
Genetic, Transcriptomic, and Epigenomic Insights into Sjögren’s Disease: An Integrative Network Investigation and Immune Diseases Comparison


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
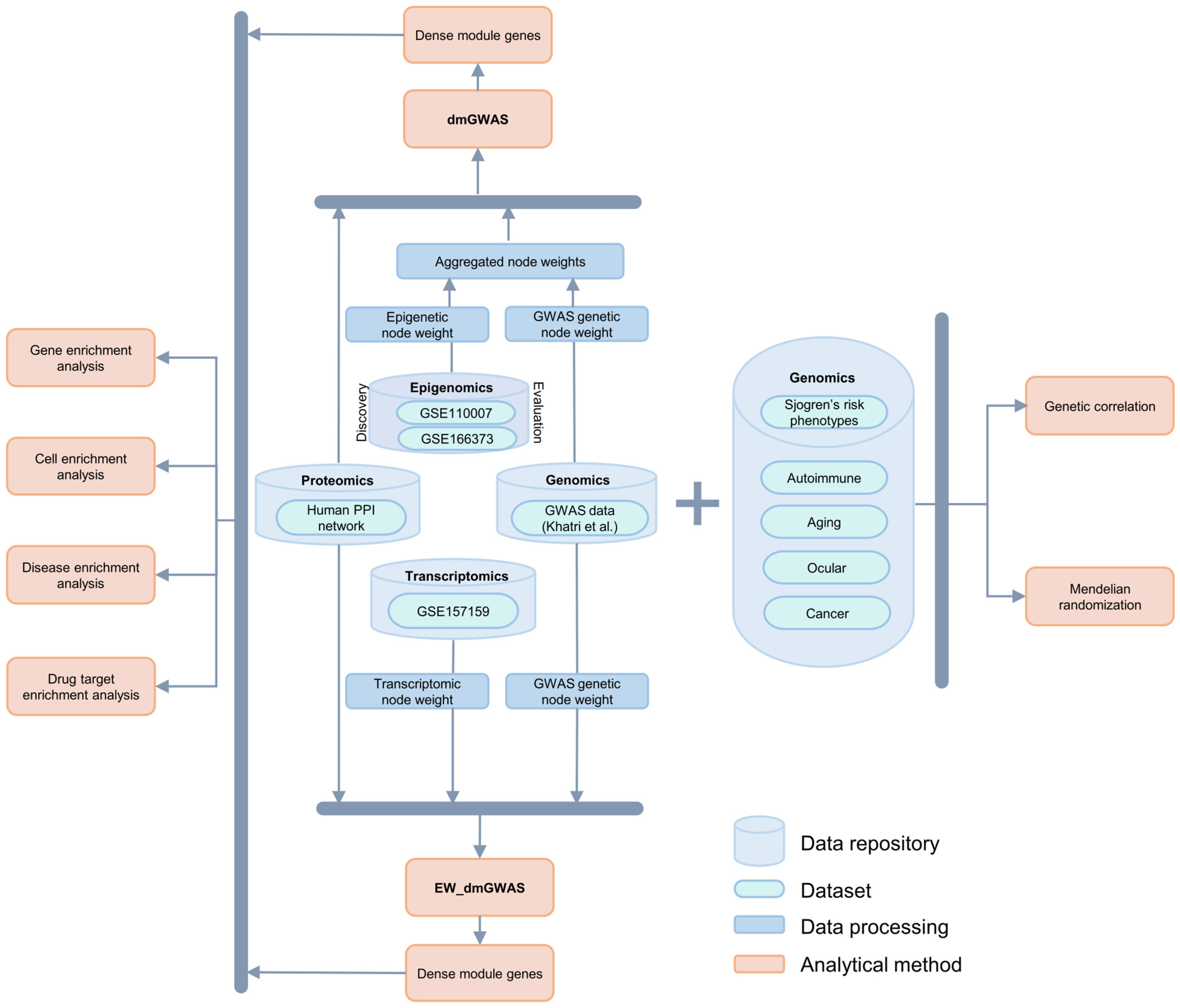
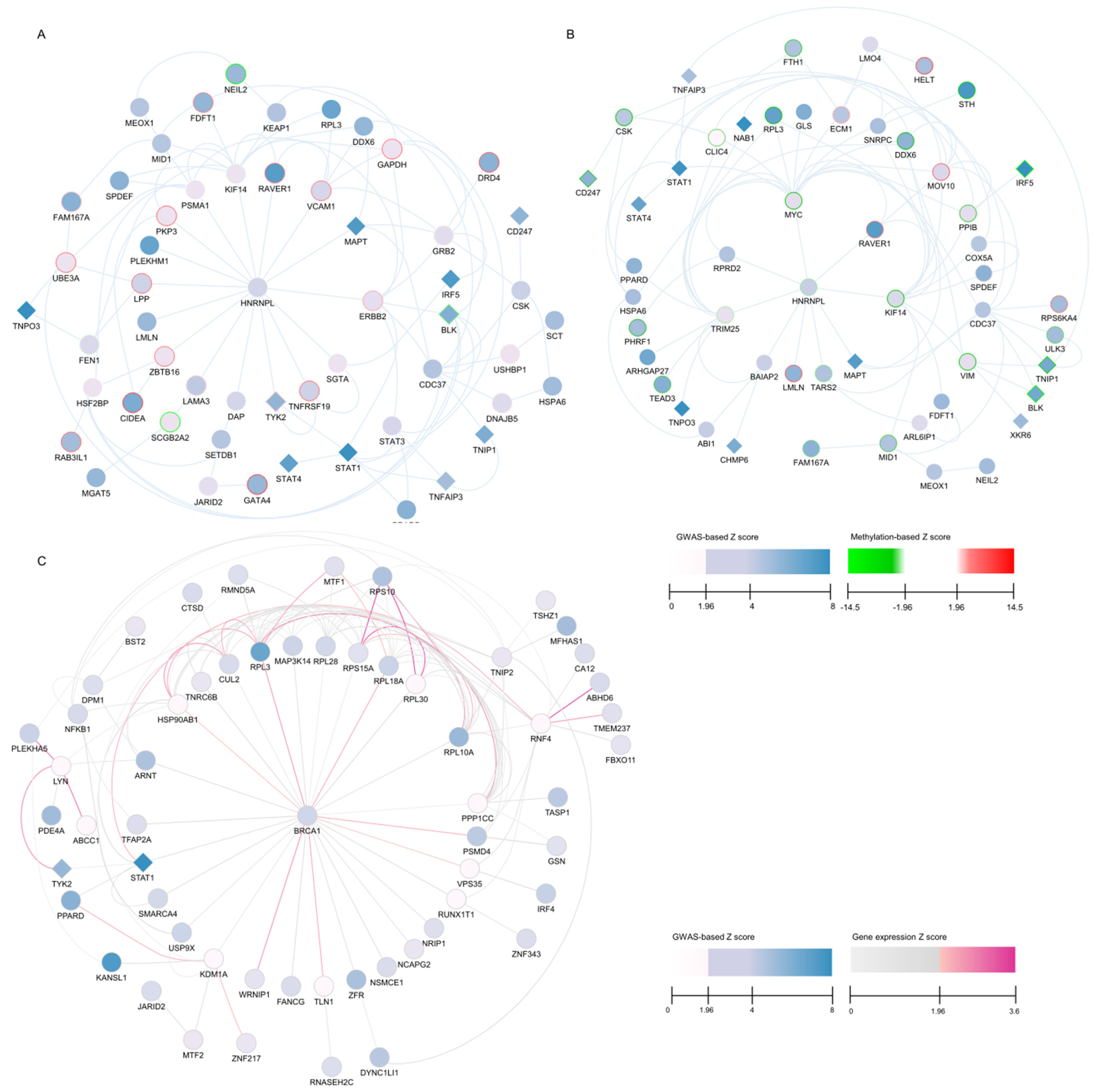
2.1. Identification of SjD-Associated Genes Using dmGWAS
2.2. Autoimmune and Cancer-Related Genes in the Top Gene Modules
2.3. Overlap Between the Discovery and Evaluation Data
2.4. EW_dmGWAS Identified a Surplus of Gene Network Modules
2.5. Selection and Visualization of TMGs from dmGWAS and EW_dmGWAS
2.6. Overlap Between dmGWAS and EW_dmGWAS Findings
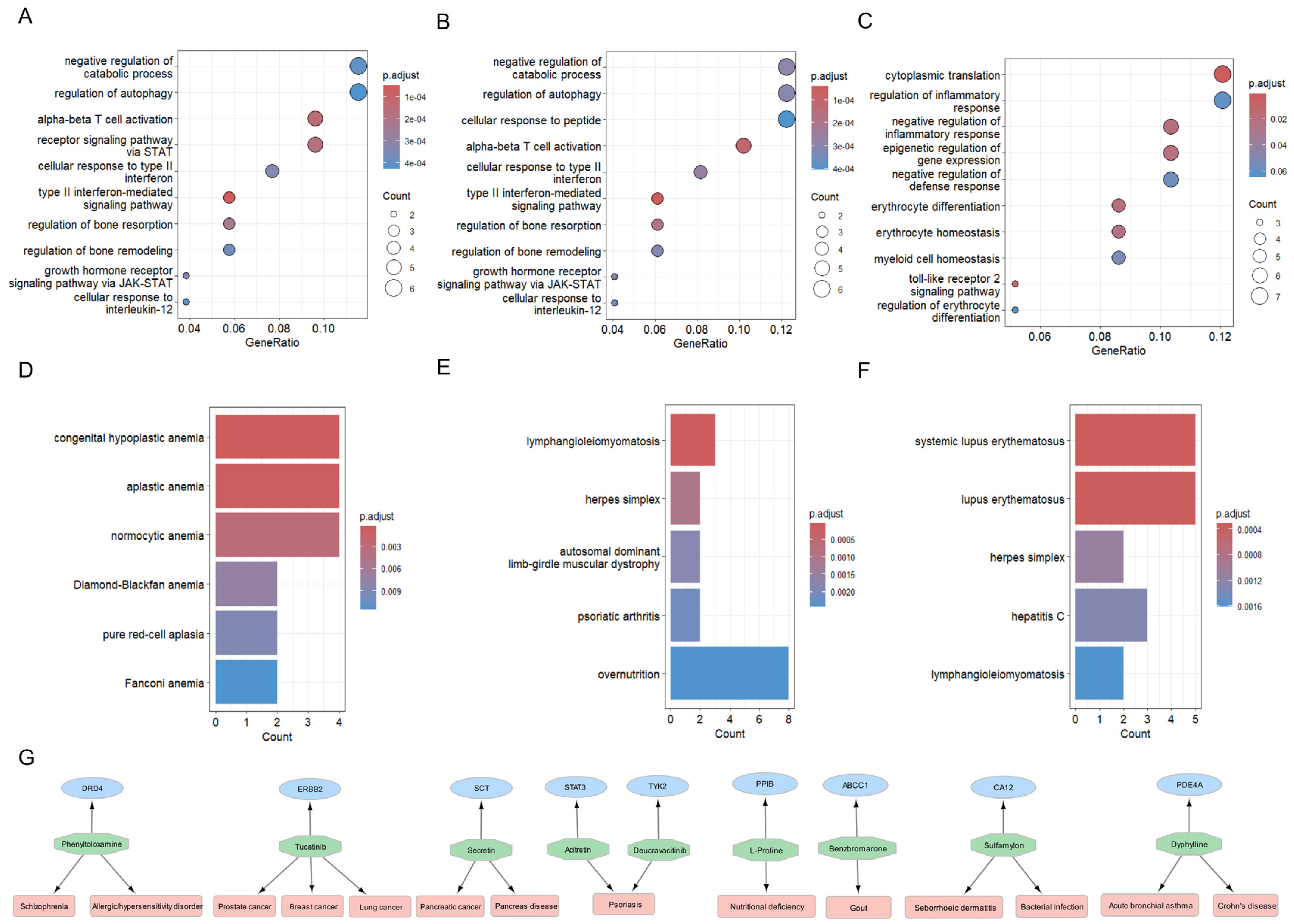
2.7. Exploration of Biological Mechanisms: Gene, Disease, and Cell Specificity Enrichment Analyses
2.8. Proposing Repurposable Drug Candidates for SjD
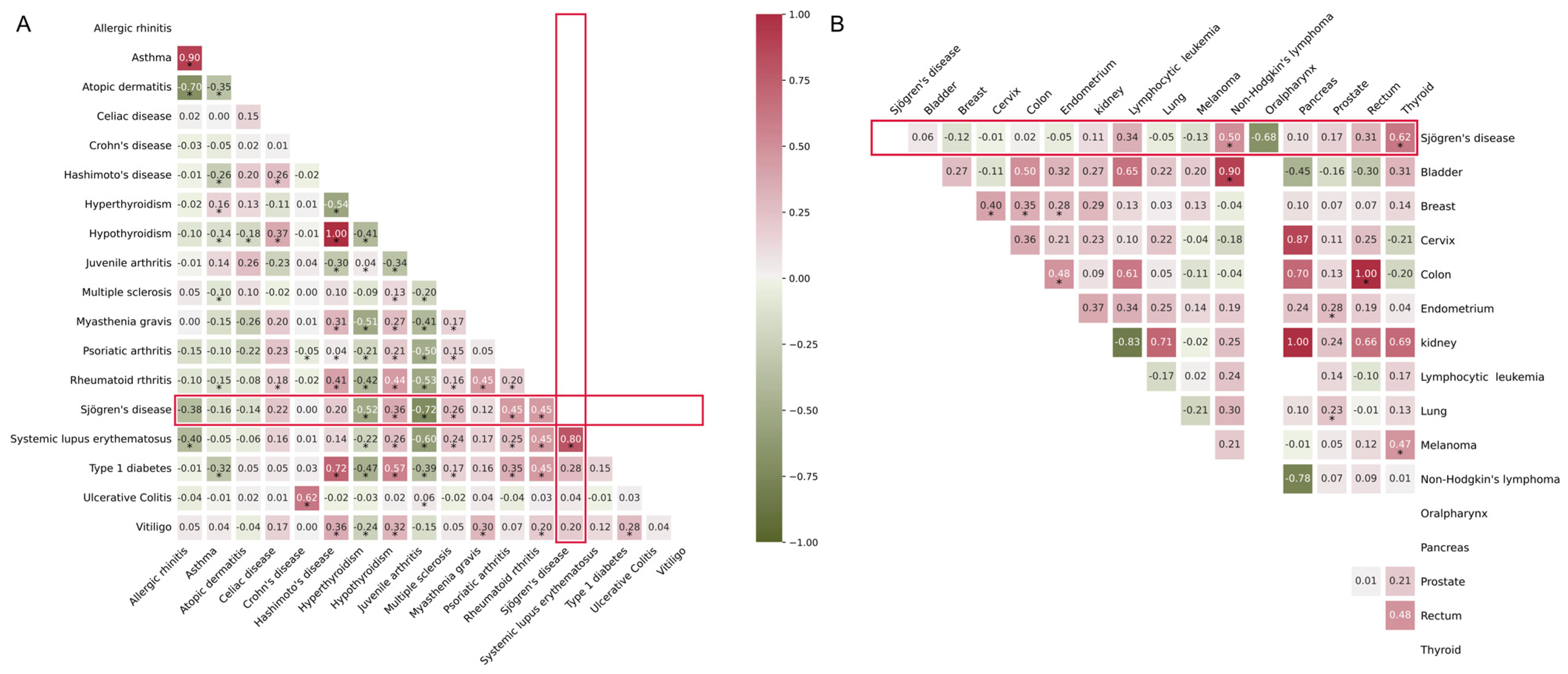
2.9. Genetic Correlation with Cancer and Other AIDs
2.10. Causal Relationship with Cancer and Other AIDs
3. Discussion
4. Materials and Methods
4.1. Multi-Omics Data Overview
4.1.1. Compilation of Sjögren’s Specific Multi-Omics Data
4.1.2. DNA Methylation Data Retrieval
4.1.3. Gene Expression Data and Analysis
4.1.4. Protein–Protein Interaction Network
4.2. Gene Network Analysis
4.2.1. dmGWAS Analysis: Discovery and Evaluation of Integrated Genetic and Epigenomic Data
4.2.2. Dense Module Search on Integrated Genetic and Epigenomic Data
4.2.3. Edge-Weighted Dense Modules Search on Integrated Genetic and Transcriptomic Data
4.2.4. Module Networks Evaluation and Selection of Top Modules
4.3. Functional Enrichment Analysis
4.3.1. Gene Ontology and Disease Enrichment
4.3.2. Cell-Type Specific Enrichment Analysis
4.3.3. Drug Target Enrichment Analysis
4.4. Genetic Correlation Analysis
4.5. Mendelian Randomization Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIDs | Autoimmune diseases |
| SjD | Sjögren’s disease |
| GWAS | Genome-wide association studies |
| dmGWAS | Dense module GWAS |
| EW_dmGWAS | Edge-weighted dmGWAS |
| MSG | Minor salivary gland |
| PPI | Protein–protein interaction |
| TMGs | Top module genes |
References
- Kassan, S.S.; Moutsopoulos, H.M. Clinical manifestations and early diagnosis of Sjogren syndrome. Arch. Intern. Med. 2004, 164, 1275–1284. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Tzioufas, A.G.; Font, J. Primary Sjogren’s syndrome: New clinical and therapeutic concepts. Ann. Rheum. Dis. 2005, 64, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Font, J. Primary Sjogren’s syndrome: Current and emergent aetiopathogenic concepts. Rheumatology 2005, 44, 1354–1367. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, A.I.; Jonsson, R. Genetic aspects of Sjogren’s syndrome. Arthritis Res. 2002, 4, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.F.; Grainge, M.J.; Valdes, A.M.; See, L.C.; Luo, S.F.; Yu, K.H.; Zhang, W.; Doherty, M. Familial Risk of Sjogren’s Syndrome and Co-aggregation of Autoimmune Diseases in Affected Families: A Nationwide Population Study. Arthritis Rheumatol. 2015, 67, 1904–1912. [Google Scholar] [CrossRef]
- Khatri, B.; Tessneer, K.L.; Rasmussen, A.; Aghakhanian, F.; Reksten, T.R.; Adler, A.; Alevizos, I.; Anaya, J.M.; Aqrawi, L.A.; Baecklund, E.; et al. Genome-wide association study identifies Sjogren’s risk loci with functional implications in immune and glandular cells. Nat. Commun. 2022, 13, 4287. [Google Scholar] [CrossRef]
- Thorlacius, G.E.; Bjork, A.; Wahren-Herlenius, M. Genetics and epigenetics of primary Sjogren syndrome: Implications for future therapies. Nat. Rev. Rheumatol. 2023, 19, 288–306. [Google Scholar] [CrossRef]
- Fox, R.I.; Kang, H.I. Pathogenesis of Sjogren’s syndrome. Rheum. Dis. Clin. North. Am. 1992, 18, 517–538. [Google Scholar] [CrossRef]
- Boumba, D.; Skopouli, F.N.; Moutsopoulos, H.M. Cytokine mRNA expression in the labial salivary gland tissues from patients with primary Sjogren’s syndrome. Br. J. Rheumatol. 1995, 34, 326–333. [Google Scholar] [CrossRef]
- Tzeng, S.J.; Kim, I.; Sun, K.H. Editorial: Single-cell analysis on the pathophysiology of autoimmune diseases. Front. Immunol. 2024, 15, 1451354. [Google Scholar] [CrossRef]
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Gunes, O.; Hall, P.; Hayhurst, J.; et al. The NHGRI-EBI GWAS Catalog: Knowledgebase and deposition resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef] [PubMed]
- Bruno, K.A.; Morales-Lara, A.C.; Bittencourt, E.B.; Siddiqui, H.; Bommarito, G.; Patel, J.; Sousou, J.M.; Salomon, G.R.; Paloka, R.; Watford, S.T.; et al. Sex differences in comorbidities associated with Sjogren’s disease. Front. Med. 2022, 9, 958670. [Google Scholar] [CrossRef] [PubMed]
- Hom, G.; Graham, R.R.; Modrek, B.; Taylor, K.E.; Ortmann, W.; Garnier, S.; Lee, A.T.; Chung, S.A.; Ferreira, R.C.; Pant, P.V.; et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N. Engl. J. Med. 2008, 358, 900–909. [Google Scholar] [CrossRef]
- Remmers, E.F.; Plenge, R.M.; Lee, A.T.; Graham, R.R.; Hom, G.; Behrens, T.W.; de Bakker, P.I.; Le, J.M.; Lee, H.S.; Batliwalla, F.; et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N. Engl. J. Med. 2007, 357, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Gateva, V.; Sandling, J.K.; Hom, G.; Taylor, K.E.; Chung, S.A.; Sun, X.; Ortmann, W.; Kosoy, R.; Ferreira, R.C.; Nordmark, G.; et al. A large-scale replication study identifies TNIP1, PRDM1, JAZF1, UHRF1BP1 and IL10 as risk loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1228–1233. [Google Scholar] [CrossRef]
- Gray, P.J., Jr.; Prince, T.; Cheng, J.; Stevenson, M.A.; Calderwood, S.K. Targeting the oncogene and kinome chaperone CDC37. Nat. Rev. Cancer 2008, 8, 491–495. [Google Scholar] [CrossRef]
- Fuller-Pace, F.V. DEAD box RNA helicase functions in cancer. RNA Biol. 2013, 10, 121–132. [Google Scholar] [CrossRef]
- Callari, M.; Sola, M.; Magrin, C.; Rinaldi, A.; Bolis, M.; Paganetti, P.; Colnaghi, L.; Papin, S. Cancer-specific association between Tau (MAPT) and cellular pathways, clinical outcome, and drug response. Sci. Data 2023, 10, 637. [Google Scholar] [CrossRef]
- Largent, A.D.; Lambert, K.; Chiang, K.; Shumlak, N.; Liggitt, D.; Oukka, M.; Torgerson, T.R.; Buckner, J.H.; Allenspach, E.J.; Rawlings, D.J.; et al. Dysregulated IFN-gamma signals promote autoimmunity in STAT1 gain-of-function syndrome. Sci. Transl. Med. 2023, 15, eade7028. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, Y.; Lian, X.; Li, F.; Wang, C.; Zhu, F.; Qiu, Y.; Chen, Y. Therapeutic target database update 2022: Facilitating drug discovery with enriched comparative data of targeted agents. Nucleic Acids Res. 2022, 50, D1398–D1407. [Google Scholar] [CrossRef]
- Kovacic, B.; Stoiber, D.; Moriggl, R.; Weisz, E.; Ott, R.G.; Kreibich, R.; Levy, D.E.; Beug, H.; Freissmuth, M.; Sexl, V. STAT1 acts as a tumor promoter for leukemia development. Cancer Cell 2006, 10, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Z. STAT1 in cancer: Friend or foe? Discov. Med. 2017, 24, 19–29. [Google Scholar]
- Borcherding, D.C.; He, K.; Amin, N.V.; Hirbe, A.C. TYK2 in Cancer Metastases: Genomic and Proteomic Discovery. Cancers 2021, 13, 4171. [Google Scholar] [CrossRef] [PubMed]
- Mine, K.; Nagafuchi, S.; Akazawa, S.; Abiru, N.; Mori, H.; Kurisaki, H.; Shimoda, K.; Yoshikai, Y.; Takahashi, H.; Anzai, K. TYK2 signaling promotes the development of autoreactive CD8(+) cytotoxic T lymphocytes and type 1 diabetes. Nat. Commun. 2024, 15, 1337. [Google Scholar] [CrossRef]
- Yoshinaga, M.; Takeuchi, O. Regulation of inflammatory diseases via the control of mRNA decay. Inflamm. Regen. 2024, 44, 14. [Google Scholar] [CrossRef]
- Pollard, K.M.; Cauvi, D.M.; Toomey, C.B.; Morris, K.V.; Kono, D.H. Interferon-gamma and systemic autoimmunity. Discov. Med. 2013, 16, 123–131. [Google Scholar]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-gamma in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Mangani, D.; Yang, D.; Anderson, A.C. Learning from the nexus of autoimmunity and cancer. Immunity 2023, 56, 256–271. [Google Scholar] [CrossRef]
- Najjar, I.; Fagard, R. STAT1 and pathogens, not a friendly relationship. Biochimie 2010, 92, 425–444. [Google Scholar] [CrossRef]
- Yin, H.; Borghi, M.O.; Delgado-Vega, A.M.; Tincani, A.; Meroni, P.L.; Alarcón-Riquelme, M.E. Association of STAT4 and BLK, but not BANK1 or IRF5, with primary antiphospholipid syndrome. Arthritis Rheum. 2009, 60, 2468–2471. [Google Scholar] [CrossRef]
- Barinotti, A.; Radin, M.; Cecchi, I.; Foddai, S.G.; Rubini, E.; Roccatello, D.; Sciascia, S.; Menegatti, E. Genetic Factors in Antiphospholipid Syndrome: Preliminary Experience with Whole Exome Sequencing. Int. J. Mol. Sci. 2020, 21, 9551. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wang, J.; Zhang, Y.; Yan, Y.; Liu, Y.; Shi, X.; Zhang, Q. Inhibition of DDX6 enhances autophagy and alleviates endoplasmic reticulum stress in Vero cells under PEDV infection. Vet. Microbiol. 2022, 266, 109350. [Google Scholar] [CrossRef]
- Tang, Z.; Shi, H.; Chen, C.; Teng, J.; Dai, J.; Ouyang, X.; Liu, H.; Hu, Q.; Cheng, X.; Ye, J.; et al. Activation of Platelet mTORC2/Akt Pathway by Anti-β2GP1 Antibody Promotes Thrombosis in Antiphospholipid Syndrome. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 1818–1832. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Pu, J.; Wang, Y.; Wu, Z.; Liang, Y.; Song, J.; Pan, S.; Han, F.; Yang, L.; Xu, X.; et al. Tofacitinib in the treatment of primary Sjogren’s syndrome-associated interstitial lung disease: Study protocol for a prospective, randomized, controlled and open-label trial. BMC Pulm. Med. 2023, 23, 473. [Google Scholar] [CrossRef]
- Gandolfo, S.; Ciccia, F. JAK/STAT Pathway Targeting in Primary Sjogren Syndrome. Rheumatol. Immunol. Res. 2022, 3, 95–102. [Google Scholar] [CrossRef]
- Zhong, H.; Liu, S.; Wang, Y.; Xu, D.; Li, M.; Zhao, Y.; Zeng, X. Primary Sjogren’s syndrome is associated with increased risk of malignancies besides lymphoma: A systematic review and meta-analysis. Autoimmun. Rev. 2022, 21, 103084. [Google Scholar] [CrossRef]
- Asherson, R.A.; Fei, H.M.; Staub, H.L.; Khamashta, M.A.; Hughes, G.R.; Fox, R.I. Antiphospholipid antibodies and HLA associations in primary Sjogren’s syndrome. Ann. Rheum. Dis. 1992, 51, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Grygiel-Gorniak, B.; Mazurkiewicz, L. Positive antiphospholipid antibodies: Observation or treatment? J. Thromb. Thrombolysis 2023, 56, 301–314. [Google Scholar] [CrossRef]
- Punnanitinont, A.; Kramer, J.M. Sex-specific differences in primary Sjögren’s disease. Front. Dent. Med. 2023, 4, 1168645. [Google Scholar] [CrossRef]
- Truglia, S.; Capozzi, A.; Mancuso, S.; Manganelli, V.; Rapino, L.; Riitano, G.; Recalchi, S.; Colafrancesco, S.; Ceccarelli, F.; Garofalo, T.; et al. Relationship Between Gender Differences and Clinical Outcome in Patients With the Antiphospholipid Syndrome. Front. Immunol. 2022, 13, 932181. [Google Scholar] [CrossRef]
- Fairweather, D.; Beetler, D.J.; McCabe, E.J.; Lieberman, S.M. Mechanisms underlying sex differences in autoimmunity. J. Clin. Investig. 2024, 134. [Google Scholar] [CrossRef] [PubMed]
- Imgenberg-Kreuz, J.; Rasmussen, A.; Sivils, K.; Nordmark, G. Genetics and epigenetics in primary Sjögren’s syndrome. Rheumatology 2021, 60, 2085–2098. [Google Scholar] [CrossRef]
- Mougeot, J.L.; Noll, B.D.; Bahrani Mougeot, F.K. Sjögren’s syndrome X-chromosome dose effect: An epigenetic perspective. Oral. Dis. 2018, 25, 372–384. [Google Scholar] [CrossRef]
- Harris, V.M.; Sharma, R.; Cavett, J.; Kurien, B.T.; Liu, K.; Koelsch, K.A.; Rasmussen, A.; Radfar, L.; Lewis, D.; Stone, D.U.; et al. Klinefelter’s syndrome (47,XXY) is in excess among men with Sjögren’s syndrome. Clin. Immunol. 2016, 168, 25–29. [Google Scholar] [CrossRef]
- Sharma, R.; Harris, V.M.; Cavett, J.; Kurien, B.T.; Liu, K.; Koelsch, K.A.; Fayaaz, A.; Chaudhari, K.S.; Radfar, L.; Lewis, D.; et al. Brief Report: Rare X Chromosome Abnormalities in Systemic Lupus Erythematosus and Sjögren’s Syndrome. Arthritis Rheumatol. 2017, 69, 2187–2192. [Google Scholar] [CrossRef] [PubMed]
- Cole, M.B.; Quach, H.; Quach, D.; Baker, A.; Taylor, K.E.; Barcellos, L.F.; Criswell, L.A. Epigenetic Signatures of Salivary Gland Inflammation in Sjogren’s Syndrome. Arthritis Rheumatol. 2016, 68, 2936–2944. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Solomon, O.; Shiboski, C.; Taylor, K.E.; Quach, H.; Quach, D.; Barcellos, L.F.; Criswell, L.A. Identification of Sjogren’s syndrome patient subgroups by clustering of labial salivary gland DNA methylation profiles. PLoS ONE 2023, 18, e0281891. [Google Scholar] [CrossRef] [PubMed]
- Oyelakin, A.; Horeth, E.; Song, E.C.; Min, S.; Che, M.; Marzullo, B.; Lessard, C.J.; Rasmussen, A.; Radfar, L.; Scofield, R.H.; et al. Transcriptomic and Network Analysis of Minor Salivary Glands of Patients with Primary Sjogren’s Syndrome. Front. Immunol. 2020, 11, 606268. [Google Scholar] [CrossRef]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F.; et al. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Manuel, A.M.; Dai, Y.; Jia, P.; Freeman, L.A.; Zhao, Z. A gene regulatory network approach harmonizes genetic and epigenetic signals and reveals repurposable drug candidates for multiple sclerosis. Hum. Mol. Genet. 2023, 32, 998–1009. [Google Scholar] [CrossRef]
- Liu, A.; Manuel, A.M.; Dai, Y.; Fernandes, B.S.; Enduru, N.; Jia, P.; Zhao, Z. Identifying candidate genes and drug targets for Alzheimer’s disease by an integrative network approach using genetic and brain region-specific proteomic data. Hum. Mol. Genet. 2022, 31, 3341–3354. [Google Scholar] [CrossRef]
- Jia, P.; Zheng, S.; Long, J.; Zheng, W.; Zhao, Z. dmGWAS: Dense module searching for genome-wide association studies in protein-protein interaction networks. Bioinformatics 2011, 27, 95–102. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef]
- Kim, S.C.; Lee, S.J.; Lee, W.J.; Yum, Y.N.; Kim, J.H.; Sohn, S.; Park, J.H.; Lee, J.; Lim, J.; Kwon, S.W. Stouffer’s test in a large scale simultaneous hypothesis testing. PLoS ONE 2013, 8, e63290. [Google Scholar] [CrossRef]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, H.; Zhao, Z.; Jia, P. EW_dmGWAS: Edge-weighted dense module search for genome-wide association studies and gene expression profiles. Bioinformatics 2015, 31, 2591–2594. [Google Scholar] [CrossRef] [PubMed]
- Khanin, R.; Wit, E. How scale-free are biological networks. J. Comput. Biol. 2006, 13, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Dai, Y.; Hu, R.; Liu, A.; Cho, K.S.; Manuel, A.M.; Li, X.; Dong, X.; Jia, P.; Zhao, Z. WebCSEA: Web-based cell-type-specific enrichment analysis of genes. Nucleic Acids Res. 2022, 50, W782–W790. [Google Scholar] [CrossRef]
- Bulik-Sullivan, B.; Finucane, H.K.; Anttila, V.; Gusev, A.; Day, F.R.; Loh, P.R.; ReproGen Consortium; Psychiatric Genomics Consortium; Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control Consortium 3; Duncan, L.; et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 2015, 47, 1236–1241. [Google Scholar] [CrossRef]
- Sakaue, S.; Kanai, M.; Tanigawa, Y.; Karjalainen, J.; Kurki, M.; Koshiba, S.; Narita, A.; Konuma, T.; Yamamoto, K.; Akiyama, M.; et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat. Genet. 2021, 53, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Rashkin, S.R.; Graff, R.E.; Kachuri, L.; Thai, K.K.; Alexeeff, S.E.; Blatchins, M.A.; Cavazos, T.B.; Corley, D.A.; Emami, N.C.; Hoffman, J.D.; et al. Pan-cancer study detects genetic risk variants and shared genetic basis in two large cohorts. Nat. Commun. 2020, 11, 4423. [Google Scholar] [CrossRef]
- Enduru, N.; Fernandes, B.S.; Bahrami, S.; Dai, Y.; Andreassen, O.A.; Zhao, Z. Genetic overlap between Alzheimer’s disease and immune-mediated diseases: An atlas of shared genetic determinants and biological convergence. Mol. Psychiatry 2024, 29, 2447–2458. [Google Scholar] [CrossRef] [PubMed]
- Enduru, N.; Fernandes, B.S.; Zhao, Z. Dissecting the shared genetic architecture between Alzheimer’s disease and frailty: A cross-trait meta-analyses of genome-wide association studies. Front. Genet. 2024, 15, 1376050. [Google Scholar] [CrossRef] [PubMed]
- Paternoster, L.; Standl, M.; Waage, J.; Baurecht, H.; Hotze, M.; Strachan, D.P.; Curtin, J.A.; Bonnelykke, K.; Tian, C.; Takahashi, A.; et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet. 2015, 47, 1449–1456. [Google Scholar] [CrossRef]
- Dubois, P.C.; Trynka, G.; Franke, L.; Hunt, K.A.; Romanos, J.; Curtotti, A.; Zhernakova, A.; Heap, G.A.; Adany, R.; Aromaa, A.; et al. Multiple common variants for celiac disease influencing immune gene expression. Nat. Genet. 2010, 42, 295–302. [Google Scholar] [CrossRef]
- de Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.G.; et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef]
- Watanabe, K.; Stringer, S.; Frei, O.; Umicevic Mirkov, M.; de Leeuw, C.; Polderman, T.J.C.; van der Sluis, S.; Andreassen, O.A.; Neale, B.M.; Posthuma, D. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet. 2019, 51, 1339–1348. [Google Scholar] [CrossRef]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef]
- Bentham, J.; Morris, D.L.; Graham, D.S.C.; Pinder, C.L.; Tombleson, P.; Behrens, T.W.; Martin, J.; Fairfax, B.P.; Knight, J.C.; Chen, L.; et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 2015, 47, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Andersen, G.; Yorgov, D.; Ferrara, T.M.; Ben, S.; Brownson, K.M.; Holland, P.J.; Birlea, S.A.; Siebert, J.; Hartmann, A.; et al. Genome-wide association studies of autoimmune vitiligo identify 23 new risk loci and highlight key pathways and regulatory variants. Nat. Genet. 2016, 48, 1418–1424. [Google Scholar] [CrossRef] [PubMed]
- Atkins, J.L.; Jylhava, J.; Pedersen, N.L.; Magnusson, P.K.; Lu, Y.; Wang, Y.; Hagg, S.; Melzer, D.; Williams, D.M.; Pilling, L.C. A genome-wide association study of the frailty index highlights brain pathways in ageing. Aging Cell 2021, 20, e13459. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Noche, R.B.; Szejko, N.; Both, C.P.; Acosta, J.N.; Leasure, A.C.; Brown, S.C.; Sheth, K.N.; Gill, T.M.; Zhao, H.; et al. A genome-wide association study of frailty identifies significant genetic correlation with neuropsychiatric, cardiovascular, and inflammation pathways. Geroscience 2023, 45, 2511–2523. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018, 7, e34408. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enduru, N.; Manuel, A.M.; Zhao, Z. Genetic, Transcriptomic, and Epigenomic Insights into Sjögren’s Disease: An Integrative Network Investigation and Immune Diseases Comparison. Int. J. Mol. Sci. 2025, 26, 4637. https://doi.org/10.3390/ijms26104637
Enduru N, Manuel AM, Zhao Z. Genetic, Transcriptomic, and Epigenomic Insights into Sjögren’s Disease: An Integrative Network Investigation and Immune Diseases Comparison. International Journal of Molecular Sciences. 2025; 26(10):4637. https://doi.org/10.3390/ijms26104637
Chicago/Turabian StyleEnduru, Nitesh, Astrid M. Manuel, and Zhongming Zhao. 2025. "Genetic, Transcriptomic, and Epigenomic Insights into Sjögren’s Disease: An Integrative Network Investigation and Immune Diseases Comparison" International Journal of Molecular Sciences 26, no. 10: 4637. https://doi.org/10.3390/ijms26104637
APA StyleEnduru, N., Manuel, A. M., & Zhao, Z. (2025). Genetic, Transcriptomic, and Epigenomic Insights into Sjögren’s Disease: An Integrative Network Investigation and Immune Diseases Comparison. International Journal of Molecular Sciences, 26(10), 4637. https://doi.org/10.3390/ijms26104637