
The Quantum Paradox in Pharmaceutical Science: Understanding Without Comprehending—A Centennial Reflection


{kind=link}
{kind=link}
{kind=link}
Abstract
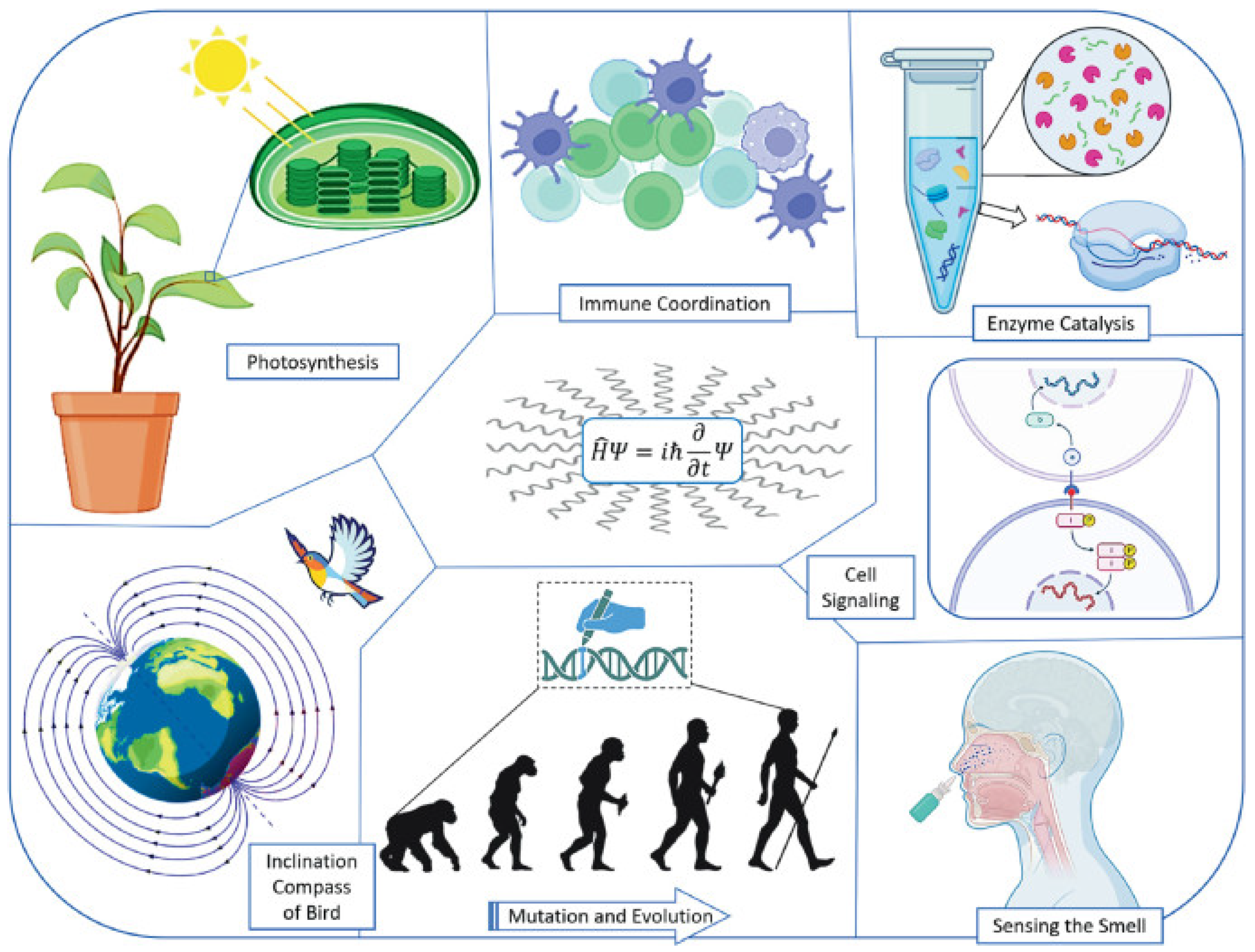
1. Introduction: The Quantum Revolution in Biology and Medicine
2. Theoretical Foundations: From Physics to Pharmaceutical Science
2.1. The Schrödinger Equation and Its Significance
2.2. Heisenberg’s Uncertainty Principle and Its Implications
2.3. The Boltzmann Distribution and Quantum Thermodynamics
3. Bridging Quantum and Molecular Scales
3.1. Electron Distributions and Chemical Bonding
3.2. Localized Quantum Events in Large Systems
3.3. Quantum-Classical Interface in Computational Drug Design
3.4. Decoherence and Environmental Protection
3.5. Measurement Problems in Drug–Target Interactions
3.6. Boltzmann Distribution and Quantum States
4. Historical Development of Quantum Applications in Drug Discovery
4.1. Pioneers in Quantum Biochemistry
4.2. Computational Advances and the Rise of In Silico Drug Design
5. Quantum Mechanics in Modern Drug Discovery
5.1. Understanding Drug–Target Interactions
5.2. Quantum Effects in Enzyme Catalysis
5.3. Density Functional Theory in Drug Design
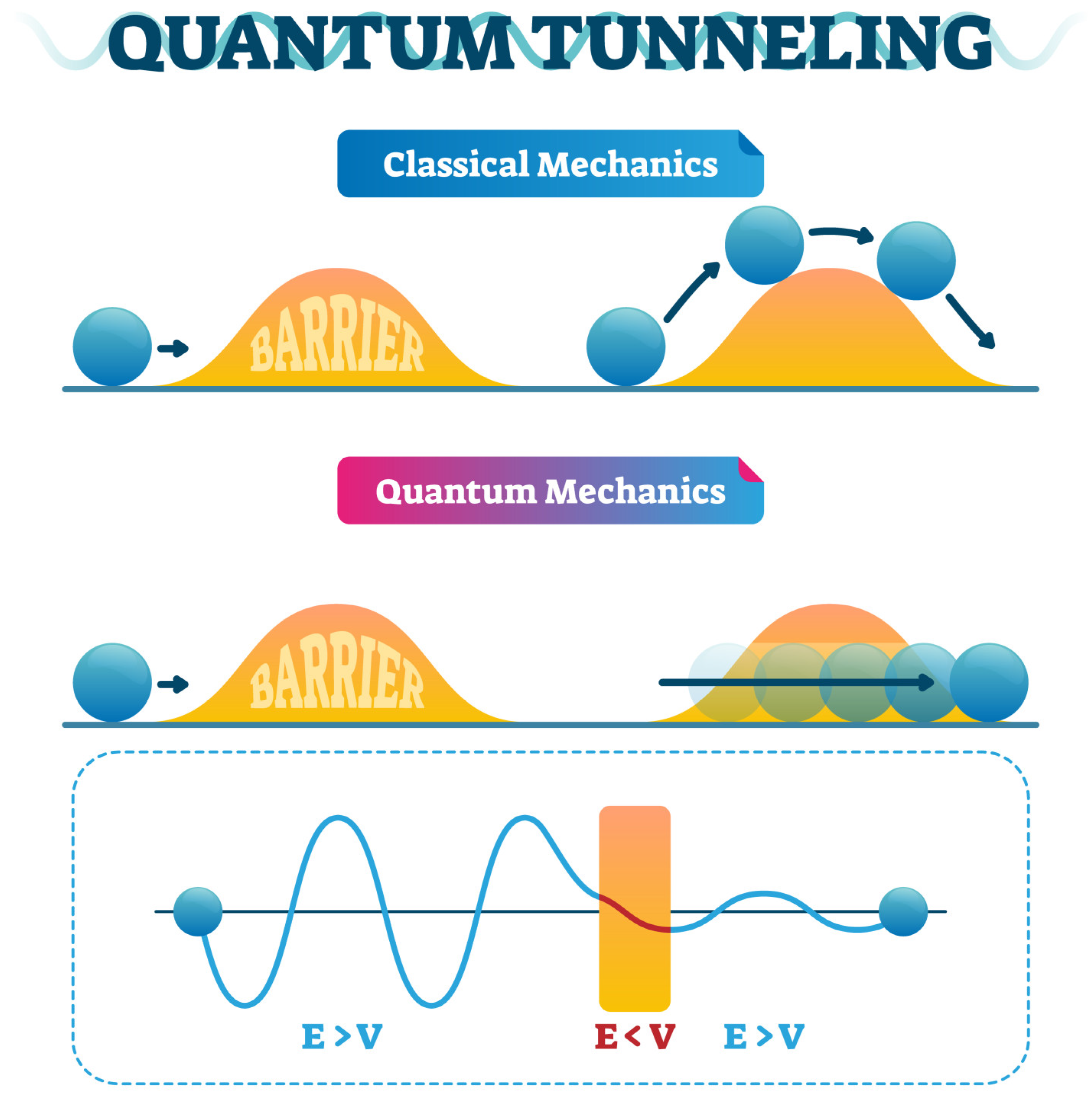
6. Quantum Tunneling in Drug Action and Development
6.1. Fundamentals of Quantum Tunneling
6.2. Tunneling in Enzymatic Reactions
6.3. Implications for Drug Design
7. Applications Across Therapeutic Areas
7.1. Neurological Drugs
7.2. Oncology Drugs
7.3. Cardiovascular Drugs
7.4. Antimicrobial Drugs
8. Computational Methods and Challenges
8.1. Hybrid QM/MM Approaches
8.2. Time-Dependent Density Functional Theory
8.3. Machine Learning and Quantum Mechanics
8.4. Current Limitations and Future Directions
9. Additional Quantum Concepts for Drug Discovery
9.1. Quantum Delocalization and Aromaticity
9.2. Quantum Phase Transitions in Allosteric Modulation
9.3. Non-Adiabatic Dynamics in Drug–Target Interactions
9.4. Quantum Confinement Effects in Nanomedicine
9.5. Vibrational Strong Coupling in Drug Activity
10. Future Perspectives: Quantum Biology and Medicine
10.1. Quantum Coherence in Biological Systems
10.2. Quantum Computing in Drug Discovery
10.3. Designing Drugs to Exploit Quantum Effects
11. The Quantum Future of Medicine
12. Philosophical and Ethical Limits of the Role of Quantum Mechanics
Funding
Data Availability Statement
Conflicts of Interest
References
- Schrödinger, E. Quantisierung als Eigenwertproblem. Ann. Phys. 1926, 384, 361–376. [Google Scholar] [CrossRef]
- Heisenberg, W. Über quantentheoretische Umdeutung kinematischer und mechanischer Beziehungen. Z. Für Phys. 1925, 33, 879–893. [Google Scholar] [CrossRef]
- Schrödinger, E. What is Life? Cambridge University Press: Cambridge, UK, 1944. [Google Scholar]
- Dirac, P.A.M.; Fowler, R.H. The quantum theory of the electron. Proc. R. Soc. London. Ser. A Contain. Pap. A Math. Phys. Character 1928, 117, 610–624. [Google Scholar]
- Pullman, A.; Pullman, B. Cancérisation Par Les Substances Chimiques et Structure Moléculaire; Masson & Cie: Paris, France, 1955. [Google Scholar]
- Pullman, A.; Pullman, B. Quantum Biochemistry; Interscience, N.Y.: New York, NY, USA, 1963. [Google Scholar]
- Pullman, B. Electronic Factors in Biochemical Evolution; Academic Press: Cambridge, MA, USA, 1970. [Google Scholar]
- Friesner, R.A. Ab initio quantum chemistry: Methodology and applications. Proc. Natl. Acad. Sci. USA 2005, 102, 6648–6653. [Google Scholar] [CrossRef]
- Zhou, H.X.; Pang, X. Electrostatic Interactions in Protein Structure, Folding, Binding, and Condensation. Chem. Rev. 2018, 118, 1691–1741. [Google Scholar] [CrossRef]
- Warshel, A.; Sharma, P.K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M.H. Electrostatic Basis for Enzyme Catalysis. Chem. Rev. 2006, 106, 3210–3235. [Google Scholar] [CrossRef]
- Cournia, Z.; Allen, B.; Sherman, W. Relative Binding Free Energy Calculations in Drug Discovery: Recent Advances and Practical Considerations. J. Chem. Inf. Model. 2017, 57, 2911–2937. [Google Scholar] [CrossRef]
- Wagner, J.R.; Lee, C.T.; Durrant, J.D.; Malmstrom, R.D.; Feher, V.A.; Amaro, R.E. Emerging Computational Methods for the Rational Discovery of Allosteric Drugs. Chem. Rev. 2016, 116, 6370–6390. [Google Scholar] [CrossRef] [PubMed]
- Lambert, N.; Chen, Y.-N.; Cheng, Y.-C.; Li, C.-M.; Chen, G.-Y.; Nori, F. Quantum Biology. Nat. Phys. 2013, 9, 10–18. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Brookes, J.C. Quantum effects in biology: Golden rule in enzymes, olfaction, photosynthesis and magnetodetection. Proc. R. Soc. A Math. Phys. Eng. Sci. 2017, 473, 20160822. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: Ithaca, NY, USA, 1939. [Google Scholar]
- Cooper, R.D.G.; Snyder, N.J. Vancomycin structure-activity relationships. ACS Symp. Ser. 1994, 569, 228–247. [Google Scholar]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Klinman, J.P.; Kohen, A. Hydrogen Tunneling Links Protein Dynamics to Enzyme Catalysis. Annu. Rev. Biochem. 2013, 82, 471–496. [Google Scholar] [CrossRef]
- Lehnert, N.; Solomon, E.I. Density-functional investigation on the mechanism of H-atom abstraction by lipoxygenase. J. Biol. Inorg. Chem. 2003, 8, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Löwdin, P.O. Proton tunneling in DNA and its biological implications. Rev. Mod. Phys. 1963, 35, 724–732. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA repair inhibition: A selective tumour targeting strategy. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Kar, P.; Knecht, V. Energetics of mutation-induced changes in potency of lersivirine against HIV-1 reverse transcriptase. J. Phys. Chem. B 2012, 116, 6269–6278. [Google Scholar] [CrossRef]
- Kulkarni, P.U.; Shah, H.; Vyas, V.K. Hybrid Quantum Mechanics/Molecular Mechanics (QM/MM) Simulation: A Tool for Structure-Based Drug Design and Discovery. Mini Rev. Med. Chem. 2022, 22, 1096–1107. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Chapsal, B.D. Design of HIV protease inhibitors targeting protein backbone: An effective strategy for combating drug resistance. Acc. Chem. Res. 2008, 41, 78–86. [Google Scholar] [CrossRef]
- Schlosshauer, M. Decoherence, the measurement problem, and interpretations of quantum mechanics. Rev. Mod. Phys. 2005, 76, 1267–1305. [Google Scholar] [CrossRef]
- Engel, G.S.; Calhoun, T.R.; Read, E.L.; Ahn, T.K.; Mančal, T.; Cheng, Y.C.; Blankenship, R.E.; Fleming, G.R. Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature 2007, 446, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Abrahamse, H.; Hamblin, M.R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Wüthrich, K. Protein structure determination in solution by NMR spectroscopy. J. Biol. Chem. 1990, 265, 22059–22062. [Google Scholar] [CrossRef]
- Tsai, J.; Lee, J.T.; Wang, W.; Zhang, J.; Cho, H.; Mamo, S.; Bremer, R.; Gillette, S.; Kong, J.; Haass, N.K.; et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent anti-melanoma activity. Proc. Natl. Acad. Sci. USA 2008, 105, 3041–3046. [Google Scholar] [CrossRef]
- Vauquelin, G.; Charlton, S.J. Long-lasting target binding and rebinding as mechanisms to prolong in vivo drug action. Br. J. Pharmacol. 2010, 161, 488–508. [Google Scholar] [CrossRef]
- Teague, S.J. Implications of protein flexibility for drug discovery. Nat. Rev. Drug Discov. 2003, 2, 527–541. [Google Scholar] [CrossRef]
- Ball, P. Physics of life: The dawn of quantum biology. Nature 2011, 474, 272–274. [Google Scholar] [CrossRef]
- Pullman, A.; Pullman, B. Les Théories Electroniques de la Chimie Organique; Masson: Paris, France, 1952. [Google Scholar]
- Pauling, L.; Wilson, E.B. Introduction to Quantum Mechanics—With Applications to Chemistry; McGraw-Hill: New York, NY, USA, 1935. [Google Scholar]
- Salem, L.; Rowland, C.S. The Electronic Properties of Diradicals. Angew. Chem. 1972, 11, 92–111. [Google Scholar] [CrossRef]
- Arodola, O.A.; Soliman, M.E. Quantum mechanics implementation in drug-design workflows: Does it help? Drug Des. Dev. Ther. 2017, 11, 2551–2564. [Google Scholar] [CrossRef]
- Kohn, W. Nobel Lecture: Electronic structure of matter—Wave functions and density functionals. Rev. Mod. Phys. 1999, 71, 1253–1266. [Google Scholar] [CrossRef]
- Karplus, M. Development of multiscale models for complex chemical systems: From H+H2 to biomolecules. Angew. Chem. Int. Ed. 2014, 53, 9992–10005. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Gan, L.; Wang, E.; Wang, J. Pocket-Based Drug Design: Exploring Pocket Space. AAPS J. 2012, 15, 228–241. [Google Scholar] [CrossRef]
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, T.; Lu, X.; Lan, X.; Chen, Z.; Lu, S. G protein-coupled receptors (GPCRs): Advances in structures, mechanisms and drug discovery. Signal Transduct. Target. Ther. 2024, 9, 88. [Google Scholar] [CrossRef]
- Sawada, R.; Sakajiri, Y.; Shibata, T.; Yamanishi, Y. Predicting therapeutic and side effects from drug binding affinities to human proteome structures. Iscience 2024, 27, 110032. [Google Scholar] [CrossRef]
- Layfield, J.P.; Hammes-Schiffer, S. Hydrogen Tunneling in Enzymes and Biomimetic Models. Chem. Rev. 2013, 114, 3466–3494. [Google Scholar] [CrossRef]
- McElheny, D.; Schnell, J.R.; Lansing, J.C.; Dyson, H.J.; Wright, P.E. Defining the role of active-site loop fluctuations in dihydrofolate reductase catalysis. Proc. Natl. Acad. Sci. USA 2005, 102, 5032–5037. [Google Scholar] [CrossRef]
- Chakravorty, D.K.; Soudackov, A.V.; Hammes-Schiffer, S. Hybrid Quantum/Classical Molecular Dynamics Simulations of the Proton Transfer Reactions Catalyzed by Ketosteroid Isomerase: Analysis of Hydrogen Bonding, Conformational Motions, and Electrostatics. Biochemistry 2009, 48, 10608–10619. [Google Scholar] [CrossRef]
- Scholes, G.D.; Fleming, G.R.; Olaya-Castro, A.; van Grondelle, R. Lessons from nature about solar light harvesting. Nat. Chem. 2011, 3, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Cavalli, A.; Carloni, P.; Recanatini, M. Target-Related Applications of First Principles Quantum Chemical Methods in Drug Design. Chem. Rev. 2006, 106, 3497–3519. [Google Scholar] [CrossRef]
- Naithani, U.; Guleria, V. Integrative computational approaches for discovery and evaluation of lead compound for drug design. Front. Drug Discov. 2024, 4, 1362456. [Google Scholar] [CrossRef]
- Al-Rawashdeh, B.M.; Qaswal, A.B.; Suleiman, A.; Zayed, F.M.; Al-Rawashdeh, S.M.; Tawalbeh, M.; Khreesha, L.; Alzubaidi, A.; Al-Zubidi, E.; Ghala, Z.; et al. The Quantum Tunneling of Ions Model Can Explain the Pathophysiology of Tinnitus. Brain Sci. 2022, 12, 426. [Google Scholar] [CrossRef]
- Hammes-Schiffer, S. Hydrogen Tunneling and Protein Motion in Enzyme Reactions. Acc. Chem. Res. 2005, 39, 93–100. [Google Scholar] [CrossRef]
- Govindjee. A role for a light-harvesting antenna complex of photosystem II in photoprotection. Plant Cell 2002, 14, 1663–1668. [Google Scholar] [CrossRef] [PubMed]
- Matarèse, B.F.E.; Rusin, A.; Seymour, C.; Mothersill, C. Quantum Biology and the Potential Role of Entanglement and Tunneling in Non-Targeted Effects of Ionizing Radiation: A Review and Proposed Model. Int. J. Mol. Sci. 2023, 24, 16464. [Google Scholar] [CrossRef] [PubMed]
- Tinoco, I. Physical chemistry of nucleic acids. Annu. Rev. Phys. Chem. 2002, 53, 1–15. [Google Scholar] [CrossRef]
- Bosi, S.; Da Ros, T.; Spalluto, G.; Prato, M. Fullerene derivatives: An attractive tool for biological applications. Eur. J. Med. Chem. 2003, 38, 913–923. [Google Scholar] [CrossRef]
- Li, Y.; Zhi, W.; Qi, B.; Wang, L.; Hu, X.; Mohamed, A.M. Update on neurobiological mechanisms of fear: Illuminating the direction of mechanism exploration and treatment development of trauma and fear-related disorders. Front. Behav. Neurosci. 2023, 17, 1216524. [Google Scholar] [CrossRef] [PubMed]
- Mäki-Marttunen, V.; Andreassen, O.A.; Espeseth, T. The role of norepinephrine in the pathophysiology of schizophrenia. Neurosci. Biobehav. Rev. 2020, 118, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Navidhamidi, M.; Rezaei, F.; Azizikia, A.; Mehranfard, N. Anxiety, and hippocampal neuronal activity: Relationship and potential mechanisms. Cogn. Affect. Behav. Neurosci. 2021, 22, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Bouras, N.N.; Mack, N.R.; Gao, W.-J. Prefrontal modulation of anxiety through a lens of noradrenergic signaling. Front. Syst. Neurosci. 2023, 17, 1173326. [Google Scholar] [CrossRef]
- Prados-Pardo, Á.; Martín-González, E.; Mora, S.; Merchán, A.; Flores, P.; Moreno, M. Increased Fear Memory and Glutamatergic Modulation in Compulsive Drinker Rats Selected by Schedule-Induced Polydipsia. Front. Behav. Neurosci. 2019, 13, 100. [Google Scholar] [CrossRef]
- Goldschen-Ohm, M.P. Benzodiazepine Modulation of GABA[A] Receptors: A Mechanistic Perspective. Biomolecules 2022, 12, 1784. [Google Scholar] [CrossRef]
- Nasir, M.; Trujillo, D.; Levine, J.; Dwyer, J.B.; Rupp, Z.W.; Bloch, M.H. Glutamate Systems in DSM-5 Anxiety Disorders: Their Role and a Review of Glutamate and GABA Psychopharmacology. Front. Psychiatry 2020, 11, 548505. [Google Scholar] [CrossRef]
- Bennett, J.P.; Onyango, I.G. Energy, Entropy and Quantum Tunneling of Protons and Electrons in Brain Mitochondria: Relation to Mitochondrial Impairment in Aging-Related Human Brain Diseases and Therapeutic Measures. Biomedicines 2021, 9, 225. [Google Scholar] [CrossRef]
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Gu, L.; Hickey, R.J.; Malkas, L.H. Therapeutic Targeting of DNA Replication Stress in Cancer. Genes 2023, 14, 1346. [Google Scholar] [CrossRef]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond ATP production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Muzammil, M.A.; Javid, S.; Afridi, A.K.; Siddineni, R.; Shahabi, M.; Haseeb, M.; Fariha, F.; Kumar, S.; Zaveri, S.; Nashwan, A.J. Artificial intelligence-enhanced electrocardiography for accurate diagnosis and management of cardiovascular diseases. J. Electrocardiol. 2024, 83, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Roy, S. Angiotensin-converting enzyme inhibitors from plants: A review of their diversity, modes of action, prospects, and concerns in managing diabetes-centric complications. J. Integr. Med. 2021, 19, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.H. Safety profiles for the HMG-CoA reductase inhibitors: Treatment and trust. Drugs 2001, 61, 197–206. [Google Scholar] [CrossRef]
- Baran, A.; Kwiatkowska, A.; Potocki, L. Antibiotics and Bacterial Resistance—A Short Story of an Endless Arms Race. Int. J. Mol. Sci. 2023, 24, 5777. [Google Scholar] [CrossRef]
- Nauta, K.M.; Ho, T.D.; Ellermeier, C.D. The Penicillin-Binding Protein PbpP Is a Sensor of β-Lactams and Is Required for Activation of the Extracytoplasmic Function σ Factor σP in Bacillus thuringiensis. Mbio 2021, 12, e00179-21. [Google Scholar] [CrossRef]
- Lade, H.; Kim, J.-S. Molecular Determinants of β-Lactam Resistance in Methicillin-Resistant Staphylococcus aureus (MRSA): An Updated Review. Antibiotics 2023, 12, 1362. [Google Scholar] [CrossRef]
- Spencer, A.C.; Panda, S.S. DNA Gyrase as a Target for Quinolones. Biomedicines 2023, 11, 371. [Google Scholar] [CrossRef]
- Wakefield, A.E.; Kozakov, D.; Vajda, S. Mapping the binding sites of challenging drug targets. Curr. Opin. Struct. Biol. 2022, 75, 102396. [Google Scholar] [CrossRef]
- Lodola, A.; De Vivo, M. The increasing role of QM/MM in drug discovery. Adv. Protein Chem. Struct. Biol. 2012, 87, 337–362. [Google Scholar]
- Casida, M.E.; Huix-Rotllant, M. Progress in Time-Dependent Density-Functional Theory. Annu. Rev. Phys. Chem. 2012, 63, 287–323. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Chu, X.; Xing, C.; Huang, X. Non-adiabatic dynamics simulations of large biological systems: Light-induced processes in the biomolecules. Phys. Chem. Chem. Phys. 2022, 24, 21019–21053. [Google Scholar]
- Reiher, M.; Wiebe, N.; Svore, K.M.; Wecker, D.; Troyer, M. Elucidating reaction mechanisms on quantum computers. Proc. Natl. Acad. Sci. USA 2017, 114, 7555–7560. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Romero, J.; Olson, J.P.; Degroote, M.; Johnson, P.D.; Kieferová, M.; Kivlichan, I.D.; Menke, T.; Peropadre, B.; Sawaya, N.P.D.; et al. Quantum Chemistry in the Age of Quantum Computing. Chem. Rev. 2019, 119, 10856–10915. [Google Scholar] [CrossRef]
- Amaro, R.E.; Mulholland, A.J. Multiscale methods in drug design bridge chemical and biological complexity in the search for cures. Nat. Rev. Chem. 2018, 2, 0148. [Google Scholar] [CrossRef]
- Schleyer, P.V.R. Introduction: Aromaticity. Chem. Rev. 2001, 101, 1115–1118. [Google Scholar] [CrossRef]
- Martinez, C.R.; Iverson, B.L. Rethinking the term “pi-stacking”. Chem. Sci. 2012, 3, 2191–2201. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Aromatic Systems as Chemical Entities: An Application of Quantum Chemical Topology. Molecules 2022, 27, 2623. [Google Scholar]
- Wand, A.J. The dark energy of proteins comes to light: Conformational entropy and collective protein dynamics in enzyme catalysis. Curr. Opin. Struct. Biol. 2023, 73, 102383. [Google Scholar]
- Cao, C.; Zhuang, X.; Kaminski, G. Simulations of G Protein-Coupled Receptors in the Era of Artificial Intelligence. Acc. Chem. Res. 2023, 56, 2493–2503. [Google Scholar]
- Pontiggia, F.; Pachov, D.V.; Clarkson, M.W.; Villali, J.; Hagan, M.F.; Pande, V.S.; Kern, D. Free energy landscape of activation in a signaling protein at atomic resolution. Nat. Commun. 2022, 13, 65. [Google Scholar]
- Szabla, R.; Šponer, J.; Góra, R.W. Electron-Driven Proton Transfer Along H₂O Wires Enables Photorelaxation of πσ* States in Chromophore-Water Clusters. J. Phys. Chem. Lett. 2022, 13, 3263–3271. [Google Scholar] [CrossRef] [PubMed]
- Tully, J.C. Perspective: Nonadiabatic dynamics theory. J. Chem. Phys. 2022, 137, A301. [Google Scholar] [CrossRef] [PubMed]
- Kagan, C.R.; Lifshitz, E.; Talapin, D.V. The Science and Applications of Quantum Dots. Science 2023, 379, eadd8655. [Google Scholar]
- Patra, J.K.; Das, G.; Fraceto, L.F.; Campos, E.V.R.; del Pilar Rodriguez-Torres, M.; Acosta-Torres, L.S.; Diaz-Torres, L.A.; Grillo, R.; Swamy, M.K.; Sharma, S.; et al. Nano based drug delivery systems: Recent developments and future prospects. J. Nanobiotechnol. 2022, 16, 71. [Google Scholar] [CrossRef]
- Langan, P.S.; Vandavasi, V.G.; Weiss, K.L.; Afonine, P.V.; El Omari, K.; Duman, R.; Wagner, A.; Coates, L. The Structure of SARS-CoV-2 Main Protease in Complex with Telaprevir Reveals a Calcium-Binding Site. Structure 2022, 30, 88–100.e6. [Google Scholar]
- Thomas, A.; Lethuillier-Karl, L.; Nagarajan, K.; Vergauwe, R.M.A.; George, J.; Chervy, T.; Shalabney, A.; Devaux, E.; Genet, C.; Moran, J.; et al. Tilting a ground-state reactivity landscape by vibrational strong coupling. Science 2022, 363, 615–619. [Google Scholar] [CrossRef]
- Lather, J.; Bhatt, P.; Thomas, A.; Ebbesen, T.W.; George, J. Cavity Catalysis by Cooperative Vibrational Strong Coupling of Reactant and Solvent Molecules. Angew. Chem. Int. Ed. 2023, 58, 10635–10638. [Google Scholar] [CrossRef]
- Hirai, K.; Takeda, R.; Hutchison, J.A.; Uji-i, H. Modulating Proton Transfer in a Medicinal Catalyst. Nat. Commun. 2023, 11, 4013. [Google Scholar]
- Marais, A.; Adams, B.; Ringsmuth, A.K.; Ferretti, M.; Gruber, J.M.; Hendrikx, R.; Schuld, M.; Smith, S.L.; Sinayskiy, I.; Krüger, T.P.J.; et al. The future of quantum biology. J. R. Soc. Interface 2018, 15, 20180640. [Google Scholar] [CrossRef] [PubMed]
- Al-Khalili, J.; McFadden, J. Life on the Edge: The Coming of Age of Quantum Biology; Bantam Press: London, UK, 2014. [Google Scholar]
- Heitler, W.; London, F. Wechselwirkung neutraler Atome und homöopolare Bindung nach der Quantenmechanik. Z. Phys. 1927, 44, 455–472. [Google Scholar] [CrossRef]
- McWeeny, R. The Density Matrix in Self-Consistent Field Theory. I. Iterative Construction of the Density Matrix. Proc. R. Soc. Lond. A Math. Phys. Sci. 1956, 235, 496–509. [Google Scholar]
- Etzkowitz, H.; Leydesdorff, L. The dynamics of innovation: From National Systems and “Mode 2” to a Triple Helix of university–industry–government relations. Res. Policy 2000, 29, 109–123. [Google Scholar] [CrossRef]
- Jasanoff, S. States of Knowledge: The Co-Production of Science and the Social Order; Routledge: London, UK, 2004. [Google Scholar]
- Aspuru-Guzik, A.; Lindh, R.; Reiher, M. The Matter Simulation (R)evolution. ACS Cent. Sci. 2018, 4, 144–152. [Google Scholar] [CrossRef]
- Santagati, R.; Wang, J.; Gentile, A.A.; Paesani, S.; Wiebe, N.; McClean, J.R.; Morley-Short, S.; Shadbolt, P.J.; Bonneau, D.; Silverstone, J.W.; et al. Witnessing eigenstates for quantum simulation of Hamiltonian spectra. Sci. Adv. 2018, 4, eaap9646. [Google Scholar] [CrossRef] [PubMed]
- Tisza, L.; Manning, I. Fluctuations and Irreversible Thermodynamics. Phys. Rev. 1957, 105, 1695–1705. [Google Scholar] [CrossRef]
- Anderson, P.W. More Is Different. Science 1972, 177, 393–396. [Google Scholar] [CrossRef]
- Hameroff, S.; Penrose, R. Consciousness in the universe: A review of the ‘Orch OR’ theory. Phys. Life Rev. 2014, 11, 39–78. [Google Scholar] [CrossRef]
- Jonas, H. The Imperative of Responsibility: In Search of an Ethics for the Technological Age; University of Chicago Press: Chicago, IL, USA, 1984. [Google Scholar]
- Kauffman, S.A. Beyond reductionism: Reinventing the sacred. Zygon 2007, 42, 903–914. [Google Scholar] [CrossRef]
- Sandel, M.J. The Case Against Perfection: Ethics in the Age of Genetic Engineering; Harvard University Press: Cambridge, MA, USA, 2007. [Google Scholar]
- Dupré, J. The Disorder of Things: Metaphysical Foundations of the Disunity of Science; Harvard University Press: Cambridge, MA, USA, 1993. [Google Scholar]
- Weber, S.; Lüthi, H.P. Access to advanced computational resources for the broader chemistry community. Int. J. Quantum Chem. 2019, 119, e25971. [Google Scholar]
- DiVincenzo, D.P. Quantum Computation. Science 1995, 270, 255–261. [Google Scholar] [CrossRef]
- Feenberg, A. Questioning Technology; Routledge: London, UK, 1999. [Google Scholar]
- Noble, D. The Music of Life: Biology Beyond Genes; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Kauffman, S.A. Investigations; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Boogerd, F.C.; Bruggeman, F.J.; Hofmeyr, J.H.S.; Westerhoff, H.V. Systems Biology: Philosophical Foundations; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Keller, E.F. Making Sense of Life: Explaining Biological Development with Models, Metaphors, and Machines; Harvard University Press: Cambridge, MA, USA, 2002. [Google Scholar]
- Kuhn, T.S. The Essential Tension: Selected Studies in Scientific Tradition and Change; University of Chicago Press: Chicago, IL, USA, 1977. [Google Scholar]
- Prigogine, I.; Stengers, I. Order out of Chaos: Man’s New Dialogue with Nature; Bantam Books: New York, NY, USA, 1984. [Google Scholar]
- Bernal, J.D. The World, the Flesh and the Devil: An Enquiry into the Future of the Three Enemies of the Rational Soul. Indiana University Press: Bloomington, IN, USA, 1969. (originally published 1929). [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, S.K. The Quantum Paradox in Pharmaceutical Science: Understanding Without Comprehending—A Centennial Reflection. Int. J. Mol. Sci. 2025, 26, 4658. https://doi.org/10.3390/ijms26104658
Niazi SK. The Quantum Paradox in Pharmaceutical Science: Understanding Without Comprehending—A Centennial Reflection. International Journal of Molecular Sciences. 2025; 26(10):4658. https://doi.org/10.3390/ijms26104658
Chicago/Turabian StyleNiazi, Sarfaraz K. 2025. "The Quantum Paradox in Pharmaceutical Science: Understanding Without Comprehending—A Centennial Reflection" International Journal of Molecular Sciences 26, no. 10: 4658. https://doi.org/10.3390/ijms26104658
APA StyleNiazi, S. K. (2025). The Quantum Paradox in Pharmaceutical Science: Understanding Without Comprehending—A Centennial Reflection. International Journal of Molecular Sciences, 26(10), 4658. https://doi.org/10.3390/ijms26104658