


ARID1A and Its Impact Across the Hallmarks of Cancer

 , ,
, , 
Abstract

1. Introduction
2. Clinical Impact
2.1. Prevalence of ARID1A Mutations Across Cancers
2.2. Overview of ARID1A Mutation Types: Frequency and Pathogenicity
2.3. Prognostic Factors Associated with ARID1A Mutations
3. Hallmarks of Cancer
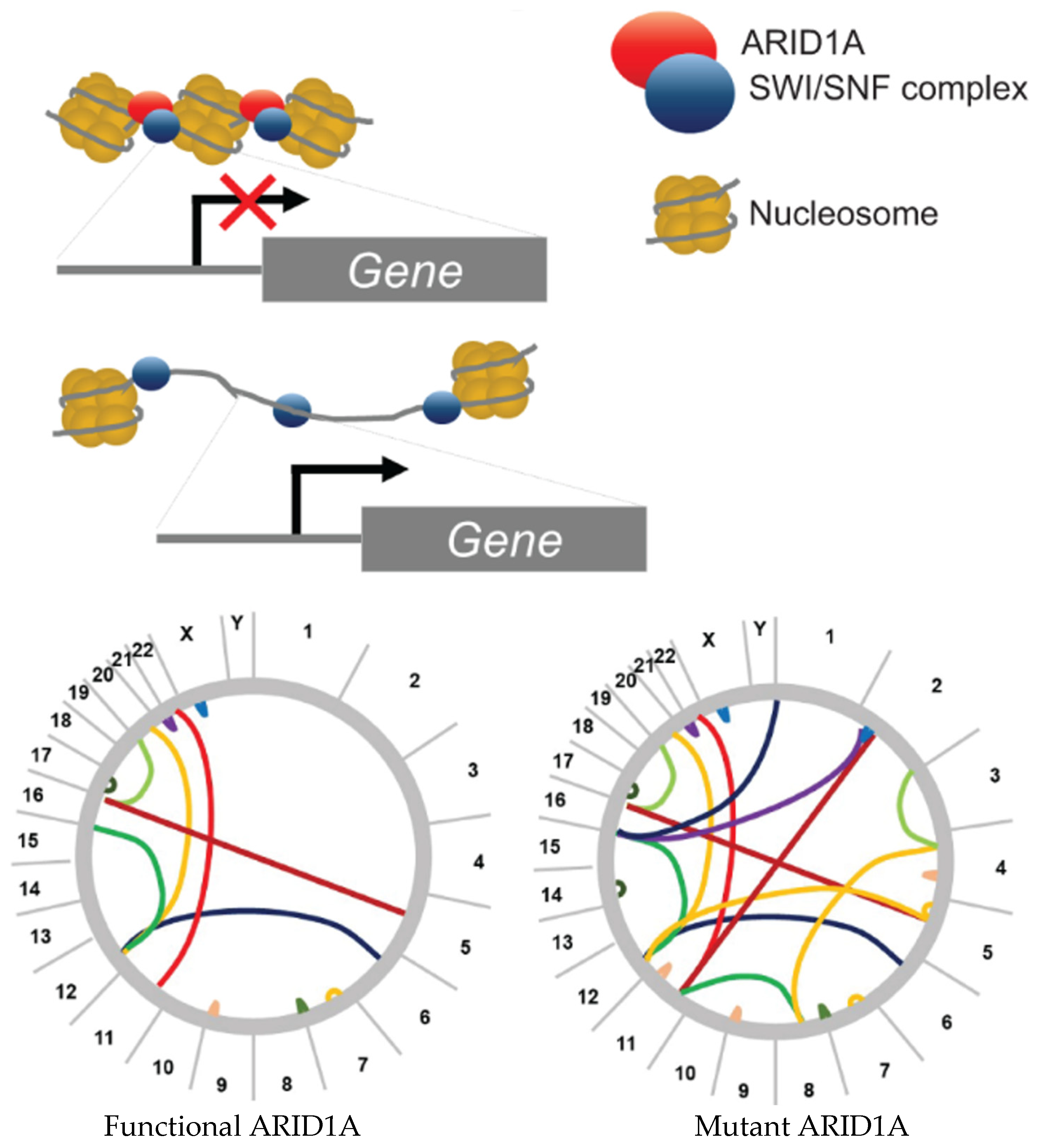
3.1. ARID1A Effect on Genomic Instability
3.2. ARID1A in the Regulation of the Cell Cycle
3.3. ARID1A Regulation of EMT and Cell Differentiation
3.4. Immune Evasion and Modulation in ARID1A-Deficient Tumors
3.5. ARID1A Loss and the Impact on Apoptosis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CDK | Cyclin-dependent Kinase |
| COSMIC | Catalog Of Somatic Mutations in Cancer |
| DSB | Double-Strand Break |
| 5-FU | 5-Fluorouracil |
| GSEA | Gene Set Enrichment Analysis |
| HDAC | Histone Deacetylase |
| EMT | Epithelial–Mesenchymal Transition |
| ICI | Immune Checkpoint Inhibitor |
| INDEL | Insertion-deletion |
| MDSC | Myeloid Derived Suppressor Cell |
| MMR | Mismatch Repair |
| OS | Overall Survival |
| RGC | Retinal Ganglion Cell |
| TAC | Transient Amplifying Cells |
| ZO-1 | Zonula Occludens-1 |
References
- Behjati, S.; Tarpey, P.S.; Haase, K.; Ye, H.; Young, M.D.; Alexandrov, L.B.; Farndon, S.J.; Collord, G.; Wedge, D.C.; Martincorena, I.; et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat. Commun. 2017, 8, 15936. [Google Scholar] [CrossRef]
- Wu, J.N.; Roberts, C.W. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.; Wang, T.L.; Shih, I.M. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011, 71, 6718–6727. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Ui, A.; Kanno, S.; Ogiwara, H.; Nagase, T.; Kohno, T.; Yasui, A. SWI/SNF factors required for cellular resistance to DNA damage include ARID1A and ARID1B and show interdependent protein stability. Cancer Res. 2014, 74, 2465–2475. [Google Scholar] [CrossRef]
- Mathur, R.; Alver, B.H.; San Roman, A.K.; Wilson, B.G.; Wang, X.; Agoston, A.T.; Park, P.J.; Shivdasani, R.A.; Roberts, C.W. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 2017, 49, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Schallenberg, S.; Bork, J.; Essakly, A.; Alakus, H.; Buettner, R.; Hillmer, A.M.; Bruns, C.; Schroeder, W.; Zander, T.; Loeser, H.; et al. Loss of the SWI/SNF-ATPase subunit members SMARCF1 (ARID1A), SMARCA2 (BRM), SMARCA4 (BRG1) and SMARCB1 (INI1) in oesophageal adenocarcinoma. BMC Cancer 2020, 20, 12. [Google Scholar] [CrossRef]
- Sen, M.; Wang, X.; Hamdan, F.H.; Rapp, J.; Eggert, J.; Kosinsky, R.L.; Wegwitz, F.; Kutschat, A.P.; Younesi, F.S.; Gaedcke, J.; et al. ARID1A facilitates KRAS signaling-regulated enhancer activity in an AP1-dependent manner in colorectal cancer cells. Clin. Epigenet. 2019, 11, 92. [Google Scholar] [CrossRef]
- Johnson, R.M.; Qu, X.; Lin, C.-F.; Huw, L.-Y.; Venkatanarayan, A.; Sokol, E.; Ou, F.-S.; Ihuegbu, N.; Zill, O.A.; Kabbarah, O.; et al. ARID1A mutations confer intrinsic and acquired resistance to cetuximab treatment in colorectal cancer. Nat. Commun. 2022, 13, 5478. [Google Scholar] [CrossRef]
- Fatema, K.; Wang, Y.; Pavek, A.; Larson, Z.; Nartker, C.; Plyler, S.; Jeppesen, A.; Mehling, B.; Capecchi, M.R.; Jones, K.B.; et al. Arid1a Loss Enhances Disease Progression in a Murine Model of Osteosarcoma. Cancers 2024, 16, 2725. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Chen, X.; Su, C.; Ren, S.; Zhou, C. Pan-cancer analysis of ARID1A Alterations as Biomarkers for Immunotherapy Outcomes. J. Cancer 2020, 11, 776–780. [Google Scholar] [CrossRef] [PubMed]
- Fontana, B.; Gallerani, G.; Salamon, I.; Pace, I.; Roncarati, R.; Ferracin, M. ARID1A in cancer: Friend or foe? Front. Oncol. 2023, 13, 1136248. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.C.; Wang, T.L.; Shih Ie, M. The emerging roles of ARID1A in tumor suppression. Cancer Biol. Ther. 2014, 15, 655–664. [Google Scholar] [CrossRef]
- Xu, S.; Tang, C. The Role of ARID1A in Tumors: Tumor Initiation or Tumor Suppression? Front. Oncol. 2021, 11, 745187. [Google Scholar] [CrossRef]
- Zhang, S.; Zhou, Y.F.; Cao, J.; Burley, S.K.; Wang, H.Y.; Zheng, X.F.S. mTORC1 Promotes ARID1A Degradation and Oncogenic Chromatin Remodeling in Hepatocellular Carcinoma. Cancer Res. 2021, 81, 5652–5665. [Google Scholar] [CrossRef]
- Clinton, T.N.; Chen, Z.; Wise, H.; Lenis, A.T.; Chavan, S.; Donoghue, M.T.A.; Almassi, N.; Chu, C.E.; Dason, S.; Rao, P.; et al. Genomic heterogeneity as a barrier to precision oncology in urothelial cancer. Cell Rep. 2022, 41, 111859. [Google Scholar] [CrossRef]
- Goutam, R.K.; Huang, G.; Medina, E.; Ding, F.; Edenfield, W.J.; Sanabria, H. Impact of frequent ARID1A mutations on protein stability provides insights into cancer pathogenesis. Sci. Rep. 2025, 15, 3072. [Google Scholar] [CrossRef]
- De Leo, A.; Ravegnini, G.; Musiani, F.; Maloberti, T.; Visani, M.; Sanza, V.; Angelini, S.; Perrone, A.M.; De Iaco, P.; Corradini, A.G.; et al. Relevance of ARID1A Mutations in Endometrial Carcinomas. Diagnostics 2022, 12, 592. [Google Scholar] [CrossRef]
- Kim, S.I.; Lee, J.W.; Lee, M.; Kim, H.S.; Chung, H.H.; Kim, J.W.; Park, N.H.; Song, Y.S.; Seo, J.S. Genomic landscape of ovarian clear cell carcinoma via whole exome sequencing. Gynecol. Oncol. 2018, 148, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhang, C.; Ren, Y.; Yi, H.; Luo, T.; Xing, F.; Bai, X.; Cui, L.; Zhu, L.; Ouyang, J.; et al. Genomic characterization of Chinese ovarian clear cell carcinoma identifies driver genes by whole exome sequencing. Neoplasia 2020, 22, 399–430. [Google Scholar] [CrossRef]
- Takeda, T.; Banno, K.; Okawa, R.; Yanokura, M.; Iijima, M.; Irie-Kunitomi, H.; Nakamura, K.; Iida, M.; Adachi, M.; Umene, K.; et al. ARID1A gene mutation in ovarian and endometrial cancers. Oncol. Rep. 2016, 35, 607–613. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Lee, C.S. The Role of the AT-Rich Interaction Domain 1A Gene (ARID1A) in Human Carcinogenesis. Genes 2023, 15, 5. [Google Scholar] [CrossRef]
- Rhee, J.K.; Yoo, J.; Kim, K.R.; Kim, J.; Lee, Y.J.; Chul Cho, B.; Kim, T.M. Identification of Local Clusters of Mutation Hotspots in Cancer-Related Genes and Their Biological Relevance. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 16, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef]
- Guan, B.; Gao, M.; Wu, C.H.; Wang, T.L.; Shih, I.M. Functional analysis of in-frame indel ARID1A mutations reveals new regulatory mechanisms of its tumor suppressor functions. Neoplasia 2012, 14, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Ying, J.; Yang, K.; Xiong, X.; Yang, N.; Wang, S.; Zhao, W.; Zhu, H.; Yu, M.; Wu, J.; et al. Deciphering the regulatory mechanisms and biological implications of ARID1A missense mutations in cancer. Cell Rep. 2024, 43, 114916. [Google Scholar] [CrossRef]
- Nan, L.; Wang, C.; Wang, J.; Zhang, S.; Bo, X.; Wang, Y.; Liu, H. ARID1A Downregulation Predicts High PD-L1 Expression and Worse Clinical Outcome in Patients With Gallbladder Cancer. Front. Oncol. 2022, 12, 787897. [Google Scholar] [CrossRef]
- Angelico, G.; Attanasio, G.; Colarossi, L.; Colarossi, C.; Montalbano, M.; Aiello, E.; Di Vendra, F.; Mare, M.; Orsi, N.; Memeo, L. ARID1A Mutations in Gastric Cancer: A Review with Focus on Clinicopathological Features, Molecular Background and Diagnostic Interpretation. Cancers 2024, 16, 2062. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.D.; Chen, Y.B.; Pan, K.; Wang, W.; Chen, S.P.; Chen, J.G.; Zhao, J.J.; Lv, L.; Pan, Q.Z.; Li, Y.Q.; et al. Decreased expression of the ARID1A gene is associated with poor prognosis in primary gastric cancer. PLoS ONE 2012, 7, e40364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lin, A.; Li, Y.; Ding, W.; Meng, H.; Luo, P.; Zhang, J. Age and Mutations as Predictors of the Response to Immunotherapy in Head and Neck Squamous Cell Cancer. Front. Cell Dev. Biol. 2020, 8, 608969. [Google Scholar] [CrossRef]
- Mandal, J.; Mandal, P.; Wang, T.L.; Shih, I.M. Treating ARID1A mutated cancers by harnessing synthetic lethality and DNA damage response. J. Biomed. Sci. 2022, 29, 71. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Shen, Z.; Gao, Z.F.; Chao, Q.; Qian, C.R.; Zheng, H.; Wang, B.C. A comprehensive analysis of the lysine acetylome reveals diverse functions of acetylated proteins during de-etiolation in Zea mays. J. Plant Physiol. 2020, 248, 153158. [Google Scholar] [CrossRef]
- Okamura, R.; Kato, S.; Lee, S.; Jimenez, R.E.; Sicklick, J.K.; Kurzrock, R. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J. Immunother. Cancer 2020, 8, e000438. [Google Scholar] [CrossRef]
- Kato, M.K.; Yoshida, H.; Tanase, Y.; Uno, M.; Ishikawa, M.; Kato, T. Loss of ARID1A Expression as a Favorable Prognostic Factor in Early-Stage Grade 3 Endometrioid Endometrial Carcinoma Patients. Pathol. Oncol. Res. 2021, 27, 598550. [Google Scholar] [CrossRef]
- D’Ambrosio, A.; Bressan, D.; Ferracci, E.; Carbone, F.; Mulè, P.; Rossi, F.; Barbieri, C.; Sorrenti, E.; Fiaccadori, G.; Detone, T.; et al. Increased genomic instability and reshaping of tissue microenvironment underlie oncogenic properties of Arid1a mutations. Sci. Adv. 2024, 10, eadh4435. [Google Scholar] [CrossRef]
- Sun, M.; Gu, Y.; Fang, H.; Shao, F.; Lin, C.; Zhang, H.; Li, H.; He, H.; Li, R.; Wang, J.; et al. Clinical outcome and molecular landscape of patients with ARID1A-loss gastric cancer. Cancer Sci. 2024, 115, 905–915. [Google Scholar] [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, F.; Tang, M.; Xu, W.; Tian, Y.; Liu, Z.; Shu, Y.; Yang, H.; Zhu, Q.; Lu, X.; et al. The ARID1A-METTL3-m6A axis ensures effective RNase H1-mediated resolution of R-loops and genome stability. Cell Rep. 2024, 43, 113779. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Chui, M.H.; Suryo Rahmanto, Y.; Yu, Z.C.; Shamanna, R.A.; Bellani, M.A.; Gaillard, S.; Ayhan, A.; Viswanathan, A.; Seidman, M.M.; et al. Loss of ARID1A in Tumor Cells Renders Selective Vulnerability to Combined Ionizing Radiation and PARP Inhibitor Therapy. Clin. Cancer Res. 2019, 25, 5584–5594. [Google Scholar] [CrossRef]
- Bakr, A.; Della Corte, G.; Veselinov, O.; Kelekçi, S.; Chen, M.M.; Lin, Y.Y.; Sigismondo, G.; Iacovone, M.; Cross, A.; Syed, R.; et al. ARID1A regulates DNA repair through chromatin organization and its deficiency triggers DNA damage-mediated anti-tumor immune response. Nucleic Acids Res. 2024, 52, 5698–5719. [Google Scholar] [CrossRef] [PubMed]
- Nacarelli, T.; Zhao, B.; Hao, X.; Zhang, R. ARID1A mutation and genomic stability. Mol. Cell. Oncol. 2020, 7, 1690923. [Google Scholar] [CrossRef] [PubMed]
- Somsuan, K.; Peerapen, P.; Boonmark, W.; Plumworasawat, S.; Samol, R.; Sakulsak, N.; Thongboonkerd, V. ARID1A knockdown triggers epithelial-mesenchymal transition and carcinogenesis features of renal cells: Role in renal cell carcinoma. FASEB J. 2019, 33, 12226–12239. [Google Scholar] [CrossRef]
- Megino-Luque, C.; Sisó, P.; Mota-Martorell, N.; Navaridas, R.; de la Rosa, I.; Urdanibia, I.; Albertí-Valls, M.; Santacana, M.; Pinyol, M.; Bonifaci, N.; et al. ARID1A-deficient cells require HDAC6 for progression of endometrial carcinoma. Mol. Oncol. 2022, 16, 2235–2259. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Zhao, J.; Wu, Y.; Zhang, N.; Shen, W. ARID1A mutations in cancer development: Mechanism and therapy. Carcinogenesis 2023, 44, 197–208. [Google Scholar] [CrossRef]
- Stubbs, F.E.; Flynn, B.P.; Rivers, C.A.; Birnie, M.T.; Herman, A.; Swinstead, E.E.; Baek, S.; Fang, H.; Temple, J.; Carroll, J.S.; et al. Identification of a novel GR-ARID1a-P53BP1 protein complex involved in DNA damage repair and cell cycle regulation. Oncogene 2022, 41, 5347–5360. [Google Scholar] [CrossRef]
- Nagl, N.G., Jr.; Patsialou, A.; Haines, D.S.; Dallas, P.B.; Beck, G.R., Jr.; Moran, E. The p270 (ARID1A/SMARCF1) subunit of mammalian SWI/SNF-related complexes is essential for normal cell cycle arrest. Cancer Res. 2005, 65, 9236–9244. [Google Scholar] [CrossRef]
- Du, J.; Jing, J.; Chen, S.; Yuan, Y.; Feng, J.; Ho, T.V.; Sehgal, P.; Xu, J.; Jiang, X.; Chai, Y. Arid1a regulates cell cycle exit of transit-amplifying cells by inhibiting the Aurka-Cdk1 axis in mouse incisor. Development 2021, 148, dev198838. [Google Scholar] [CrossRef]
- Wang, T.; Gao, X.; Zhou, K.; Jiang, T.; Gao, S.; Liu, P.; Zuo, X.; Shi, X. Role of ARID1A in epithelial-mesenchymal transition in breast cancer and its effect on cell sensitivity to 5-FU. Int. J. Mol. Med. 2020, 46, 1683–1694. [Google Scholar] [CrossRef] [PubMed]
- Tomihara, H.; Carbone, F.; Perelli, L.; Huang, J.K.; Soeung, M.; Rose, J.L.; Robinson, F.S.; Lissanu Deribe, Y.; Feng, N.; Takeda, M.; et al. Loss of ARID1A Promotes Epithelial-Mesenchymal Transition and Sensitizes Pancreatic Tumors to Proteotoxic Stress. Cancer Res. 2021, 81, 332–343. [Google Scholar] [CrossRef]
- Wanna-Udom, S.; Aluksanasuwan, S.; Somsuan, K.; Mongkolwat, W.; Sakulsak, N. ARID1A overexpression inhibits colorectal cancer cell migration through the regulation of epithelial-mesenchymal transition. Mol. Med. Rep. 2024, 30, 201. [Google Scholar] [CrossRef]
- Wilson, M.R.; Reske, J.J.; Holladay, J.; Wilber, G.E.; Rhodes, M.; Koeman, J.; Adams, M.; Johnson, B.; Su, R.W.; Joshi, N.R.; et al. ARID1A and PI3-kinase pathway mutations in the endometrium drive epithelial transdifferentiation and collective invasion. Nat. Commun. 2019, 10, 3554. [Google Scholar] [CrossRef]
- Xu, S.; Zhu, C.; Xu, Q.; An, Z.; Xu, S.; Xuan, G.; Lin, C.; Tang, C. ARID1A restrains EMT and stemness of ovarian cancer cells through the Hippo pathway. Int. J. Oncol. 2024, 65, 76. [Google Scholar] [CrossRef]
- Lu, W.C.; Liu, C.J.; Tu, H.F.; Chung, Y.T.; Yang, C.C.; Kao, S.Y.; Chang, K.W.; Lin, S.C. miR-31 targets ARID1A and enhances the oncogenicity and stemness of head and neck squamous cell carcinoma. Oncotarget 2016, 7, 57254–57267. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, S.; Gao, H.; Han, L.; Chu, X.; Sheng, Y.; Shou, W.; Wang, Y.; Liu, Y.; Wan, J.; et al. Genome-wide studies reveal the essential and opposite roles of ARID1A in controlling human cardiogenesis and neurogenesis from pluripotent stem cells. Genome Biol. 2020, 21, 169. [Google Scholar] [CrossRef]
- Sun, D.; Qian, H.; Wang, J.; Xie, T.; Teng, F.; Li, J.; Xing, P. ARID1A deficiency reverses the response to anti-PD(L)1 therapy in EGFR-mutant lung adenocarcinoma by enhancing autophagy-inhibited type I interferon production. Cell Commun. Signal. 2022, 20, 156. [Google Scholar] [CrossRef] [PubMed]
- Ashizawa, M.; Saito, M.; Min, A.K.T.; Ujiie, D.; Saito, K.; Sato, T.; Kikuchi, T.; Okayama, H.; Fujita, S.; Endo, H.; et al. Prognostic role of ARID1A negative expression in gastric cancer. Sci. Rep. 2019, 9, 6769. [Google Scholar] [CrossRef]
- Samartzis, E.P.; Gutsche, K.; Dedes, K.J.; Fink, D.; Stucki, M.; Imesch, P. Loss of ARID1A expression sensitizes cancer cells to PI3K- and AKT-inhibition. Oncotarget 2014, 5, 5295–5303. [Google Scholar] [CrossRef]
- Tokunaga, R.; Xiu, J.; Goldberg, R.M.; Philip, P.A.; Seeber, A.; Battaglin, F.; Arai, H.; Lo, J.H.; Naseem, M.; Puccini, A.; et al. The impact of ARID1A mutation on molecular characteristics in colorectal cancer. Eur. J. Cancer 2020, 140, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Cai, Q.; Yu, S.; Ji, J.; Zhu, Z.; Yan, C.; Zhang, J. Combinatorial Analysis of AT-Rich Interaction Domain 1A and CD47 in Gastric Cancer Patients Reveals Markers of Prognosis. Front. Cell Dev. Biol. 2021, 9, 745120. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, Q.; Han, Y.; Pei, S.; Cheng, B.; Xu, J.; Miao, X.; Pan, Q.; Wang, H.; Guo, J.; et al. ARID1A loss induces polymorphonuclear myeloid-derived suppressor cell chemotaxis and promotes prostate cancer progression. Nat. Commun. 2022, 13, 7281. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.Q.; Dai, S.K.; Li, C.P.; Liu, P.P.; Wang, Z.M.; Du, H.Z.; Teng, Z.Q.; Yang, S.G.; Liu, C.M. Loss of Arid1a Promotes Neuronal Survival Following Optic Nerve Injury. Front. Cell. Neurosci. 2020, 14, 131. [Google Scholar] [CrossRef]
- Bitler, B.G.; Wu, S.; Park, P.H.; Hai, Y.; Aird, K.M.; Wang, Y.; Zhai, Y.; Kossenkov, A.V.; Vara-Ailor, A.; Rauscher, F.J., III; et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat. Cell Biol. 2017, 19, 962–973. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| AA Position | R1989 * | R1276 * | R1721 * | R693 * | G2087R | R1335 * |
|---|---|---|---|---|---|---|
| cDNA Position | 5965C>T | 3826C>T | 5161C>T | 2077C>T | 6259G>A | 4003C>T |
| Total number of mutations out of 550 samples | 65 | 38 | 31 | 31 | 24 | 24 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kearns, B.; McKell, A.; Steveson, I.; Worley, P.; Barton, B.; Bennett, J.; Anderson, D.; Harris, J.; Christensen, J.; Barrott, J.J. ARID1A and Its Impact Across the Hallmarks of Cancer. Int. J. Mol. Sci. 2025, 26, 4644. https://doi.org/10.3390/ijms26104644
Kearns B, McKell A, Steveson I, Worley P, Barton B, Bennett J, Anderson D, Harris J, Christensen J, Barrott JJ. ARID1A and Its Impact Across the Hallmarks of Cancer. International Journal of Molecular Sciences. 2025; 26(10):4644. https://doi.org/10.3390/ijms26104644
Chicago/Turabian StyleKearns, Bridger, Andralyn McKell, Isaac Steveson, Peyton Worley, Braeden Barton, Jordan Bennett, DeLaney Anderson, Jacob Harris, James Christensen, and Jared J. Barrott. 2025. "ARID1A and Its Impact Across the Hallmarks of Cancer" International Journal of Molecular Sciences 26, no. 10: 4644. https://doi.org/10.3390/ijms26104644
APA StyleKearns, B., McKell, A., Steveson, I., Worley, P., Barton, B., Bennett, J., Anderson, D., Harris, J., Christensen, J., & Barrott, J. J. (2025). ARID1A and Its Impact Across the Hallmarks of Cancer. International Journal of Molecular Sciences, 26(10), 4644. https://doi.org/10.3390/ijms26104644