
Genetic Diversity, Admixture, and Selection Signatures in a Rarámuri Criollo Cattle Population Introduced to the Southwestern United States

 , , , , , , ,
, , , , , , ,
Abstract
1. Introduction
2. Results
2.1. Genetic Diversity
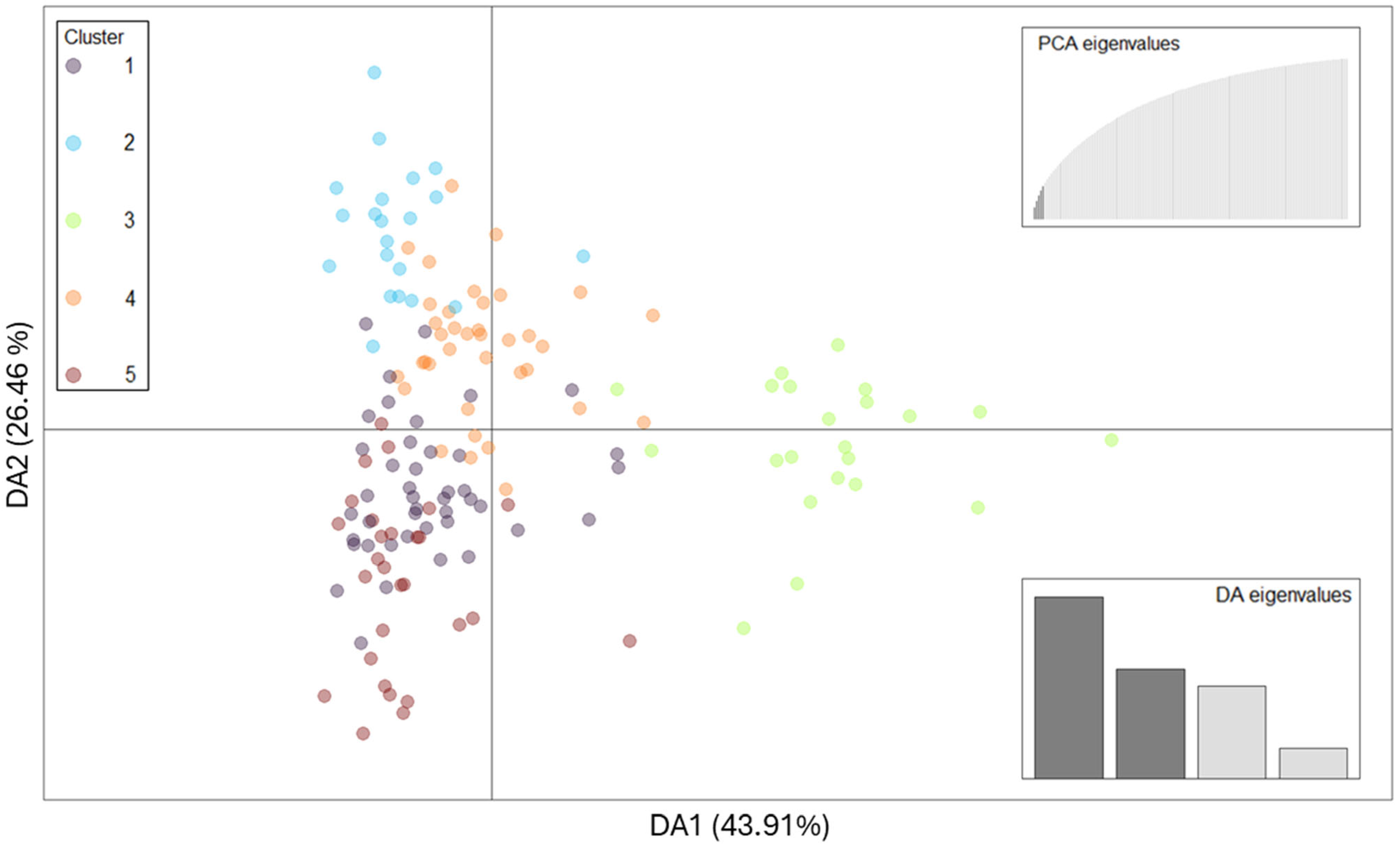
2.2. Population Structure and Genetic Relationships
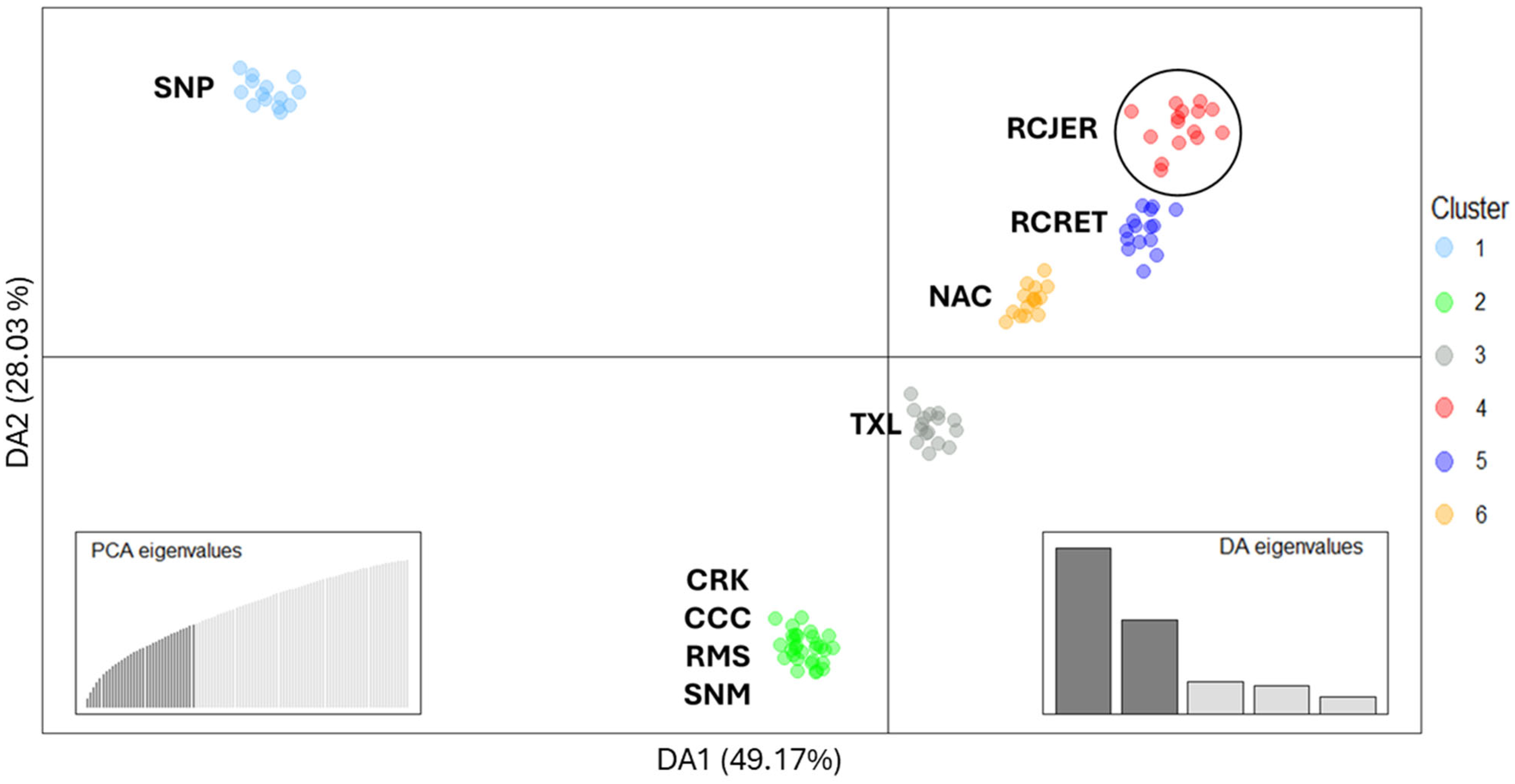
2.3. Relationship with Other Criollo Cattle Populations
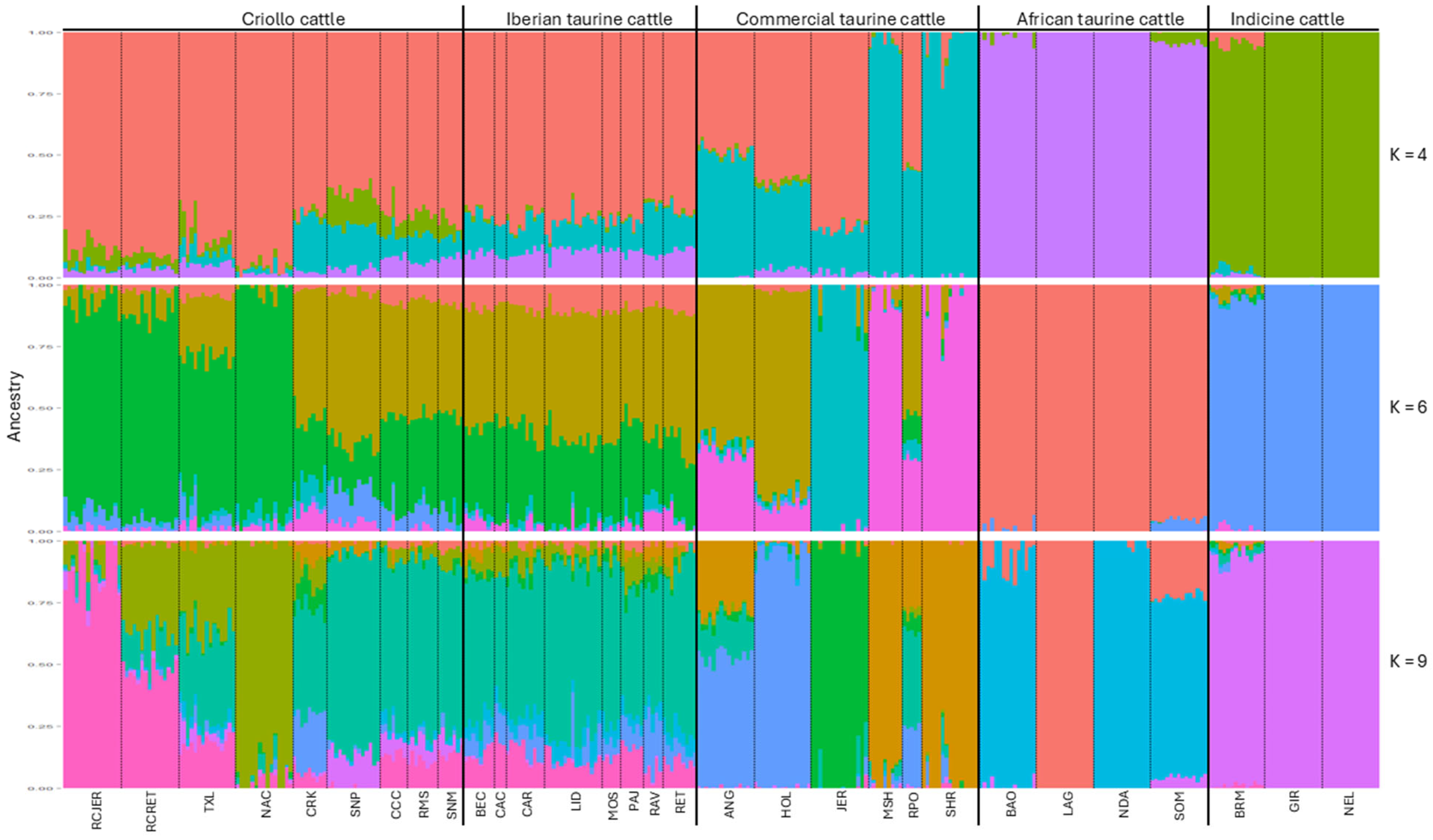
2.4. Ancestral Composition
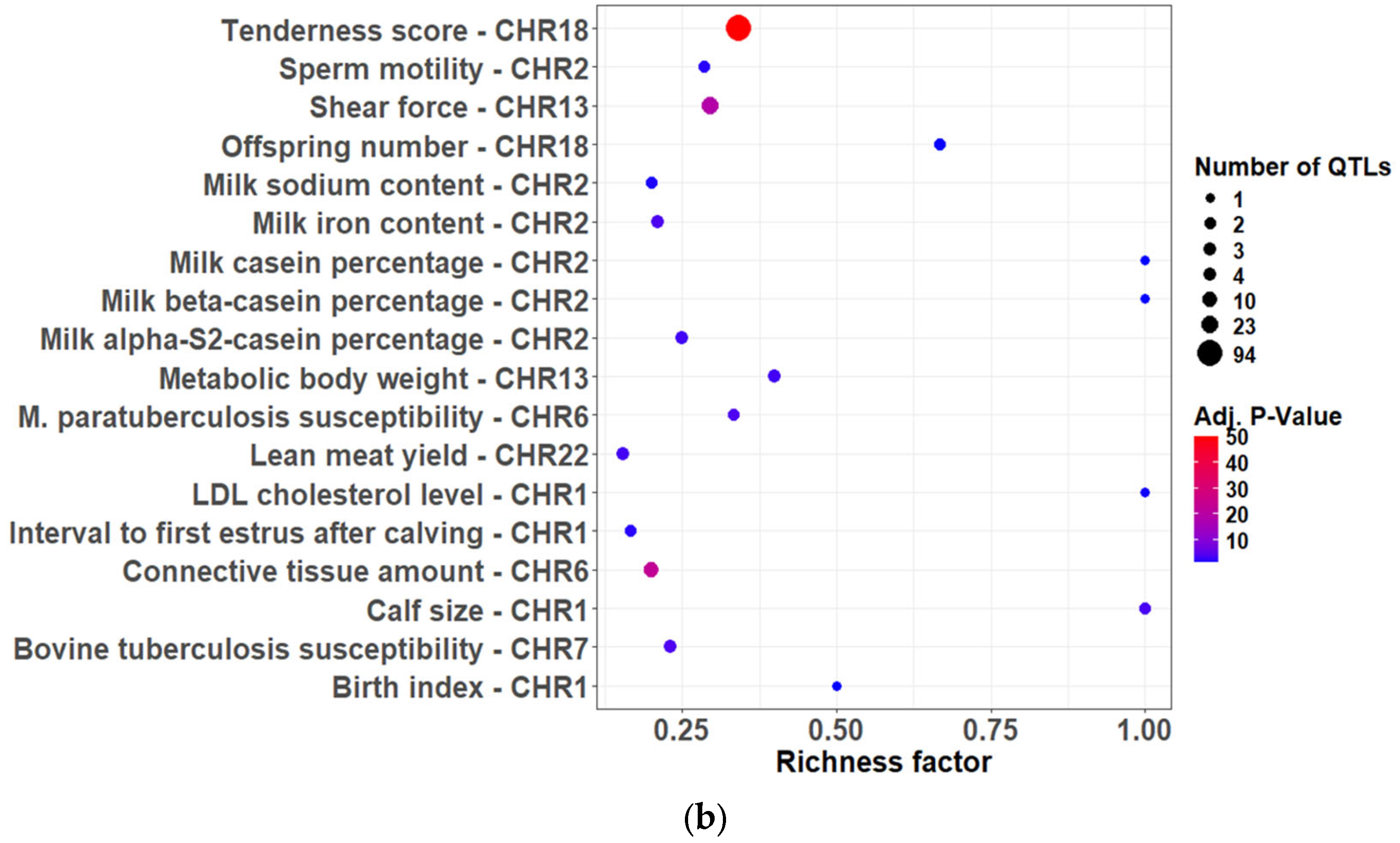
2.5. Selection Signature Analysis
2.6. Identification of Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Sampling, Genotyping and Quality Control
4.2. Genetic Diversity
4.3. Population Structure and Genetic Relationships
4.4. Relationship with Other Criollo Cattle Populations
4.5. Ancestral Composition
4.6. Selection Signature Analysis
4.7. Identification of Candidate Genes
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| RC | Rarámuri Criollo |
| JER | Jornada Experimental Range |
| RCJER | Rarámuri Criollo from the Jornada Experimental Range |
| HO | Observed heterozygosity |
| HE | Expected heterozygosity |
| ROH | Runs of homozygosity |
| FROH | Genomic inbreeding coefficient based on runs of homozygosity |
| Ne | Effective population size |
| PCA | Principal component analysis |
| DAPC | Discriminant Analysis of Principal Components |
| K | Cluster |
| PC | Principal components |
| DA | Discriminant Axis |
| iHS | Integrated haplotype score |
| QTL | Quantitative trait loci |
| NAC | North American Corriente |
| TXL | Texas Longhorn |
| RCRET | Rarámuri Criollo from the Rancho Experimental Teseachi |
| CRK | Florida Cracker |
| SNP | Senepol |
| CCC | Costeño con Cuernos |
| RMS | Romosinuano |
| SNM | San Martinero |
| BEC | Berrenda en Colorado |
| CAC | Cachena |
| CAR | Cardena Andaluza |
| LID | Lidia |
| MOS | Mostrenca |
| PAJ | Pajuna |
| RAV | Asturiana de los Valles |
| RET | Retinta |
| ANG | Angus |
| HOL | Holstein |
| JER | Jersey |
| MSH | Milking Shorthorn |
| RPO | Red Poll |
| SHR | Beef Shorthorn |
| BAO | Baoule |
| LAG | Lagune |
| NDA | N’Dama |
| SOM | Somba |
| BRM | Brahman |
| GIR | Gir |
| NEL | Nelore |
References
- Rodero Serrano, E.; Rodero Franganillo, A.; Delgado-Bermejo, J.V. Primitive Andalusian livestock and their implications in the discovery of America. Arch. Zootec. 1992, 41, 383–400. [Google Scholar]
- Primo, A. El ganado bovino ibérico en las Américas: 500 años después. Arch. Zootec. 1992, 41, 421–432. [Google Scholar]
- Delgado, J.V.; Martínez, A.M.; Acosta, A.; Álvarez, L.A.; Armstrong, E.; Camacho, E.; Cañón, J.; Cortés, O.; Dunner, S.; Landi, V.; et al. Genetic characterization of Latin-American Creole cattle using microsatellite markers. Anim. Genet. 2012, 43, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.M.; Gama, L.T.; Cañón, J.; Ginja, C.; Delgado, J.V.; Dunner, S.; Landi, V.; Martín-Burriel, I.; Penedo, M.C.T.; Rodellar, C.; et al. Genetic footprints of Iberian cattle in America 500 years after the arrival of Columbus. PLoS ONE 2012, 7, e49066. [Google Scholar] [CrossRef]
- McTavish, E.J.; Decker, J.E.; Schnabel, R.D.; Taylor, J.F.; Hillis, D.M. New World cattle show ancestry from multiple independent domestication events. Proc. Natl. Acad. Sci. USA 2013, 110, 1398–1406. [Google Scholar] [CrossRef]
- Ginja, C.; Gama, L.T.; Cortés, O.; Burriel, I.M.; Vega-Pla, J.L.; Penedo, C.; Sponenberg, P.; Cañón, J.; Sanz, A.; Alves, A.; et al. The genetic ancestry of American Creole cattle inferred from uniparental and autosomal genetic markers. Sci. Rep. 2019, 9, 11486. [Google Scholar] [CrossRef]
- Rouse, J.E. (Ed.) The Criollo: Spanish Cattle in the Americas, 1st ed.; University of Oklahoma Press: Norman, OK, USA, 1977. [Google Scholar]
- Anderson, D.M.; Estell, R.E.; Gonzalez, A.L.; Cibils, A.F.; Torell, L.A. Criollo cattle: Heritage genetics for arid landscapes. Rangelands 2015, 37, 62–67. [Google Scholar] [CrossRef]
- Pitt, D.; Bruford, M.W.; Barbato, M.; Orozco-terWengel, P.; Martínez, R.; Sevane, N. Demography and rapid local adaptation shape Creole cattle genome diversity in the tropics. Evol. Appl. 2019, 12, 105–122. [Google Scholar] [CrossRef]
- Armstrong, E.; Rodriguez Almeida, F.A.; McIntosh, M.M.; Poli, M.; Cibils, A.F.; Martínez-Quintana, J.A.; Félix-Portillo, M.; Estell, R.E. Genetic and productive background of Criollo cattle in Argentina, Mexico, Uruguay and the United States. J. Arid Environ. 2022, 200, 104722. [Google Scholar] [CrossRef]
- de Alba Martínez, J. (Ed.) El Libro de los Bovinos Criollos de América, 1st ed.; Ediciones Papiro Omega, S.A de C.V.: Morelia, Mexico, 2011. [Google Scholar]
- Rubio-Tabarez, E.; Pérez-Eguia, E. El bovino criollo de la Sierra Tarahumara. AICA 2015, 6, 485–494. [Google Scholar]
- Hernandez Sandoval, R.M. Caracterización Fenotípica y del Sistema de Producción del Ganado Criollo de Rodeo de la Sierra de Chihuahua. Master’s Thesis, Universidad Autónoma de Chihuahua, Chihuahua, Mexico, 2001. [Google Scholar]
- Russell, N.D.; Rios, J.; Erosa, G.; Remmenga, M.D.; Hawkins, D.E. Genetic differentiation among geographically isolated populations of Criollo cattle and their divergence from other Bos taurus breeds. J. Anim. Sci. 2000, 78, 2314–2322. [Google Scholar] [CrossRef] [PubMed]
- Estell, R. The genesis of the Jornada criollo cattle program. J. Arid Environ. 2021, 193, 104563. [Google Scholar] [CrossRef]
- McIntosh, M.; Gonzalez, A.; Cibils, A.; Estell, R.; Nyamuryekunge, S.; Almeida, F.R.; Spiegal, S. A phenotypic characterization of Rarámuri Criollo cattle introduced into the southwestern United States. Arch. Latinoam. Prod. Anim. 2020, 28, 111–119. [Google Scholar] [CrossRef]
- Spiegal, S.; Estell, R.E.; Cibils, A.F.; James, D.K.; Peinetti, H.R.; Browning, D.M.; Romig, K.B.; Gonzalez, A.L.; Lyons, A.J.; Bestelmeyer, B.T. Seasonal divergence of landscape use by heritage and conventional cattle on desert rangeland. Rangel. Ecol. Manag. 2019, 72, 590–601. [Google Scholar] [CrossRef]
- Nyamuryekung’e, S.; Cibils, A.F.; Estell, R.E.; VanLeeuwen, D.; Spiegal, S.; Steele, C.; González, A.L.; McIntosh, M.M.; Gong, Q.; Cao, H. Movement, activity, and landscape use patterns of heritage and commercial beef cows grazing Chihuahuan Desert rangeland. J. Arid Environ. 2022, 199, 104704. [Google Scholar] [CrossRef]
- Duni, D.; McIntosh, M.M.; Nyamuryekung’e, S.; Cibils, A.F.; Duniway, M.C.; Estell, R.E.; Spiegal, S.; Gonzalez, A.L.; Gedefaw, M.G.; Redd, M.; et al. Foraging behavior of Raramuri criollo vs. Angus cattle grazing California chaparral and Colorado Plateau shrublands. J. Arid Environ. 2023, 213, 104975. [Google Scholar] [CrossRef]
- Roacho Estrada, O.; Rodríguez Almeida, F.A.; Utsumi, S.A.; Fredrickson, E.L.; Bezanilla Enríquez, G.A.; Cibils, A.F.; Estell, R.E.; Gonzalez, A.L. Foraging behavior of Raramuri Criollo vs. commercial crossbred cows on rangelands of the southwestern United States and Northern Mexico. J. Arid Environ. 2023, 212, 104943. [Google Scholar] [CrossRef]
- Nyamuryekung’e, S.; Cibils, A.F.; Estell, R.E.; McIntosh, M.; VanLeeuwen, D.; Steele, C.; González, A.L.; Spiegal, S.; Reyes, L.A.; Rodríguez Almeida, F.A. Foraging behavior and body temperature of heritage vs. commercial beef cows in relation to desert ambient heat. J. Arid Environ. 2021, 193, 104565. [Google Scholar] [CrossRef]
- Estell, R.E.; Nyamuryekung’e, S.; James, D.K.; Spiegal, S.; Cibils, A.F.; Gonzalez, A.L.; McIntosh, M.M.; Romig, K. Diet selection of Raramuri criollo and Angus x Hereford crossbred cattle in the Chihuahuan Desert. J. Arid Environ. 2022, 205, 104823. [Google Scholar] [CrossRef]
- Castaño-Sánchez, J.P.; Rotz, C.A.; McIntosh, M.M.; Tolle, C.; Gifford, C.A.; Duff, G.C.; Spiegal, S.A. Grass finishing of Criollo cattle can provide an environmentally preferred and cost effective meat supply chain from United States drylands. Agric. Syst. 2023, 210, 103694. [Google Scholar] [CrossRef]
- Spiegal, S.; Estell, R.E.; Cibils, A.F.; Armstrong, E.; Blanco, L.J.; Bestelmeyer, B.T. Can heritage Criollo cattle promote sustainability in a changing world? J. Arid Environ. 2023, 216, 104980. [Google Scholar] [CrossRef]
- Torell, G.L.; Torell, L.A.; Enyinnaya, J.; Spiegal, S.; Estell, R.E.; Cibils, A.F.; Anderson, D.D.; Gonzalez, A.L. Economics of Raramuri Criollo and British crossbred cattle production in the Chihuahuan desert: Effects of foraging distribution and finishing strategy. J. Arid Environ. 2023, 211, 104922. [Google Scholar] [CrossRef]
- Sponenberg, D.P. Conservation of criollo livestock in the United States: Challenges and solutions. ALPA 2020, 28, 103–109. [Google Scholar] [CrossRef]
- Meyermans, R.; Gorssen, W.; Buys, N.; Janssens, S. How to study runs of homozygosity using PLINK? A guide for analyzing medium density SNP data in livestock and pet species. BMC Genom. 2020, 21, 94. [Google Scholar] [CrossRef]
- Manichaikul, A.; Mychaleckyj, J.C.; Rich, S.S.; Daly, K.; Sale, M.; Chen, W.M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010, 26, 2867–2873. [Google Scholar] [CrossRef]
- Corredor, F.A.; Figueroa, D.; Estrada, R.; Salazar, W.; Quilcate, C.; Vásquez, H.V.; Gonzales, J.; Maicelo, J.L.; Medina, P.; Arbizu, C.I. Genetic diversity and population structure of a Peruvian cattle herd using SNP data. Front. Genet. 2023, 14, 1073843. [Google Scholar] [CrossRef]
- Martinez, R.; Bejarano, D.; Ramírez, J.; Ocampo, R.; Polanco, N.; Perez, J.E.; Onofre, H.G.; Rocha, J.F. Genomic variability and population structure of six Colombian cattle breeds. Trop. Anim. Health Prod. 2023, 55, 185. [Google Scholar] [CrossRef]
- Marcuzzi, O.; Calcaterra, F.; Loza Vega, A.; Ortega Masagué, M.F.; Armstrong, E.; Pereira Rico, J.A.; Jara, E.; Olivera, L.H.; Peral García, P.; Giovambattista, G. Genomic analysis of inbreeding level, kinship and breed relationships in Creole cattle from South America. Anim. Genet. 2024, 55, 527–539. [Google Scholar] [CrossRef]
- Martínez Ruiz, P.C. Estimación de la Diversidad Genética Mediante Marcadores SNP en Bovino Criollo Coreño (Bos taurus). Bachelor’s Thesis, Universidad de Guadalajara, Guadalajara, Mexico, 2015. [Google Scholar]
- Armstrong, E.; Iriarte, A.; Martínez, A.M.; Feijoo, M.; Vega-Pla, J.L.; Delgado, J.V.; Postiglioni, A. Genetic diversity analysis of the Uruguayan Creole cattle breed using microsatellites and mtDNA markers. Genet. Mol. Res. 2013, 12, 1119–1131. [Google Scholar] [CrossRef]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef]
- FAO. In Vivo Conservation of Animal Genetic Resources; FAO Animal Production and Health Guidelines, Commission on Genetic Resources for food and Agriculture; FAO: Rome, Italy, 2013; No. 14. Available online: https://www.fao.org/4/i3327e/i3327e.pdf (accessed on 27 January 2025).
- Meuwissen, T. Genetic management of small populations: A review. Acta Agric. Scand. A 2009, 59, 71–79. [Google Scholar] [CrossRef]
- Frankham, R.; Bradshaw, C.J.; Brook, B.W. Genetics in conservation management: Revised recommendations for the 50/500 rules, Red List criteria and population viability analyses. Biol. Conserv. 2014, 170, 56–63. [Google Scholar] [CrossRef]
- Caivio-Nasner, S.; López-Herrera, A.; González-Herrera, L.G.; Rincón, J.C. Diversity analysis, runs of homozygosity and genomic inbreeding reveal recent selection in Blanco Orejinegro cattle. J. Anim. Breed. Genet. 2021, 138, 613–627. [Google Scholar] [CrossRef]
- Hidalgo, J.; Cesarani, A.; Garcia, A.; Sumreddee, P.; Larios, N.; Mancin, E.; García, J.G.; Núñez, R.; Ramírez, R. Genetic background and inbreeding depression in Romosinuano cattle breed in Mexico. Animals 2021, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Frankham, R.; Ballou, J.D.; Ralls, K.; Eldridge, M.D.B.; Dubash, M.; Fenster, C.B.; Lacy, R.C.; Sunnucks, P. (Eds.) Genetic Management of Fragmented Animal and Plant Populations, 1st ed.; Oxford University Press: Oxford, UK, 2017. [Google Scholar] [CrossRef]
- Rodríguez Almeida, F.A. (Facultad de Zootecnia y Ecología, Universidad Autónoma de Chihuahua, Chihuahua, México). Personal communication, 2025. [Google Scholar]
- Ulloa-Arvizu, R.; Gayosso-Vázquez, A.; Ramos-Kuri, M.; Estrada, F.J.; Montaño, M.; Alonso, R.A. Genetic analysis of Mexican Criollo cattle populations. J. Anim. Breed. Genet. 2008, 125, 351–359. [Google Scholar] [CrossRef]
- Decker, J.E.; McKay, S.D.; Rolf, M.M.; Kim, J.; Molina Alcalá, A.; Sonstegard, T.S.; Hanotte, O.; Götherström, A.; Seabury, C.M.; Praharani, L.; et al. Worldwide patterns of ancestry, divergence, and admixture in domesticated cattle. PLoS Genet. 2014, 10, e1004254. [Google Scholar] [CrossRef]
- Michiels, B.; Pardo, A.M.; Ortega Masague, M.F.; Giovambattista, G.; Corva, P.M. Characterization of experimental cattle populations from Argentina with a low-density SNP genotyping panel. Anim. Genet. 2023, 54, 68–72. [Google Scholar] [CrossRef]
- Venturini, G.C.; Cardoso, D.F.; Baldi, F.; Freitas, A.C.; Aspilcueta-Borquis, R.R.; Santos, D.J.A.; Camargo, G.M.F.; Stafuzza, N.B.; Albuquerque, L.G.; Tonhati, H. Association between single-nucleotide polymorphisms and milk production traits in buffalo. Genet. Mol. Res. 2014, 13, 10256–10268. [Google Scholar] [CrossRef]
- Buaban, S.; Lengnudum, K.; Boonkum, W.; Phakdeedindan, P. Genome-wide association study on milk production and somatic cell score for Thai dairy cattle using weighted single-step approach with random regression test-day model. J. Dairy Sci. 2022, 105, 468–494. [Google Scholar] [CrossRef]
- Raschia, M.A.; Nani, J.P.; Carignano, H.A.; Amadio, A.F.; Maizon, D.O.; Poli, M.A. Weighted single-step genome-wide association analyses for milk traits in Holstein and Holstein x Jersey crossbred dairy cattle. Livest. Sci. 2020, 242, 104294. [Google Scholar] [CrossRef]
- Illa, S.K.; Mukherjee, S.; Nath, S.; Mukherjee, A. Genome-wide scanning for signatures of selection revealed the putative genomic regions and candidate genes controlling milk composition and coat color traits in Sahiwal cattle. Front. Genet. 2021, 12, 699422. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.P.; Govignon-Gion, A.; Croiseau, P.; Fritz, S.; Hozé, C.; Miranda, G.; Martin, P.; Barbat-Leterrier, A.; Letaïef, R.; Rocha, D.; et al. Within-breed and multi-breed GWAS on imputed whole-genome sequence variants reveal candidate mutations affecting milk protein composition in dairy cattle. Genet. Sel. Evol. 2017, 49, 68. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, M.P.; Ramayo-Caldas, Y.; Wolf, V.; Laithier, C.; El Jabri, M.; Michenet, A.; Boussaha, M.; Taussat, S.; Fritz, S.; Delacroix-Buchet, A.; et al. Sequence-based GWAS, network and pathway analyses reveal genes co-associated with milk cheese-making properties and milk composition in Montbéliarde cows. Genet. Sel. Evol. 2019, 51, 34. [Google Scholar] [CrossRef]
- Macciotta, N.P.P.; Gaspa, G.; Bomba, L.; Vicario, D.; Dimauro, C.; Cellesi, M.; Ajmone-Marsan, P. Genome-wide association analysis in Italian Simmental cows for lactation curve traits using a low-density (7K) SNP panel. J. Dairy Sci. 2015, 98, 8175–8185. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, G.; Yang, L.; Zhu, B.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Liu, G.E.; Li, J. Genomic patterns of homozygosity in Chinese local cattle. Sci. Rep. 2019, 9, 16977. [Google Scholar] [CrossRef]
- Bionaz, M.; Loor, J.J. Gene networks driving bovine milk fat synthesis during the lactation cycle. BMC Genom. 2008, 9, 366. [Google Scholar] [CrossRef]
- Zhang, F.; Luo, J.; Shi, C.; Zhu, L.; He, Q.; Tian, H.; Wu, J.; Zhao, J.; Li, C. Genome-wide analysis of the acyl-coenzyme A synthetase family and their association with the formation of goat milk flavour. Front. Genet. 2022, 13, 980463. [Google Scholar] [CrossRef]
- de Camargo, G.M.F.; Aspilcueta-Borquis, R.R.; Fortes, M.R.S.; Porto-Neto, R.; Cardoso, D.F.; Santos, D.J.A.; Lehnert, S.A.; Reverter, A.; Moore, S.S.; Tonhati, H. Prospecting major genes in dairy buffaloes. BMC Genom. 2015, 16, 872. [Google Scholar] [CrossRef]
- Li, X.; Yuan, L.; Wang, W.; Zhang, D.; Zhao, Y.; Chen, J.; Xu, D.; Zhao, L.; Li, F.; Zhang, X. Whole genome re-sequencing reveals artificial and natural selection for milk traits in east Friesian sheep. Front. Vet. Sci. 2022, 9, 1034211. [Google Scholar] [CrossRef]
- Araújo, D.N.; de Camargo, G.M.F.; Fonseca, P.D.d.S.; Cardoso, D.F.; Hurtado-Lugo, N.A.; Aspilcueta-Borquis, R.R.; Tonhati, H. Polymorphisms in oxytocin and α1a adrenergic receptor genes and their effects on production traits in dairy buffaloes. Anim. Biotechnol. 2015, 26, 165–168. [Google Scholar] [CrossRef]
- de Simoni Gouveia, J.J.; Silva, M.V.G.B.D.; Paiva, S.R.; Oliveira, S.M.P.D. Identification of selection signatures in livestock species. Genet. Mol. Biol. 2014, 37, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Barbato, M.; Hailer, F.; Upadhyay, M.; Del Corvo, M.; Colli, L.; Negrini, R.; Kim, E.-S.; Crooijmans, R.P.M.A.; Sonstegard, T.; Ajmone-Marsan, P. Adaptive introgression from indicine cattle into white cattle breeds from Central Italy. Sci. Rep. 2020, 10, 1279. [Google Scholar] [CrossRef] [PubMed]
- Ceccobelli, S.; Landi, V.; Senczuk, G.; Mastrangelo, S.; Sardina, M.T.; Ben-Jemaa, S.; Persichilli, C.; Karsli, T.; Bâlteanu, V.-A.; Raschia, M.A.; et al. A comprehensive analysis of the genetic diversity and environmental adaptability in worldwide Merino and Merino-derived sheep breeds. Genet. Sel. Evol. 2023, 55, 24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhuang, Z.; Yang, M.; Ding, R.; Quan, J.; Zhou, S.; Gu, T.; Xu, Z.; Zheng, E.; Cai, G.; et al. Genome-Wide detection of genetic loci and candidate genes for body conformation traits in Duroc × Landrace × Yorkshire crossbred pigs. Front. Genet. 2021, 12, 664343. [Google Scholar] [CrossRef]
- Yang, R.; Zhou, D.; Tan, X.; Zhao, Z.; Lv, Y.; Tian, X.; Ren, L.; Wang, Y.; Li, J.; Zhao, Y.; et al. Genome-Wide Association study of body conformation traits in Tashi goats (Capra hircus). Animals 2024, 14, 1145. [Google Scholar] [CrossRef]
- Fernandes, G.A.; Costa, R.B.; de Camargo, G.M.F.; Carvalheiro, R.; Rosa, G.J.M.; Baldi, F.; Garcia, D.A.; Gordo, D.G.M.; Espigolan, R.; Takada, L.; et al. Genome scan for postmortem carcass traits in Nellore cattle. J. Anim. Sci. 2016, 94, 4087–4095. [Google Scholar] [CrossRef]
- Lee, Y.S.; Shin, D.; Song, K.D. Dominance effects of ion transport and ion transport regulator genes on the final weight and backfat thickness of landrace pigs by dominance deviation analysis. Genes Genom. 2018, 40, 1331–1338. [Google Scholar] [CrossRef]
- Edea, Z.; Jeoung, Y.H.; Shin, S.S.; Ku, J.; Seo, S.; Kim, I.H.; Kim, S.W.; Kim, K.S. Genome-wide association study of carcass weight in commercial Hanwoo cattle. Asian-Austr. J. Anim. Sci. 2017, 31, 327–334. [Google Scholar] [CrossRef]
- Dakhlan, A.; Moghaddar, N.; Gondro, C.; van der Werf, J.H.J. Gene by birth type interaction in merino lamb. Proc. Assoc. Advmt. Anim. Breed. Genet. 2018, 22, 45–48. [Google Scholar]
- Li, J.; Peng, S.; Zhong, L.; Yan, G.; Xiao, S.; Ma, J.; Huang, L. Identification and validation of a regulatory mutation upstream of the BMP2 gene associated with carcass length in pigs. Genet. Sel. Evol. 2021, 53, 94. [Google Scholar] [CrossRef]
- Colacchio, A.M. Association Between Carcass Maturity Grade and Genes Involved in Bone Growth, in Young Heifers. Master’s Thesis, University of Idaho, Moscow, ID, USA, 2019. [Google Scholar]
- Shira, K.A.; Murdoch, B.M.; Davenport, K.M.; Becker, G.M.; Xie, S.; Colacchio, A.M.; Bass, P.D.; Colle, M.J.; Murdoch, G.K. Advanced skeletal ossification is associated with genetic variants in chronologically young beef heifers. Genes 2023, 14, 1629. [Google Scholar] [CrossRef] [PubMed]
- Murgiano, L.; Jagannathan, V.; Benazzi, C.; Bolcato, M.; Brunetti, B.; Muscatello, L.V.; Dittmer, K.; Piffer, C.; Gentile, A.; Drögemüller, C. Deletion in the EVC2 gene causes chondrodysplastic dwarfism in Tyrolean Grey cattle. PLoS ONE 2014, 9, e94861. [Google Scholar] [CrossRef] [PubMed]
- Muscatello, L.V.; Benazzi, C.; Dittmer, K.E.; Thompson, K.G.; Murgiano, L.; Drögemüller, C.; Avallone, G.; Gentile, A.; Edwards, J.F.; Piffer, C.; et al. Ellis–van Creveld Syndrome in Grey Alpine cattle: Morphologic, immunophenotypic, and molecular characterization. Vet. Pathol. 2015, 52, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Nakada, S.; Yoshihara, T.; Nara, T.; Furuya, N.; Miida, T.; Hattori, N.; Arikawa-Hirasawa, E. Perlecan, a heparan sulfate proteoglycan, regulates systemic metabolism with dynamic changes in adipose tissue and skeletal muscle. Sci. Rep. 2018, 8, 7766. [Google Scholar] [CrossRef]
- Mei, C.; Li, S.; Abbas, S.H.; Tian, W.; Wang, H.; Li, Y.; Gui, L.; Zhang, Y.; Wu, X.; Zan, L. Performance measurement and comparative transcriptome analysis revealed the efforts on hybrid improvement of Qinchuan cattle. Anim. Biotechnol. 2019, 30, 13–20. [Google Scholar] [CrossRef]
- Lehnert, S.A.; Reverter, A.; Byrne, K.A.; Wang, Y.; Nattrass, G.S.; Hudson, N.J.; Greenwood, P.L. Gene expression studies of developing bovine longissimus muscle from two different beef cattle breeds. BMC Dev. Biol. 2007, 7, 95. [Google Scholar] [CrossRef]
- Berton, M.P.; Fonseca, L.F.S.; Gimenez, D.F.J.; Utembergue, B.L.; Cesar, A.S.M.; Coutinho, L.L.; de Lemos, M.V.A.; Aboujaoude, C.; Pereira, A.S.C.; de O Silva, R.M.; et al. Gene expression profile of intramuscular muscle in Nellore cattle with extreme values of fatty acid. BMC Genom. 2016, 17, 972. [Google Scholar] [CrossRef]
- Picard, B.; Gagaoua, M.; Jammas, M.A.; Bonnet, M. Beef tenderness and intramuscular fat proteomic biomarkers: Effect of gender and rearing practices. J. Proteom. 2019, 200, 1–10. [Google Scholar] [CrossRef]
- Crespo-Piazuelo, D.; Criado-Mesas, L.; Revilla, M.; Castelló, A.; Noguera, J.L.; Fernández, A.I.; Ballester, M.; Folch, J.M. Identification of strong candidate genes for backfat and intramuscular fatty acid composition in three crosses based on the Iberian pig. Sci. Rep. 2020, 10, 13962. [Google Scholar] [CrossRef]
- Tian, D.; Sun, D.; Ren, Q.; Zhang, P.; Zhang, Z.; Zhang, W.; Luo, H.; Li, X.; Han, B.; Liu, D.; et al. Genome-wide identification of candidate copy number polymorphism genes associated with complex traits of Tibetan-sheep. Sci. Rep. 2023, 13, 17283. [Google Scholar] [CrossRef]
- Kim, J.B.; Zeng, Z.X.; Nam, Y.J.; Kim, Y.; Yang, S.L.; Wu, X.; Lee, C. Association of mahogany/attractin gene (ATRN) with porcine growth and fat. Asian-Aust. J. Anim. Sci. 2005, 18, 1383–1386. [Google Scholar] [CrossRef]
- Liu, Y.; Albrecht, E.; Schering, L.; Kuehn, C.; Yang, R.; Zhao, Z.; Maak, S. Agouti signaling protein and its receptors as potential molecular markers for intramuscular and body fat deposition in cattle. Front. Physiol. 2018, 9, 172. [Google Scholar] [CrossRef] [PubMed]
- Supakankul, P.; Kumchoo, T.; Mekchay, S. Identification and characterization of novel single nucleotide polymorphism markers for fat deposition in muscle tissue of pigs using amplified fragment length polymorphism. Asian-Aust. J. Anim. Sci. 2017, 30, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Liu, J.; Han, J.; Yang, B. Association between BMP2 functional polymorphisms and sheep tail type. Animals 2020, 10, 739. [Google Scholar] [CrossRef]
- Yang, L.; Hao, W.; Wang, H.; Ren, W.; Yan, P.; Wei, S. BMP2 increases hyperplasia and hypertrophy of bovine subcutaneous preadipocytes via BMP/SMAD signaling. In Vitro Cell. Dev. Biol. Anim. 2022, 58, 210–219. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, W.; Zhang, D.; Zhang, Y.; Zhao, Y.; Li, X.; Zhao, L.; Lin, C.; Wang, J.; Zhou, B.; et al. Ovine RAP1GAP and rBAT gene polymorphisms and their association with tail fat deposition in Hu sheep. Front. Vet. Sci. 2022, 9, 974513. [Google Scholar] [CrossRef]
- Yilmaz, O.; Kizilaslan, M.; Arzik, Y.; Behrem, S.; Ata, N.; Karaca, O.; Elmaci, C.; Cemal, I. Genome-wide association studies of preweaning growth and in vivo carcass composition traits in Esme sheep. J. Anim. Breed. Genet. 2022, 139, 26–39. [Google Scholar] [CrossRef]
- Miao, Y.; Zhao, Y.; Wan, S.; Mei, Q.; Wang, H.; Fu, C.; Li, X.; Zhao, S.; Xu, X.; Xiang, T. Integrated analysis of genome-wide association studies and 3D epigenomic characteristics reveal the BMP2 gene regulating loin muscle depth in Yorkshire pigs. PLoS Genet. 2023, 19, e1010820. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, T.; Du, L.; Li, K.; Zhang, L.; Li, H.; Gao, X.; Xu, L.; Li, J.; Gao, H. Transcriptomic analysis reveals diverse expression patterns underlying the fiber diameter of oxidative and glycolytic skeletal muscles in steers. Meat Sci. 2024, 207, 109350. [Google Scholar] [CrossRef]
- McIntosh, M.M.; Cibils, A.F.; Estell, R.E.; Nyamuryekung’e, S.; González, A.L.; Gong, Q.; Cao, H.; Spiegal, S.A.; Soto-Navarro, S.A.; Blair, A.D. Weight gain, grazing behavior and carcass quality of desert grass-fed Rarámuri Criollo vs. crossbred steers. Livest. Sci. 2021, 249, 104511. [Google Scholar] [CrossRef]
- Audoin, F. Seasonal Grazing Behavior, Diet Selection, and Meat Characteristics of Range-Fed Raramuri Criollo Cattle (Cows, Heifers, and Steers) in Southeastern Arizona. Ph.D. Thesis, The University of Arizona, Tucson, AZ, USA, 2022. [Google Scholar]
- de Araujo Neto, F.R.; Takada, L.; Dos Santos, D.J.A.; Aspilcueta-Borquis, R.R.; Cardoso, D.F.; do Nascimento, A.V.; Martins Leão, K.; Nunes de Oliveira, H.; Tonhati, H. Identification of genomic regions related to age at first calving and first calving interval in water buffalo using single-step GBLUP. Reprod. Domest. Anim. 2020, 55, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Reding, J.J.; van der Westhuizen, R.R.; Berry, D.P.; van Marle-Köste, E. Understanding the underlying genetic mechanisms for age at first calving, inter-calving period and scrotal circumference in Bonsmara cattle. BMC Genom. 2023, 24, 480. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, R.J.; Irving-Rodgers, H.F.; Russell, D.L. Extracellular matrix of the developing ovarian follicle. Semin. Reprod. Med. 2003, 126, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Hwang, S.C.; Park, Y.S.; Oh, J.S. Cdc25B phosphatase participates in maintaining metaphase II arrest in mouse oocytes. Mol. Cells 2013, 35, 514–518. [Google Scholar] [CrossRef]
- Sánchez-Ramos, R.; Trujano-Chavez, M.Z.; Gallegos-Sánchez, J.; Becerril-Pérez, C.M.; Cadena-Villegas, S.; Cortez-Romero, C. Detection of candidate genes associated with fecundity through genome-wide selection signatures of Katahdin ewes. Animals 2023, 13, 272. [Google Scholar] [CrossRef]
- Kamiński, S.; Hering, D.M.; Oleński, K.; Lecewicz, M.; Kordan, W. Genome-wide association study for sperm membrane integrity in frozen-thawed semen of Holstein-Friesian bulls. Anim. Reprod. Sci. 2016, 170, 135–140. [Google Scholar] [CrossRef]
- Melo, T.P.D.; De Camargo, G.M.F.; De Albuquerque, L.G.; Carvalheiro, R. Genome-Wide association study provides strong evidence of genes affecting the reproductive performance of Nellore beef cows. PLoS ONE 2017, 12, e0178551. [Google Scholar] [CrossRef]
- Seabury, C.M.; Smith, J.L.; Wilson, M.L.; Bhattarai, E.; Santos, J.E.P.; Chebel, R.C.; Galvão, K.N.; Schuenemann, G.M.; Bicalho, R.C.; Gilbert, R.O. Genome-Wide association and genomic prediction for a reproductive index summarizing fertility outcomes in U.S. Holsteins. G3 Genes Genomes Genet. 2023, 13, jkad043. [Google Scholar] [CrossRef]
- Michailidou, S.; Tsangaris, G.T.; Tzora, A.; Skoufos, I.; Banos, G.; Argiriou, A.; Arsenos, G. Analysis of genome-wide DNA arrays reveals the genomic population structure and diversity in autochthonous Greek goat breeds. PLoS ONE 2016, 14, e0226179. [Google Scholar] [CrossRef]
- Neves, H.H.R.; Vargas, G.; Brito, L.F.; Schenkel, F.S.; Albuquerque, L.G.; Carvalheiro, R. Genetic and genomic analyses of testicular hypoplasia in Nellore cattle. PLoS ONE 2019, 14, e0211159. [Google Scholar] [CrossRef]
- Hering, D.M.; Olenski, K.; Kaminski, S. Genome-wide association study for poor sperm motility in Holstein-Friesian bulls. Anim. Reprod. Sci. 2014, 146, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Floriano-López, A.; Rodríguez-Almeida, F.A.; Martinez-Quintana, J.A.; Sandoval-Villalobos, J.; Roacho-Estrada, O.; Villareal-Balderrama, J.; Antillón-Ruiz, J. Productive performance of Raramuri Criollo, Angus x Raramuri Criollo and Hereford x Angus cows in a low-input cow-calf system in northern Mexico. J. Anim. Sci. 2024, 102, 456–457. [Google Scholar] [CrossRef]
- Andersson, E.A.; Harder, M.N.; Pilgaard, K.; Pisinger, C.; Stančáková, A.; Kuusisto, J.; Grarup, N.; Færch, K.; Poulsen, P.; Witte, D.R.; et al. The birth weight lowering C-allele of rs900400 near LEKR1 and CCNL1 associates with elevated insulin release following an oral glucose challenge. PLoS ONE 2011, 6, e27096. [Google Scholar] [CrossRef]
- Mook-Kanamori, D.O.; Marsh, J.A.; Warrington, N.M.; Taal, H.R.; Newnham, J.P.; Beilin, L.J.; Lye, S.J.; Palmer, L.J.; Hofman, A.; Steegers, E.A.P.; et al. Variants near CCNL1/LEKR1 and in ADCY5 and fetal growth characteristics in different trimesters. J. Clin. Endocrinol. Metab. 2011, 5, 810–815. [Google Scholar] [CrossRef]
- Hu, C.J.; Iyer, S.; Sataur, A.; Covello, K.L.; Chodosh, L.A.; Simon, M.C. Differential regulation of the transcriptional activities of hypoxia-inducible factor 1 alpha (HIF-1α) and HIF-2α in stem cells. Mol. Cell. Biol. 2006, 26, 3514–3526. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Verma, P.; Sharma, A.; Sodhi, M.; Thakur, K.; Bharti, V.K.; Kumar, P.; Giri, A.; Kalia, S.; Kumar Swami, S.; Mukesh, M. Overexpression of genes associated with hypoxia in cattle adapted to Trans Himalayan region of Ladakh. Cell Biol. Int. 2018, 42, 1141–1148. [Google Scholar] [CrossRef]
- Lopez-Ongil, S.; Saura, M.; Zaragoza, C.; Gonzalez-Santiago, L.; Rodrıguez-Puyol, M.; Lowenstein, C.J.; Rodrıguez-Puyol, D. Hydrogen peroxide regulation of bovine endothelin-converting enzyme-1. Free Radic. Biol. Med. 2002, 32, 406–413. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Li, H.; Zhang, S.; Li, F.; Qin, L. NLRX1 attenuates apoptosis and inflammatory responses in myocardial ischemia by inhibiting MAVS-dependent NLRP3 inflammasome activation. Mol. Immunol. 2016, 76, 90–97. [Google Scholar] [CrossRef]
- Hum, N.R.; Bourguet, F.A.; Sebastian, A.; Lam, D.; Phillips, A.M.; Sanchez, K.R.; Rasley, A.; Loots, G.G.; Weilhammer, D.R. MAVS mediates a protective immune response in the brain to Rift Valley fever virus. PLoS Pathog. 2022, 18, e1010231. [Google Scholar] [CrossRef] [PubMed]
- Suchocki, T.; Egger-Danner, C.; Schwarzenbacher, H.; Szyda, J. Two-stage genome-wide association study for the identification of causal variants underlying hoof disorders in cattle. J. Dairy Sci. 2020, 103, 4483–4494. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, D.; Ginja, C.; Carolino, N.; Leão, C.; Monteiro, H.; Tábuas, L.; Branco, S.; Padre, L.; Caetano, P.; Romão, R.; et al. Genome-wide association study identifies genetic variants underlying footrot in Portuguese Merino sheep. BMC Genom. 2024, 25, 100. [Google Scholar] [CrossRef]
- Hartati, H.; Putra, W.P.B.; Handiwirawan, E.; Ramon, E.; Firison, J.; Zubir, Z.; Suretno, N.D.; Mariyono, M.; Yusriani, Y.; Robba, D.K.; et al. Genome-wide association study of genetic markers of coat color patterns in Sumatran native cattle. Vet. World 2024, 17, 2537–2543. [Google Scholar] [CrossRef]
- Gunn, T.M.; Inui, T.; Kitada, K.; Ito, S.; Wakamatsu, K.; He, L.; Bouley, D.M.; Serikawa, T.; Barsh, G.S. Molecular and phenotypic analysis of Attractin mutant mice. Genetics 2001, 158, 1683–1695. [Google Scholar] [CrossRef]
- Seo, K.; Mohanty, T.R.; Choi, T.; Hwang, I. Biology of epidermal and hair pigmentation in cattle: A mini-review. Vet. Dermatol. 2007, 18, 392–400. [Google Scholar] [CrossRef]
- Pausch, H.; Wang, X.; Jung, S.; Krogmeier, D.; Edel, C.; Emmerling, R.; Götz, K.-U.; Fries, R. Identification of QTL for UV-protective eye area pigmentation in cattle by progeny phenotyping and genome-wide association analysis. PLoS ONE 2012, 7, e36346. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Barbato, M. Bioinformatics pipeline for quality control of genomic data. In Genomic Characterization of Animal Genetic Resources—Practical Guide; Ajmone-Marsan, P., Boettcher, P.J., Colli, L., Ginja, C., Kantanen, J., Lenstra, J.A., Eds.; FAO Animal Production and Health Guidelines; FAO: Rome, Italy, 2023; pp. 147–161. [Google Scholar] [CrossRef]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsam, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2014, 46, 110–121. [Google Scholar] [CrossRef]
- Biscarini, F.; Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes. R Package Version 0.9.5. 2019. Available online: https://cran.r-project.org/web/packages/detectRUNS/index.html (accessed on 13 January 2024).
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front. Genet. 2015, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Santiago, E.; Novo, I.; Pardiñas, A.F.; Saura, M.; Wang, J.; Caballero, A. Recent demographic history inferred by high-resolution analysis of linkage disequilibrium. Mol. Biol. Evol. 2020, 37, 3642–3653. [Google Scholar] [CrossRef]
- Sved, J.A.; Feldman, M.W. Correlation and probability methods for one and two loci. Theor. Popul. Biol. 1973, 4, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. (Ed.) ggplot2: Elegant Graphics for Data Analysis, 1st ed.; Springer: New York, NY, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 29 October 2024).
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, Y.; Ma, Q.; Liu, S.; Ma, Y.; Jiang, L. Analysis of family structure and paternity test of Tan sheep in Yanchi area, China. Animals 2022, 12, 3099. [Google Scholar] [CrossRef]
- Jombart, T.; Devillard, S.; Balloux, F. Discriminant analysis of principal components: A new method for the analysis of genetically structured populations. BMC Genet. 2010, 11, 94. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, 7. [Google Scholar] [CrossRef]
- Milanesi, M.; Capomaccio, S.; Vajana, E.; Bomba, L.; Garcia, J.F.; Ajmone-Marsan, P.; Colli, L. BITE: An R package for biodiversity analyses. BioRxiv 2017, 18161. [Google Scholar] [CrossRef]
- Pembleton, L.; Cogan, N.; Forster, J. StAMPP: An R package for calculation of genetic differentiation and structure of mixed-ploidy level populations. Mol. Ecol. Resour. 2013, 13, 946–952. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Novembre, J. Pritchard, Stephens, and Donnelly on population structure. Genetics 2016, 204, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Utsunomiya, Y.T.; Pérez O’Brien, A.M.; Sonstegard, T.S.; Van Tassell, C.P.; do Carmo, A.S.; Meszaros, G.; Sölkner, J.; Garcia, J.F. Detecting loci under recent positive selection in dairy and beef cattle by combining different genome-wide scan methods. PLoS ONE 2013, 8, e64280. [Google Scholar] [CrossRef] [PubMed]
- Saravanan, K.A.; Panigrahi, M.; Kumar, H.; Bhushan, B.; Dutt, T.; Mishra, B.P. Selection signatures in livestock genome: A review of concepts, approaches and applications. Livest. Sci. 2020, 241, 104257. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Rajawat, D.; Panigrahi, M.; Kumar, H.; Nayak, S.S.; Parida, S.; Bhushan, B.; Gaur, G.K.; Dutt, T.; Mishra, B.P. Identification of important genomic footprints using eight different selection signature statistics in domestic cattle breeds. Gene 2022, 816, 146165. [Google Scholar] [CrossRef]
- Xu, L.; Shi, L.; Liu, L.; Liang, R.; Li, Q.; Li, J.; Han, B.; Sun, S.D. Analysis of liver proteome and identification of critical proteins affecting milk fat, protein, and lactose metabolism in dairy cattle with iTRAQ. Proteomics 2019, 19, 1800387. [Google Scholar] [CrossRef]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar] [CrossRef]
- Gautier, M.; Klassmann, A.; Vitalis, R. rehh 2.0: A reimplementation of the R package rehh to detect positive selection from haplotype structure. Mol. Ecol. Resour. 2017, 17, 78–90. [Google Scholar] [CrossRef]
- Browning, B.L.; Tian, X.; Zhou, Y.; Browning, S.R. Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 2021, 108, 1880–1890. [Google Scholar] [CrossRef]
- Turner, S.D. qqman: An R package for visualizing GWAS results using Q-Q and manhattan plots. J. Open Source Softw. 2018, 3, 731. [Google Scholar] [CrossRef]
- Maiorano, A.M.; Lourenco, D.L.; Tsuruta, S.; Ospina, A.M.T.; Stafuzza, N.B.; Masuda, Y.; Eugenio, A.; Filho, V.; dos Santos Goncalves Cyrillo, J.N.; Abdallah Curi, R.; et al. Assessing genetic architecture and signatures of selection of dual purpose Gir cattle populations using genomic information. PLoS ONE 2018, 13, e0200694. [Google Scholar] [CrossRef] [PubMed]
- Neumann, G.B.; Korkuć, P.; Arends, D.; Wolf, M.J.; May, K.; König, S.; Brockmann, G.A. Genomic diversity and relationship analyses of endangered German Black Pied cattle (DSN) to 68 other taurine breeds based on whole-genome sequencing. Front. Genet. 2023, 13, 993959. [Google Scholar] [CrossRef]
- Elsik, C.G.; Unni, D.R.; Diesh, C.M.; Tayal, A.; Emery, M.L.; Nguyen, H.N.; Hagen, D.E. Bovine Genome Database: New tools for gleaning function from the Bos taurus genome. Nucleic Acids Res. 2016, 44, 834–839. [Google Scholar] [CrossRef]
- Fonseca, P.A.; Suarez-Vega, A.; Marras, G.; Cánovas, Á. GALLO: An R package for genomic annotation and integration of multiple data sources in livestock for positional candidate loci. Gigascience 2020, 9, giaa149. [Google Scholar] [CrossRef]
- Hu, Z.L.; Park, C.A.; Reecy, J.M. Building a livestock genetic and genomic information knowledgebase through integrative developments of Animal QTLdb and CorrDB. Nucleic Acids Res. 2018, 47, 701–710. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class (Mb) | ROH | FROH | |||
|---|---|---|---|---|---|
| Number | % | Mean | SD | Range | |
| >1–2 | 7620 | 63.25 | 0.106 | 0.062 | 0.023–0.402 |
| >2–4 | 2533 | 21.03 | 0.078 | 0.064 | 0.004–0.382 |
| >4–8 | 909 | 7.55 | 0.062 | 0.062 | 0.002–0.367 |
| >8–16 | 574 | 4.76 | 0.068 | 0.056 | 0.003–0.358 |
| >16 | 411 | 3.41 | 0.048 | 0.041 | 0.006–0.274 |
| Chr | SNPs | Position (bp) | Candidate Genes | Trait |
|---|---|---|---|---|
| 1 | 1 | 110,375,430–110,875,430 | RF00026, CCNL1, LOC112447734, LEKR1, TRNAW-CCA, LOC101902535 | reproduction |
| 1 | 6 | 137,774,984–138,733,932 | CPNE4, MIR2288, MRPL3, LOC104971058, NUDT16, NEK11, RF00026 | milk, growth |
| 2 | 8 | 130,797,318–131,607,803 | LOC101905607, LOC515042, CELA3B, LOC100847958, LOC789612, HSPG2, LDLRAD2, USP48, RAP1GAP, TRNAC-GCA, ALPL, RF00026, LOC101906756, ECE1, LOC112443420, LOC112443419, EIF4G3 | milk, growth, meat, reproduction |
| 6 | 1 | 103,360,904–103,860,904 | CRMP1, EVC, EVC2, RF00026, TRNAG-CCC, STK32B | meat, bone development |
| 7 | 4 | 69,034,944–69,695,703 | CYFIP2, NIPAL4, ADAM19, SOX30, THG1L, LSM11, CLINT1 | milk, coat color |
| 13 | 5 | 42,300,967–43,138,553 | SYNDIG1, TRNAG-CCC, LOC112449290, CST7, LOC107133049, APMAP, ACSS1, LOC112449375, VSX1, MIR2285df, ENTPD6, PYGB, ABHD12, LOC112449292, TRNAC-ACA, ANKRD16, LOC112449291, GDI2, FAM208B, RF00322, ASB13, LOC104973792 | milk, meat |
| 13 | 1 | 49,119,652–49,619,652 | BMP2, LOC104973807 | growth, meat |
| 13 | 9 | 50,252,256–51,412,240 | HAO1, ADRA1D, SMOX, LOC104973937, RNF24, PANK2, MIR103A2, MIR103-2, MAVS, AP5S1, CDC25B | meat, health |
| 13 | 1 | 51,530,574–52,030,574 | GFRA4, ATRN, C13H20orf194, SLC4A11, ITPA, DDRGK1, LZTS3 | meat, health |
| 18 | 1 | 24,264,845–24,764,845 | GNAO1, LOC112442287, CES5A, TRNAS-GGA, BREH1 | meat |
| 22 | 5 | 13,892,598–14,508,727 | ULK4, LOC107131659, TRAK1 | neurogenesis |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spetter, M.J.; Utsumi, S.A.; Armstrong, E.M.; Rodríguez Almeida, F.A.; Ross, P.J.; Macon, L.; Jara, E.; Cox, A.; Perea, A.R.; Funk, M.; et al. Genetic Diversity, Admixture, and Selection Signatures in a Rarámuri Criollo Cattle Population Introduced to the Southwestern United States. Int. J. Mol. Sci. 2025, 26, 4649. https://doi.org/10.3390/ijms26104649
Spetter MJ, Utsumi SA, Armstrong EM, Rodríguez Almeida FA, Ross PJ, Macon L, Jara E, Cox A, Perea AR, Funk M, et al. Genetic Diversity, Admixture, and Selection Signatures in a Rarámuri Criollo Cattle Population Introduced to the Southwestern United States. International Journal of Molecular Sciences. 2025; 26(10):4649. https://doi.org/10.3390/ijms26104649
Chicago/Turabian StyleSpetter, Maximiliano J., Santiago A. Utsumi, Eileen M. Armstrong, Felipe A. Rodríguez Almeida, Pablo J. Ross, Lara Macon, Eugenio Jara, Andrew Cox, Andrés R. Perea, Micah Funk, and et al. 2025. "Genetic Diversity, Admixture, and Selection Signatures in a Rarámuri Criollo Cattle Population Introduced to the Southwestern United States" International Journal of Molecular Sciences 26, no. 10: 4649. https://doi.org/10.3390/ijms26104649
APA StyleSpetter, M. J., Utsumi, S. A., Armstrong, E. M., Rodríguez Almeida, F. A., Ross, P. J., Macon, L., Jara, E., Cox, A., Perea, A. R., Funk, M., Redd, M., Cibils, A. F., Spiegal, S. A., & Estell, R. E. (2025). Genetic Diversity, Admixture, and Selection Signatures in a Rarámuri Criollo Cattle Population Introduced to the Southwestern United States. International Journal of Molecular Sciences, 26(10), 4649. https://doi.org/10.3390/ijms26104649