IGHV1-46 | IGKV3-11 | ADD2 | CIR1 | GCLC | LMO2 | PGLS | SLC4A1 | XK |
| ALB | IGHV1-69 | IGKV3-15 | ADIPOR1 | CLCN3 | GCLM | LPIN2 | PICALM | SLC66A2 | XPO7 |
| UROS | IGHV2-5 | IGKV3-20 | AGPAT4 | CLIC2 | GDE1 | LRP10 | PIGQ | SLC6A8 | YPEL5 |
| HMBS | IGHV2-70 | IGKV3D-20 | AHSP | CROCCP2 | GLRX5 | MAP2K3 | PPP2R5B | SLC6A9 | |
| ABCC2 | IGHV3-11 | IGKV4-1 | ALDH1L1 | CTNS | GMPS | MARCHF2 | PRDX2 | SLC7A11 | |
| BLVRB | IGHV3-13 | IGKV5-2 | ALDH6A1 | CTSB | GYPA | MARCHF8 | PSMD9 | SMOX | |
| HMOX1 | IGHV3-23 | IGLC2 | ANK1 | CTSE | GYPB | MARK3 | RAD23A | SNCA | |
| ABCC1 | IGHV3-30 | IGLC3 | AQP3 | DAAM1 | GYPC | MBOAT2 | RANBP10 | SPTA1 | |
| HMOX2 | IGHV3-33 | IGLV1-40 | ARHGEF12 | DCAF10 | GYPE | MFHAS1 | RAP1GAP | SPTB | |
| BLVRA | IGHV3-48 | IGLV1-44 | ARL2BP | DCAF11 | H1-0 | MGST3 | RBM38 | SYNJ1 | |
| SLCO1B3 | IGHV3-53 | IGLV1-47 | ASNS | DCUN1D1 | H4C3 | MINPP1 | RBM5 | TAL1 | |
| SLCO1B1 | IGHV3-7 | IGLV1-51 | ATG4A | DMTN | HAGH | MKRN1 | RCL1 | TCEA1 | |
| SLCO2B1 | IGHV4-34 | IGLV2-11 | ATP6V0A1 | E2F2 | HBBP1 | MOCOS | RHAG | TENT5C | |
| FABP1 | IGHV4-39 | IGLV2-14 | BACH1 | EIF2AK1 | HBD | MOSPD1 | RHCE | TFDP2 | |
| UGT1A1 | IGHV4-59 | IGLV2-23 | BCAM | ELL2 | HBQ1 | MPP1 | RHD | TFRC | |
| GSTA1 | IGKV1-12 | IGLV2-8 | BMP2K | ENDOD1 | HBZ | MXI1 | RIOK3 | TMCC2 | |
| UGT1A4 | IGKV1-16 | IGLV3-1 | BNIP3L | EPB41 | HDGF | MYL4 | RNF123 | TMEM9B | |
| AMBP | IGKV1-17 | IGLV3-19 | BPGM | EPB42 | HEBP1 | NARF | RNF19A | TNRC6B | |
| APOA1 | IGKV1-33 | IGLV3-21 | BSG | EPOR | HTATIP2 | NCOA4 | SDCBP | TNS1 | |
| APOL1 | IGKV1-39 | IGLV3-25 | BTG2 | ERMAP | HTRA2 | NEK7 | SEC14L1 | TOP1 | |
| Gene | HR | Coef | p Value | LowerCI | UpperCI |
|---|---|---|---|---|---|
| PPOX | 0.752504 | −0.284348 | 0.028362 | 0.58358 | 0.970326 |
| HMBS | 1.379057 | 0.3214 | 0.009022 | 1.083454 | 1.75531 |
| ABCC2 | 1.160223 | 0.148612 | 0.000087 | 1.077248 | 1.249589 |
| ABCC1 | 1.218173 | 0.197352 | 0.043659 | 1.005626 | 1.475644 |
| SLCO1B3 | 1.15296 | 0.142333 | 0.026213 | 1.016984 | 1.307117 |
| SLCO2B1 | 0.873744 | −0.134968 | 0.043410 | 0.766482 | 0.996015 |
| FABP1 | 1.407514 | 0.341825 | 0.006982 | 1.097981 | 1.804308 |
| APOL1 | 1.189261 | 0.173332 | 0.019232 | 1.028612 | 1.375 |
| JCHAIN | 0.870827 | −0.138312 | 0.002853 | 0.795181 | 0.95367 |
| ACKR1 | 0.885668 | −0.121414 | 0.021099 | 0.798835 | 0.981939 |
| ACP5 | 0.825942 | −0.19123 | 0.026820 | 0.697319 | 0.978291 |
| AQP3 | 0.883456 | −0.123914 | 0.005288 | 0.809776 | 0.96384 |
| ARL2BP | 1.583942 | 0.459917 | 0.022251 | 1.067797 | 2.349579 |
| BCAM | 0.867207 | −0.142478 | 0.027419 | 0.76407 | 0.984265 |
| BSG | 1.328363 | 0.283948 | 0.040976 | 1.011707 | 1.74413 |
| BTG2 | 0.80163 | −0.221108 | 0.002304 | 0.695385 | 0.924108 |
| CAT | 0.76955 | −0.261949 | 0.010607 | 0.629476 | 0.940794 |
| CCDC28A | 0.701313 | −0.354801 | 0.011501 | 0.532601 | 0.923467 |
| CDC27 | 1.516675 | 0.416521 | 0.012826 | 1.092501 | 2.10554 |
| CLCN3 | 1.362124 | 0.309045 | 0.014418 | 1.063403 | 1.744758 |
| DCUN1D1 | 1.469944 | 0.385224 | 0.005040 | 1.122989 | 1.924093 |
| DMTN | 0.835953 | −0.179183 | 0.020993 | 0.717961 | 0.973337 |
| EIF2AK1 | 1.571036 | 0.451735 | 0.003926 | 1.155738 | 2.135566 |
| EZH1 | 0.753368 | −0.283201 | 0.026424 | 0.586699 | 0.967384 |
| FBXO9 | 0.744254 | −0.295373 | 0.026388 | 0.573441 | 0.965947 |
| GATA1 | 0.475254 | −0.743907 | 0.006836 | 0.277209 | 0.814786 |
| GCLC | 1.146746 | 0.136928 | 0.007932 | 1.036491 | 1.268728 |
| GCLM | 1.176157 | 0.162253 | 0.037816 | 1.00917 | 1.370776 |
| GLRX5 | 1.553487 | 0.440502 | 0.007061 | 1.127513 | 2.140394 |
| GMPS | 1.528959 | 0.424587 | 0.000510 | 1.203396 | 1.942598 |
| HDGF | 1.318635 | 0.276597 | 0.031325 | 1.025095 | 1.696231 |
| HTATIP2 | 1.520409 | 0.418979 | 0.000264 | 1.21393 | 1.904264 |
| KAT2B | 0.754193 | −0.282107 | 0.011658 | 0.60573 | 0.939044 |
| KLF3 | 1.30228 | 0.264116 | 0.048514 | 1.00172 | 1.693021 |
| LRP10 | 1.368367 | 0.313618 | 0.010557 | 1.075973 | 1.740218 |
| MAP2K3 | 1.342451 | 0.294497 | 0.039175 | 1.014722 | 1.776028 |
| MGST3 | 1.372603 | 0.316709 | 0.036875 | 1.019481 | 1.848037 |
| NFE2 | 0.842092 | −0.171867 | 0.028823 | 0.721826 | 0.982395 |
| NNT | 0.799911 | −0.223255 | 0.043716 | 0.643894 | 0.993731 |
| RAD23A | 1.833232 | 0.606081 | 0.001152 | 1.272041 | 2.642006 |
| RBM5 | 0.743686 | −0.296136 | 0.016172 | 0.584221 | 0.946678 |
| SELENBP1 | 0.873031 | −0.135784 | 0.013918 | 0.78349 | 0.972806 |
| SLC10A3 | 1.348278 | 0.298829 | 0.042569 | 1.010058 | 1.799754 |
| SLC2A1 | 1.29581 | 0.259136 | 0.000020 | 1.150278 | 1.459753 |
| SLC6A8 | 1.165467 | 0.153122 | 0.024519 | 1.019865 | 1.331856 |
| SLC7A11 | 1.123212 | 0.116192 | 0.019410 | 1.018945 | 1.238148 |
| SMOX | 1.369945 | 0.314771 | 0.000072 | 1.172704 | 1.600361 |
| TAL1 | 0.715952 | −0.334143 | 0.043638 | 0.517495 | 0.990514 |
| TENT5C | 0.7314 | −0.312794 | 0.000116 | 0.62387 | 0.857464 |
Appendix B


References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Leiter, A.; Veluswamy, R.R.; Wisnivesky, J.P. The Global Burden of Lung Cancer: Current Status and Future Trends. Nat. Rev. Clin. Oncol. 2023, 20, 624–639. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sohoni, S.; Ghosh, P.; Wang, T.; Kalainayakan, S.P.; Vidal, C.; Dey, S.; Konduri, P.C.; Zhang, L. Elevated Heme Synthesis and Uptake Underpin Intensified Oxidative Metabolism and Tumorigenic Functions in Non-Small Cell Lung Cancer Cells. Cancer Res. 2019, 79, 2511–2525. [Google Scholar] [CrossRef]
- Martínez-Reyes, I.; Chandel, N.S. Cancer Metabolism: Looking Forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef]
- Cai, X.; Miao, J.; Sun, R.; Wang, S.; Molina-Vila, M.A.; Chaib, I.; Rosell, R.; Cao, P. Dihydroartemisinin Overcomes the Resistance to Osimertinib in EGFR-Mutant Non-Small-Cell Lung Cancer. Pharmacol. Res. 2021, 170, 105701. [Google Scholar] [CrossRef]
- Suresh, S.; Chen, B.; Zhu, J.; Golden, R.J.; Lu, C.; Evers, B.M.; Novaresi, N.; Smith, B.; Zhan, X.; Schmid, V.; et al. eIF5B Drives Integrated Stress Response-Dependent Translation of PD-L1 in Lung Cancer. Nat. Cancer 2020, 1, 533–545. [Google Scholar] [CrossRef]
- Torres, Á.; Quintanilla, F.; Barnafi, E.; Sánchez, C.; Acevedo, F.; Walbaum, B.; Merino, T. Dietary Interventions for Cancer Prevention: An Update to ACS International Guidelines. Nutrients 2024, 16, 2897. [Google Scholar] [CrossRef]
- Tasevska, N.; Sinha, R.; Kipnis, V.; Subar, A.F.; Leitzmann, M.F.; Hollenbeck, A.R.; Caporaso, N.E.; Schatzkin, A.; Cross, A.J. A Prospective Study of Meat, Cooking Methods, Meat Mutagens, Heme Iron, and Lung Cancer Risks. Am. J. Clin. Nutr. 2009, 89, 1884–1894. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in Cancer Therapy: A Novel Approach to Reversing Drug Resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Stockwell, B.R. Ferroptosis Turns 10: Emerging Mechanisms, Physiological Functions, and Therapeutic Applications. Cell 2022, 185, 2401–2421. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, X.; Ren, Z.; Li, Y.; Zou, W.; Chen, J.; Wang, H. Overcoming Cancer Chemotherapy Resistance by the Induction of Ferroptosis. Drug Resist. Updat. 2023, 66, 100916. [Google Scholar] [CrossRef] [PubMed]
- Lei, G.; Zhuang, L.; Gan, B. Targeting Ferroptosis as a Vulnerability in Cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef]
- Campbell, N.K.; Fitzgerald, H.K.; Dunne, A. Regulation of Inflammation by the Antioxidant Haem Oxygenase 1. Nat. Rev. Immunol. 2021, 21, 411–425. [Google Scholar] [CrossRef]
- Yu, F.; Wang, Z.; Zhang, Z.; Zhou, J.; Li, J.; Chen, J.; Du, G.; Zhao, X. Biosynthesis, Acquisition, Regulation, and Upcycling of Heme: Recent Advances. Crit. Rev. Biotechnol. 2024, 44, 1422–1438. [Google Scholar] [CrossRef]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in Pathophysiology: A Matter of Scavenging, Metabolism and Trafficking across Cell Membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef]
- Swenson, S.A.; Moore, C.M.; Marcero, J.R.; Medlock, A.E.; Reddi, A.R.; Khalimonchuk, O. From Synthesis to Utilization: The Ins and Outs of Mitochondrial Heme. Cells 2020, 9, 579. [Google Scholar] [CrossRef]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, Function, and Assembly of Heme Centers in Mitochondrial Respiratory Complexes. Biochim. Biophys. Acta. 2012, 1823, 1604–1616. [Google Scholar] [CrossRef]
- Kelland, L. The Resurgence of Platinum-Based Cancer Chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Jordan, M.A.; Wilson, L. Microtubules as a Target for Anticancer Drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Topoisomerase I Inhibitors: Camptothecins and Beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Majeed, U.; Manochakian, R.; Zhao, Y.; Lou, Y. Targeted Therapy in Advanced Non-Small Cell Lung Cancer: Current Advances and Future Trends. J. Hematol. Oncol. 2021, 14, 108. [Google Scholar] [CrossRef] [PubMed]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018, 378, 1976–1986. [Google Scholar] [CrossRef]
- Guo, J.; Xu, B.; Han, Q.; Zhou, H.; Xia, Y.; Gong, C.; Dai, X.; Li, Z.; Wu, G. Ferroptosis: A Novel Anti-Tumor Action for Cisplatin. Cancer Res. Treat. 2018, 50, 445–460. [Google Scholar] [CrossRef]
- Liu, F.; Tang, L.; Liu, H.; Chen, Y.; Xiao, T.; Gu, W.; Yang, H.; Wang, H.; Chen, P. Cancer-Associated Fibroblasts Secrete FGF5 to Inhibit Ferroptosis to Decrease Cisplatin Sensitivity in Nasopharyngeal Carcinoma through Binding to FGFR2. Cell Death Dis. 2024, 15, 279. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, H.; Yang, S.; Su, D. Increased ABCC2 Expression Predicts Cisplatin Resistance in Non-Small Cell Lung Cancer. Cell Biochem. Funct. 2021, 39, 277–286. [Google Scholar] [CrossRef]
- Hooda, J.; Shah, A.; Zhang, L. Heme, an Essential Nutrient from Dietary Proteins, Critically Impacts Diverse Physiological and Pathological Processes. Nutrients 2014, 6, 1080–1102. [Google Scholar] [CrossRef]
- Bonkovsky, H.L.; Guo, J.-T.; Hou, W.; Li, T.; Narang, T.; Thapar, M. Porphyrin and Heme Metabolism and the Porphyrias. Compr. Physiol. 2013, 3, 365–401. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Yang, Y.; Cai, C.-Y.; Teng, Q.-X.; Cui, Q.; Lin, J.; Assaraf, Y.G.; Chen, Z.-S. Multidrug Resistance Proteins (MRPs): Structure, Function and the Overcoming of Cancer Multidrug Resistance. Drug Resist. Updates 2021, 54, 100743. [Google Scholar] [CrossRef]
- Mao, Y.-X.; Chen, Z.-P.; Wang, L.; Wang, J.; Zhou, C.-Z.; Hou, W.-T.; Chen, Y. Transport Mechanism of Human Bilirubin Transporter ABCC2 Tuned by the Inter-Module Regulatory Domain. Nat. Commun. 2024, 15, 1061. [Google Scholar] [CrossRef] [PubMed]
- Zhou, N.; Yuan, X.; Du, Q.; Zhang, Z.; Shi, X.; Bao, J.; Ning, Y.; Peng, L. FerrDb V2: Update of the Manually Curated Database of Ferroptosis Regulators and Ferroptosis-Disease Associations. Nucleic Acids Res. 2023, 51, D571–D582. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gan, X.; Cheng, X.; Jia, Y.; Wang, G.; Tang, X.; Du, H.; Li, X.; Liu, X.; Xing, X.; et al. ABCC2 Induces Metabolic Vulnerability and Cellular Ferroptosis via Enhanced Glutathione Efflux in Gastric Cancer. Clin. Transl. Med. 2024, 14, e1754. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.-L.; Fitzgerald, B.G.; Paz-Ares, L.; Cappuzzo, F.; Jänne, P.A.; Peters, S.; Hirsch, F.R. New Promises and Challenges in the Treatment of Advanced Non-Small-Cell Lung Cancer. Lancet 2024, 404, 803–822. [Google Scholar] [CrossRef]
- Zou, Y.; Xie, J.; Zheng, S.; Liu, W.; Tang, Y.; Tian, W.; Deng, X.; Wu, L.; Zhang, Y.; Wong, C.-W.; et al. Leveraging Diverse Cell-Death Patterns to Predict the Prognosis and Drug Sensitivity of Triple-Negative Breast Cancer Patients after Surgery. Int. J. Surg. 2022, 107, 106936. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Han, H.; Zhou, X.; Gong, T.; Zhu, Y.; Xiao, B.; Liu, S.; Zhao, W.; Wu, N. Integrating Bulk and Single-Cell Transcriptomics with Machine Learning Reveals a Heme Metabolism-Based Panel for Lung Adenocarcinoma Chemotherapy Resistance. Int. J. Mol. Sci. 2025, 26, 4685. https://doi.org/10.3390/ijms26104685
Zhao L, Han H, Zhou X, Gong T, Zhu Y, Xiao B, Liu S, Zhao W, Wu N. Integrating Bulk and Single-Cell Transcriptomics with Machine Learning Reveals a Heme Metabolism-Based Panel for Lung Adenocarcinoma Chemotherapy Resistance. International Journal of Molecular Sciences. 2025; 26(10):4685. https://doi.org/10.3390/ijms26104685
Chicago/Turabian StyleZhao, Lin, Haibo Han, Xuantong Zhou, Tongyang Gong, Yuge Zhu, Bufan Xiao, Shuchang Liu, Wei Zhao, and Nan Wu. 2025. "Integrating Bulk and Single-Cell Transcriptomics with Machine Learning Reveals a Heme Metabolism-Based Panel for Lung Adenocarcinoma Chemotherapy Resistance" International Journal of Molecular Sciences 26, no. 10: 4685. https://doi.org/10.3390/ijms26104685
APA StyleZhao, L., Han, H., Zhou, X., Gong, T., Zhu, Y., Xiao, B., Liu, S., Zhao, W., & Wu, N. (2025). Integrating Bulk and Single-Cell Transcriptomics with Machine Learning Reveals a Heme Metabolism-Based Panel for Lung Adenocarcinoma Chemotherapy Resistance. International Journal of Molecular Sciences, 26(10), 4685. https://doi.org/10.3390/ijms26104685